Abstract
Previously, we showed that in utero exposure to mercury induced phenotypic changes in fetal immune cells. Here, we sought to determine whether the effects of in utero exposure on immune cells persisted in the adult. After overnight breeding to DBA/1 males, pregnant BALB/c dams were given either mercuric chloride in drinking water at 10 mg/L ad libitum for the duration of gestation or plain water. At the time of parturition, all dams were placed on regular drinking water. The pups (DBF1) were weaned and thymic and splenic tissues were harvested at 10 wk-of-age to assess T-cell phenotypes and function. Significant changes in the CD4/CD8 subsets in the thymus and spleen among mercury-exposed male and female mice were not observed. However, there was a significant reduction in splenic CD4+CD25+ cells in mercury-exposed female, but not in male, mice. ConA-stimulated splenocytes from mercury-exposed mice showed significant increases in proliferative responses relative to cells from control mice, regardless of sex. Cytokine secretion was also modulated in the mercury-exposed mice. In particular, the production of IL-4 and IFN by ConA-stimulated splenocytes from mercury-exposed male and female mice was significantly increased, while IL-2 and IL-10 levels were unaffected. The results of our study revealed that exposure of the developing immune system to relatively low levels of inorganic mercury could lead to persistent alterations in adult immune cell phenotypes and functions. These changes could pose a relevant health risk, if they contribute to impaired responses to pathogens and/or an increased risk for the development of atopy, asthma or possibly autoimmune diseases.
Keywords::
Introduction
It has been proposed that perturbations during the sensitive period of ontological development may underlie the unique susceptibility of the fetal immune system to chemical- induced toxicities. Developmental immunotoxicologists have long speculated that xenobiotic exposure in utero could have consequential effects in immune phenotypes and function in the offspring (Holladay and Smialowicz, Citation2000; Bunn et al., Citation2001a; Dietert et al., Citation2002; Holsapple, Citation2003). The persistence of the immunotoxic effects of lead, for instance, has been demonstrated across several in vivo animal systems (Bunn et al., Citation2000, Citation2001a, Citation2001b). Gestational exposure to other environmental xenobiotics with known immunotoxic effects, such as dioxins, has shown even more profound consequences on offspring immune function, including the suppression of hypersensitivity responses (Gehrs and Smialowicz, Citation1997) and impairment of cellular and humoral responses to viral infection (Vorderstrasse et al., Citation2006).
The persistent low-level exposure of the general population to environmental xenobiotics and contaminants, including heavy metals such as mercury, presents a potential and continuing public health problem as a result of ingestion of contaminated water or mercury-tainted fish (Koos and Longo, Citation1976; Davidson et al., Citation1998; Axtell et al., Citation2000; Risher et al., Citation2002; Myers et al., Citation2003). However, the health risks involved in low-level mercury exposure remain largely confined to its effects on neurodevelopment despite its known toxicity to the immune system as well (Axtell et al., Citation2000; Myers et al., Citation2003). In fact, it has been suggested that immunotoxicity testing may constitute a more sensitive indicator of low-level perinatal mercury exposure (Dietert et al., Citation2002; Luebke et al., 2004; Dietert and Piepenbrink, Citation2006). The immunotoxic effects of mercury are determined by a number of factors, including the dose and mercury species as well as the route and timing of exposure, with the intrauterine period especially sensitive to disruption of critical events during immune development. It is possible that modulations of the immune phenotypes that occur in utero could persist, leading to altered immune functions in the adult. Indeed, there has been evidence to suggest that suppression of abnormal immune responses (i.e., hypersensitivity) may occur in adulthood as a result of disrupted immune development (Gehrs and Smialowicz, Citation1997). Alternatively, others have suggested that environmental xenobiotics can induce suppression of cellular and humoral responses during viral infection (Vorderstrasse et al., Citation2006).
A recent 4-year survey has shown that as many as 5% of women of childbearing age had blood mercury levels above the oral reference dose (Rfd) of 5.8 μg/L, putting at least 1 in every 6 children born at significant risk for mercury-related toxicity as a result of in utero exposure (Schober et al., Citation2003). However, data concerning the long-term risks of exposure in humans is limited and inconsistent. While Bilrha and co-workers (2003) found no differences in the expression of activation markers in cord blood lymphocytes from exposed neonates, others have reported immune alterations that included reductions in the numbers of naïve CD4+ cells and decreased mitogen-induced proliferative responses (Belles-Isles et al., Citation2002). On the other hand, data in animal models have demonstrated more consistently the adverse effects associated with gestational exposure to mercury. For example, injection of pregnant BALB/c mice with mercuric chloride resulted in modulation of cytokine production in adult offspring in a sex-dependent manner, strongly suggesting that immune dysfunction after in utero mercury exposure persisted long after termination of exposure (Silva et al., Citation2005). Further, persistent effects of gestational exposures to other known immunomodulating xenobiotics, including lead (Miller et al., 1998) and TCDD (Vordestrasse et al., Citation2006), have also been reported. Based on these observations, it is very likely that mercury, which has well-described immunomodulating effects and can induce an autoimmune-like condition in genetically susceptible adult mice (Hultman et al., Citation1992, Citation1993; Pollard and Hultman, Citation1997), could also lead to similar effects.
We have recently reported that prenatal exposure to 10 ppm mercuric chloride resulted in acute changes in immune phenotypes in the gestational day 16 fetus, that included increased numbers of double positive thymocytes, fetal hepatic B-cells, and expression of a known biomarker of autoimmune disease (Pilones et al., Citation2007). In this study, we used a similar regimen of exposure to mercuric chloride as in the prenatal study, so that pregnant dams received 10 ppm mercury in drinking water. This exposure level was recently shown to modulate immune phenotypes in adult BALB/c mice (Kim et al., Citation2003) and also induced autoimmunity when injected in H-2s mice (Hultman et al., Citation1995; Hultman and Nielsen, Citation2001). In the present study, we have extended our investigations to include tests of immune function and histological characterization of kidney sections that may indicate acceleration of autoimmune nephritis. Notably, we found that prenatal exposure to mercuric chloride resulted in a significant increase in the numbers of splenic CD4+ T-cells reactive with the IdLNF1 idiotype, which is a biomarker of autoimmunity in the (SWR × NZB)F1 (SNF1) model for lupus nephritis. Immune functions were also modified as a result of prenatal exposure, and included increased proliferation to mitogen and increased secretion of interleukin (IL)-4 and interferon (IFN)-γ. The consequences of prenatal mercury exposure on the immune function of the adult offspring, particularly its possible role as an environmental trigger for postnatal immune dysfunction, are discussed.
Materials and methods
Animals
Eight-week-old female BALB/c and male DBA/1 mice were obtained from The Jackson Labs (Bar Harbor, ME) and housed separately under standard conditions of temperature, humidity and 12-hr light/dark cycle. Harems consisted of three females for each male and pregnant dams were housed individually. Mice were treated in accordance with animal use and care guidelines set by the Cornell University Center for Animal Resources and Education (CARE).
Experimental design
Pregnant dams were identified by the presence of vaginal plugs and arbitrarily assigned to either a control or mercury treatment group. Starting at gestation day 0 (gd 0, the day after overnight breeding), the mercury-treated group received 10 mg mercuric chloride (HgCl2)/L in drinking water while control dams received plain water, both supplied ad libitum. In the course of the studies, there were no differences noted in the consumption by the mice of the mercury-treated water as compared to plain water. To monitor for daily imbibement levels, residual water in each cage water bottle was measured at the time of daily bottle exchange; the results indicated that ≈ 5 ml/day/mouse was consumed in both the mercury-treated and control groups. At parturition, the dams were shifted to plain water. DBF1 offspring mice were weaned at Day 21 and segregated by sex. All mice were observed at least once daily; no mortalities or morbidities were noted prior to sacrifice. At 10 weeks-of-age, mice were individually examined for immunophenotype and function of thymus and spleen cells. The experiment was replicated three times yielding a total of 26 control mice (11 males, 15 females) and 23 mercury-exposed mice (11 males, 12 females) ().
Table 1. Litter statistics from 3 independent timed-breed experiments to evaluate the persistent immune effects of gestational exposure to 10 ppm HgCl2.
Reagents
Mercuric chloride (HgCl2, Sigma, St. Louis, MO) at 10 mg/L (ppm) was prepared in solution using endotoxin-free water. RPMI 1640 media, 200 mM L-glutamine, fetal bovine serum (FBS), and ammonium chloride (NH4Cl) were obtained from Sigma. Non-essential amino acids supplement was obtained from Cellgro (Mediatech, Manassas, VA).
Preparation of single cell suspensions
Thymus and spleen from individual mice were aseptically dissected and placed in cold complete media (RPMI 1640 media supplemented with 10% FBS, 2 mM β-mercaptoethanol, 1 mM L-glutamine, and 0.1 M non-essential amino acids). Cell suspensions were prepared and erythrocytes were lysed with 0.2 M Tris-buffered NH4Cl solution. The remaining cells were then washed, resuspended in complete media, and viable cells were then counted by trypan blue exclusion on a hemocytometer.
Immunophenotyping
Flow cytometry was used to identify lymphocyte populations in the isolated thymus or spleen from each mouse. Briefly, cells were washed in phosphate-buffered saline (PBS) buffer containing 0.5% bovine serum albumin (BSA) and 0.1% NaN3, and re-suspended in aliquots of 2 × 105 cells. The cells were subsequently stained with fluorochrome- or biotin-conjugated monoclonal antibodies at optimum concentrations. After a 20-min incubation in the dark, the stained cells were washed in PBS and incubated with APC Cy7 conjugated to streptavidin for 20 min. After a final washing, the cells were fixed with 1% paraformaldehyde and stored at 4°C until analysis. The following monoclonal antibodies were obtained from BD Pharmingen (San Diego, CA): PE,FITC- or APC-conjugated rat anti-mouse CD8; Biotin-conjugated rat anti-mouse CD25; PeCy7-conjugated rat anti-mouse CD3; APC Cy7-,PE- or APC-conjugated rat anti-mouse CD4; APC-conjugated rat anti-mouse CD44; PE-conjugated rat anti-mouse CD45RB, and Biotin-conjugated rat anti-mouse B220. FITC-conjugated rabbit anti-mouse IdLNF1 was prepared as previously described (Knupp et al., Citation1995). Twenty thousand cells/samples were acquired using a FACS LSR II Flow Cytometer (Becton Dickinson, Mountain View, CA). The lymphocytic gate was drawn based on plot of forward versus side angle light scatter. Negative and positive Ig isotype controls were used in each run to set the analysis gates and quadrants. For quantitative analysis of specific lymphocyte populations, WinMDI software was used.
Cell cultures
Splenic cells were suspended in complete media to a final concentration of 107 cells/ml and 100 μl of cell suspensions were seeded per well in 24-well culture plates (Costar, Corning, NY). The cells were stimulated with the following: Concanavalin-A (ConA, Sigma) at a final concentration of 2 μg/ml (per well), lipopolysaccharide (LPS) at 20 μg/ml, or idiopeptide (aa62-73) at 15 μg/ml, and then cultured at 37°C in an atmosphere of 5% CO2. The idiopeptide (KFKGKATLTSDK) was derived from the heavy chain variable region of an IdLNF1 autoantibody (Dudek, Citation1996). After 72 hr incubation ( 24 hr for IFN-γ), well cultured supernatants were collected and frozen at -70°C until assayed. Assays for each mouse were performed in duplicate.
Proliferation assay
Splenic lymphocytes obtained from water or mercury- exposed mice were tested for their ability to proliferate after mitogen stimulation in vitro using the Promega CellTiter96® Non-Radioactive Cell Proliferation Assay Kit (Promega, Madison, WI), as per manufacturer’s instructions. Briefly, 5 × 105 cells/well were cultured in triplicate in complete media in a 96-well flat-bottomed cell culture plate (Costar). ConA (Sigma) was added at 2 μg/ml and LPS at 20 μg/ml. Response to idiopeptide 62-73 (15 μg/ml), to detect functional responses of pathogenic IdLNF1-reactive phenotypes (Knupp et al., Citation1993), was also tested. After a 72-hr incubation at 37°C in an atmosphere of 5% CO2, 15 μl of the MTT dye solution was added to each well, after which the plates were re-incubated for 4 hr, followed by the addition of 50 μl of the stop solution. After 1– 4 hr of additional incubation to allow complete solubilization of the formazan crystals, any absorbance in each well was read at 600 nm using a BioRad Model 2550 plate reader (BioRad, Hercules, CA). The data was expressed as an index of proliferation (PI), calculated as follows:
ELISA for cytokine production
Cytokine levels in the culture supernatants were measured using a direct binding ELISA. Briefly, 96-well NUNC-Immuno® Maxi-Sorp plates (Nalgene, Rochester, NY) were coated with 50 μl of purified rat anti-mouse IL-2, IL-4, or IFNγ (Pharmingen, Carlsbad, CA) in 0.1 M carbonate coating buffer (0.2 M phosphate buffer [pH 6.5] for IL-10) overnight at 4°C. The plates were washed twice with 100 μl of PBS/0.05% Tween-20 (Tw) and subsequently blocked with 50 μl of PBS/1% FBS for 1 hr at room temperature. After a second wash in 0.05% PBS-Tw, 50 μl of each culture supernatant was dispensed in triplicate. Standards were prepared and plated in the following range of concentrations: 2– 200 pg/ml (IL-2), 4–500 pg/ml (IL-4), 8–1000 pg/ml (IFNγ), and 15-2000 pg/ml (IL-10) in PBS/10% FBS. The plates were incubated overnight at 4°C. They were then washed five times in PBS/0.5% Tw and 50 μl of either biotinylated anti-mouse IL-2, -4, -10 or IFNγ (Pharmingen) (diluted 1:500 in assay diluent) was added to each well. After another overnight incubation at 4°C, the plates were washed 5 times and then 50 μl of avidin-horseradish peroxidase (HRP) diluted 1:1000 in PBS/1% FBS was added to the wells, and incubated for 30 min at room temperature.
Finally, bound enzyme was visualized by the addition of 50 μl 3,3’,5,5’-tetramethylbenzidine (TMB) substrate (Sigma) per well. After 15–30 min, the reaction was stopped by the addition of 50 μl of 1.0 M H3PO4. Absorbance at 450 nm was then assessed using an ELISA plate reader (BioRad). Cytokine protein levels in the culture supernatant, expressed in picogram per milliliter (pg/ml), were extrapolated based on the linear region of the standard curve. The correlation coefficient for each standard curve was computed and only those assays with values greater than 0.95 were included in the data analysis. Samples with concentrations below the limits of detection of the assay were reported as the lowest concentration detectable on the standard curve multiplied by the dilution factor. The intra-assay coefficient of variation ranged between 4.7% and 13.4%.
Statistics
Data were expressed as mean ± SD, and experimental groups were compared to sex-matched control groups. Results were analyzed using one-way Analysis of Variance with Bonferroni post-test analysis (GraphPad InStat) and significance set at p < 0.05.
Results
Prenatal exposure to mercuric chloride did not modulate body weights at 10 weeks
Consistent with previously published data (Pilones et al., Citation2007), exposure to 10 ppm HgCl2 did not induce significant toxicity to mercury-exposed dams, since gestational parameters were comparable to those in unexposed mice (). From three separate timed-breeding experiments, we obtained 26 unexposed pups (11 males, 15 females) and 23 mercury-exposed pups (11 males, 12 females). No fetal deaths were seen and all offspring remained viable until harvest at 10 wk. For each set of experiments, body weights in mercury-exposed mice were comparable to those of the unexposed controls, ().
Prenatal exposure to mercuric chloride did not modulate adult thymic or splenic cellularity
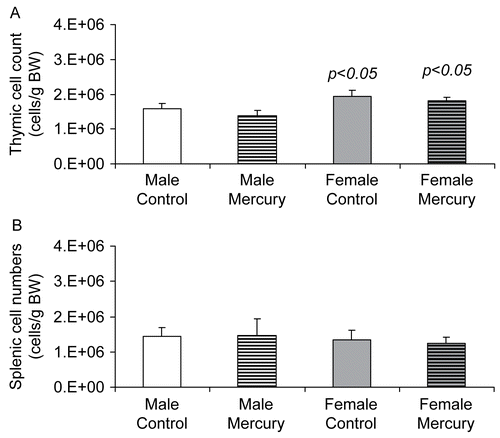
We evaluated whether in utero mercuric chloride exposure altered thymic cellularity in DBF1 mice at 10 weeks-of-age. Total thymic cell counts, adjusted to body weights, were compared for mercury-exposed mice to those from sex-matched unexposed controls. Among unexposed mice, thymus cell numbers were significantly higher in female mice compared to male mice (p = 0.02, ). This finding was also seen in the mercury-exposed group, where female mice had significantly higher adjusted thymic cells counts (p = 0.02). However, the average number of thymus cells in mercury-exposed mice was similar to gender-matched unexposed controls. There also were no significant differences in relative splenic cellularity among mercury-exposed mice when compared to unexposed controls for both males and females ().
Figure 1. The effects of prenatal HgCl2 exposure on thymic and splenic cellularity. Cell numbers in the (A) thymus and (B) spleen of 10-wk-old DBF1 mice exposed to plain water (control) or 10 ppm mercuric chloride during gestation were enumerated. Data were collected from six control mice (3 female and 3 male DBF1 mice) and 12 mercury-exposed mice (6 female and 6 male DBF1 mice). The differences between mercury-exposed mice and sex-matched control mice were considered significant when p < 0.05.
Prenatal mercury exposure did not modulate immune cell phenotypes in the thymus
We next evaluated whether exposure to mercury in utero modulated CD4+ and CD8+ expressing subsets in the thymus. For control mice, there were no statistically significant differences among the subsets examined between male and female mice. This was true for mercury-exposed mice as well when males were compared to females. We found that exposure to mercury did not affect the percentage composition in any of the T-cell subsets in the thymus in either male or female mice (). The CD4:CD8 ratios remained comparable for both mercury-exposed and unexposed mice.
Table 2. The effect of prenatal mercuric chloride exposure on CD4/CD8-expressing cells in the thymus.
Prenatal mercury exposure decreased the numbers of splenic CD4+CD25+ cells in female DBF1 mice
We found that mercury exposure resulted in a significant reduction in the total numbers of splenic CD4+ cells co-expressing CD25+ in female mice (). On the other hand, mercury-exposed male mice showed a tendency towards increased numbers of these cells, although this increase was not statistically significant.
Table 3. The effect of prenatal mercuric chloride exposure on CD4/25-expressing cells in the thymus.
Prenatal mercury exposure modulated in vitro proliferative responses
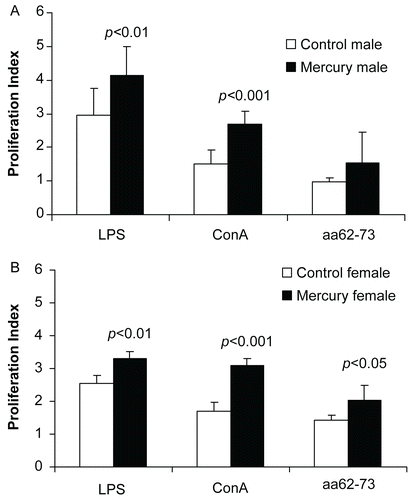
The effects of gestational mercury exposure on the proliferation of adult splenocytes to the T-cell mitogen ConA, the B-cell mitogen LPS, and idiopeptide aa62-73 were measured. We found that splenocytes from mercury-exposed mice showed significantly higher proliferation indices in response to Con-A compared to those of cells from gender-matched control mice in both males () and females (). Proliferative responses to LPS were also significantly increased in cells from mercury- exposed males () and females (). Proliferative responses to idiotypic peptide aa62-73 were significantly increased in cells from mercury-exposed females (), but not in those from mercury-exposed males ().
Figure 2. Prenatal HgCl2 exposure leads to increased proliferative responses in lymphocytes. Splenic lymphocytes (5 × 105 cells) from mercury-exposed and unexposed (A) male and (B) female mice were stimulated with ConA (2 μg/ml), LPS (15 μg/ml) or idiopeptide (15 μg/ml) for 72 hr. Data were expressed as the proliferation index, computed as the ratio of the proliferative response of stimulated cells (measured as absorbance) to that of the unstimulated cells. Data were collected from 18 control mice (9 female and 9 male DBF1 mice) and 23 mercury-exposed mice (12 female and 11 male DBF1 mice) and shown as mean ± SD. The differences between mercury-exposed mice and sex-matched unexposed mice were considered significant when p < 0.05.
Effects of prenatal mercury exposure on TH1 cytokine production
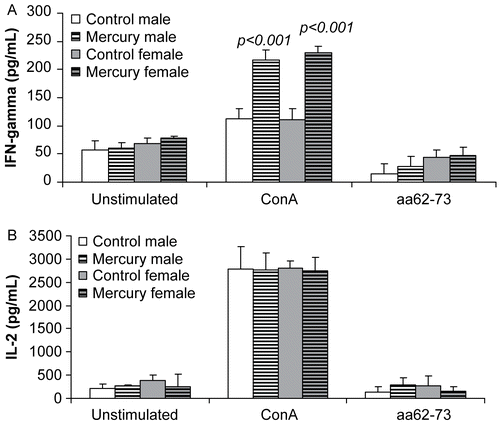
The effects of prenatal mercury exposure on the production of TH1 cytokines were also measured. We found that in vitro production of IFNγ was significantly increased in Con-A stimulated splenocytes from mercury-exposed mice (), regardless of sex. Levels of IL-2, on the other hand, were not affected by mercury exposure ().
Figure 3. The effects of prenatal mercury exposure on the production of TH1 cytokines. In vitro production of (A) IFNγ and (B) IL-2 by lymphocytes (106 cells/well) from mercury-exposed and unexposed mice that were stimulated with ConA (2 μg/ml) or idiopeptide (15 μg/ml) was measured after 24 hr (IFNγ) or 72 hr (IL-2). Data were collected from 18 control mice (9 female and 9 male DBF1 mice) and 23 mercury-exposed mice (12 female and 11 male DBF1 mice) and shown as mean ± SD. The differences between mercury-exposed mice and sex-matched unexposed mice were considered significant when p < 0.05.
Effects of prenatal mercury exposure on TH2 cytokine production
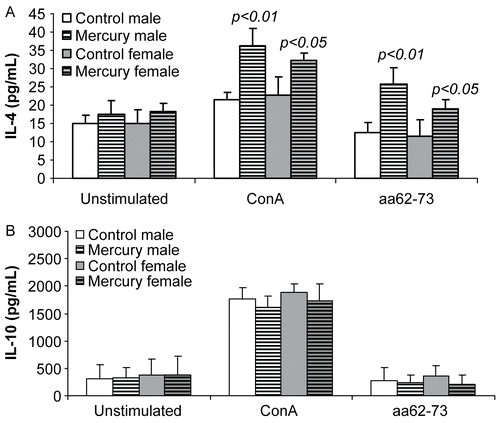
In vitro production of TH2 cytokines was determined by ELISA. We found that in unstimulated cultures, the production of IL-4 or IL-10 was comparable in both mercury- exposed and unexposed mice. However, with Con-A stimulation, the production of IL-4 in mercury-exposed mice was increased significantly compared to unexposed sex-matched control mice (). The production of IL-10, while also up-regulated after Con-A stimulation, remained comparable regardless of exposure (). Thus, sex differences did not appear to play a role in modulating TH2 cytokine production, since sex differences were not seen.
Figure 4. The effects of prenatal mercury exposure on the production of TH2 cytokines. In vitro production of (A) IL-4 and (B) IL-10 by lymphocytes (106 cells/well) from mercury-exposed and unexposed mice after stimulation with ConA (2 μg/ml) or idiopeptide (15 μg/ml) was measured after 72 hr. Data were collected from 18 control mice (9 female and 9 male DBF1 mice) and 23 mercury-exposed mice (12 female and 11 male DBF1 mice) and shown as mean ± SD. The differences between mercury- exposed mice and sex-matched unexposed mice were considered significant when p < 0.05.
Effects of prenatal exposure to mercury on autoimmune-associated immune responses and clinical disease
In this study, we found that mercury exposure did not affect the IdLNF1 reactive CD4+ T-cell population in the thymus of mice of either gender (). However, splenic populations of IdLNF1-reactive CD4+ T-cells were significantly higher in mercury-exposed female, but not male, DBF1 mice, compared to controls (). The numbers of IdLNF1-reactive B-cells were not affected by mercury exposure in utero (data not shown). In previous work, we have found that pathogenic IdLNF1-reactive T-cell clones proliferated in the presence of the idiopeptide aa62-73 and vaccination of pre-nephritic SNF1 mice with the IdLNF1 peptide resulted in a decrease in the proliferation of T-cells to aa62-73, decreased levels of IdLNF1-Ig and IgG, and significantly delayed the onset and reduced severity of nephritis, thereby improving survival rates (Knupp et al., Citation1993; Uner et al., Citation1994). In the present study, mercury exposure also correlated with an increase in the number of IdLNF1-reactive CD4+ T-cells and increased proliferation in splenic lymphocytes in female mice in response to idiopeptide aa62-73 (). Increased proliferation to aa62-73 was also observed in cells from male mercury-exposed mice but the levels did not achieve statistical significance ().
Table 4. The effect of prenatal mercuric chloride exposure on IdLNF1-reactivity in splenic CD4+ T-cells.
In SNF1 mice and estrogen-treated DBF1 mice, autoimmune nephritic disease may be accompanied by increasing levels of proteinuria. In this study, we tested whether early developmental exposure in DBF1 mice would induce postnatal nephritic disease. We found that prenatal mercury exposure had not compromised renal function at 10 wk-of-age, since protein levels in the urine, although increased in some of the mercury-exposed mice, were not significantly different from levels in controls and histopathological examination of kidneys showed no evidence of glomerulonephritis in mercury-exposed mice (data not shown).
Discussion
Mercury is a well-known immunotoxicant that can induce both suppression and stimulation of the immune system. Immunomodulation due to exposure to mercury is influenced by several factors that include the chemical form of mercury, the exposure level, and the route of exposure. The timing of the exposure is also relevant, as it is speculated that exposure during the prenatal period, a critical window of development, could significantly alter postnatal immune responses even at concentrations too low to have any immunological consequence in adults.
In a recent study, we found that phenotypic modulations occurred as a result of intra-uterine exposure to a relatively low concentration of mercury (Pilones et al., Citation2007). Additional tests of immune functions, however, could not be properly assessed since these do not achieve “adult levels” until after birth. To effectively determine whether gestational exposure to mercury can lead to long-term effects in the pups, pregnant dams were exposed to a similar dose of mercury throughout gestation, but were shifted to plain drinking water as soon as their pups were born. Whereas the prior study looked at the fetal thymus and liver, the current study investigated changes in immune phenotypes and function when the gestationally exposed mice reached 10 weeks of age.
In the present study, we found that developmental exposure to mercury had persistent effects on the immune cell phenotypes and functions in the adult offspring. While low-level mercury exposure did not induce thymic atrophy, immune phenotypes in the thymus and spleen were modulated, and included significant reductions in the numbers of splenic CD4+CD25+ cells, a subset that has been identified phenotypically with naturally occurring regulatory T-cells (Tregs; Apostolou et al., Citation2002). The decrease in the numbers of CD4+CD25+ T-cells in mice exposed to mercury in utero, which could be detected as early as gestational day 16 (Pilones et al., Citation2007), was found to persist in adult mice. This finding provides compelling evidence that some cell phenotypes may be more sensitive to the immunotoxic effects of mercury exposure in utero which may have important consequences in determining adult immune responses. Specifically, since CD4+CD25+ subsets have been strongly implicated in the maintenance of peripheral tolerance, the persistent down-modulation of CD4+CD25+ T-cells in the adult offspring may potentially increase the risk of autoimmune disease.
Tregs have been shown to be key players in maintaining peripheral tolerance in T-cell-mediated autoimmune models such as experimental autoimmune encephalomyelitis (EAE) and spontaneous diabetes in transgenic mice (reviewed in Wraith et al., Citation2004). Further, emerging data chiefly from the work of Kosuda and co-workers (Kosuda et al., Citation1991) have given support for modulations in Treg function in the pathogenesis of metal-induced autoimmune diseases. In susceptible Brown–Norway (BN) rats treated with mercury, depletion of immunoregulatory RT6+ T-lymphocytes was found to coincide with the appearance of autoantibodies in the serum and immune complex glomerulonephritis (Kosuda et al., Citation1991, Citation1993, Citation1994, Citation1998). Further, in H-2S mice, the adoptive transfer of CD4+CD25+ T-cell fractions abrogated the formation of anti-nuclear antibodies in mercury- treated H-2s mice (Layland et al., Citation2004). These findings and the results of our study suggest that prenatal mercury exposure may have long-lasting inhibitory effects on regulatory networks that could possibly enhance autoimmunity.
Our data also found that female, but not male, mice were preferentially affected, suggesting that mercury-associated phenotypic changes may show gender differences. This was also noted in another recent study that found TH2 cytokines were increased in mercury-exposed females in contrast to a decrease in males (Silva et al., Citation2005). Although the basis of such divergent effects between the sexes is unclear, there are data to support the possibility that metal xenobiotics may interact directly with endogenous maternally derived steroid hormones to prevent activation or bind to steroid hormone receptors, resulting in activation of downstream pathways. Supporting evidence for the latter has been derived from in vitro studies demonstrating estrogen-like effects of mercury, suggesting that metals may comprise an emerging group of environmental pollutants with endocrine-like activities (Martin et al., Citation2003). Our data showing sex differences in modulations of immune phenotypes in females suggests the possibility that estrogen levels may be critical during the rapid cellular cycling in fetal lymphopoiesis. Since sex differences were not seen in the functional immune assays, we speculate that estrogenic influence involved during these developmental events might not be as profound.
On the other hand, we previously reported comparable trends in the modulations of immune phenotypes in the gestational day 16 fetus prenatally exposed to mercury, regardless of fetal sex (Pilones et al., Citation2007). It is therefore likely that while prenatal exposure to mercury can modulate postnatal immune phenotypes and function, the final outcome is significantly impacted by endogenous factors, including sex hormones. It remains speculative whether the phenotypic changes could be mediated through direct action of mercury on estrogen receptors on immune cells.
We found that in vivo perinatal exposure to HgCl2 led to significantly increased proliferative responses to ConA by the cells of both male and female adult mice. This suggested that early exposure to mercury may prime peripheral lymphocytes to proliferate, and is consistent with other observations of mercury-associated T-cell hyper-reactivity (Pollard and Landberg, Citation2001). In contrast, Silva and co-workers did not find changes in proliferative responses to ConA-stimulated splenocytes from 10-, 14-, or 21-day-old mice that were exposed to mercuric chloride in utero (Silva et al., Citation2005). We suspect that the stimulatory effects of mercury in enhancing splenocyte proliferation found in our study might be a latent effect. Alternatively, the lack of definitive changes in ConA responsiveness as reported by Silva et al. could be related to the functional immaturity of immune cells in pre-weaned pups, since ConA responsiveness does not typically achieve adult levels until after weaning (Mosier, Citation1977), and in the present study, proliferation studies were conducted in adult mice. Furthermore, we cannot discount strain differences that could explain the differential responses. While the Silbergeld group used BALB/c dams of the H-2d haplotype (Silva et al., Citation2005), we used a hybrid strain that included haplotype (H-2q/d) (Hultman et al., Citation1996; Johansson et al., Citation1997) that is more susceptible to the effects of mercury.
We saw markedly increased secretion of IFNγ by ConA-stimulated splenocytes from both male and female mercury- exposed mice. These results contrasted with in vitro studies by Silva et al (Citation2005) that reported no differences in IFNγ production by splenocytes from adult mice exposed to mercury in utero. It is unlikely that the differences between these studies were due to differences in the level of mercury exposure. Our study employed an oral regimen of 10 mg/L (ppm) mercuric chloride, which was calculated at 208 μg/kg/day for the duration of pregnancy, given an average water intake at 5 ml/day and 10% absorption of inorganic mercury across the gastrointestinal tract. This dose was in the range used by Silva and co-workers (200 μg/kg). However, some of the differences that we observed could possibly be attributed to differences in the route and regimen of exposure; specifically, the mice in the present study were exposed to mercury orally, while the mice in the Silva et al. study were injected subcutaneously. This is especially relevant since the route of exposure will determine the organs in which mercury will accumulate (Tchounwou et al., Citation2003). Further, in the Silva et al. study, mice were treated every other day for 11 days beginning at the fifth day of gestation, while the mice in the present study were exposed to mercury throughout the duration of gestation, which would more closely mimic actual human exposure to mercury.
IL-4 levels were significantly increased by cells from mercury-exposed mice, consistent with the classical TH2-type bias observed after mercury treatment in susceptible mice (Hultman et al., Citation1995, Citation1996; Johansson et al., Citation1997, Citation1998; Pollard et al., Citation2004). This finding is also in agreement with Silva et al.; both IL-4 and IL-10 secretion were found to be significantly increased by cells from mercury-exposed males, while IL-10 levels were found to be lower by cells of exposed females (Silva et al., Citation2005). However, in the present study, while IL-4 secretion was comparatively increased among the cells from the exposed group for both sexes, the cells from males exhibited a tendency towards higher levels than females. Further, we did not detect mercury-induced modulations in IL-10 levels by cells from either males or females. Again, the treatment regimen could possibly account for the differences in TH2 responses in the two studies. Finally, as mentioned before, the variation in the results of the two studies could be due to differences in MHC background of the mice that were tested. Additional studies, such as ELISPOT assays, may be necessary to further distinguish whether the increase in the production of IL-4 and IFNγ by the cells from prenatally-exposed male and female mice is due directly to the increased proliferation of cells or is secondary to the up-regulation of cytokine production.
It has been suggested that environmental agents may act as possible triggers of autoimmune diseases in humans (Mayes, Citation1999) and epidemiologic studies have found a close correlation between significant mercury exposure and increased titers of biomarkers of autoimmunity (Dantas and Quieroz, Citation1997; Silva et al., Citation2004). Further, compelling evidence to support a connection between exposure to metals and autoimmunity has been derived from animal models of chemical-induced autoimmunity (Hultman and Enestrom, Citation1992; CitationBagenstose et al., 1999; Hultman and Hansson-Georgiadis, Citation1999), where mercury exposure induced autoantibody production in genetically-susceptible mice. This led us to investigate whether we could detect phenotypic changes that are known to be associated with the autoimmune response that develops spontaneously in the lupus prone mouse model SNF1 and in estrogen-treated DBF1 mice, both of which develop renal pathology similar to that seen in human systemic lupus erythematosus (SLE) nephritis.
In prior studies using autoimmune SNF1 mice, the expansion of IdLNF1-reactive CD4+ T-cells has been shown to be a crucial event that ultimately leads to the increased production of nephritogenic IdLNF1 autoantibodies that cause disease (Knupp et al., Citation1992, Citation1993; Uner et al., Citation1994). This pattern of idiotypic dysregulation has further been demonstrated in estrogen-induced autoimmunity in DBF1 mice as well, since IdLNF1-reactive cells are expanded in MHC-matched DBF1 mice after treatment with pharmacologic doses of estrogen (Feng et al., submitted). In several autoimmune mouse models, exposure to mercury has resulted in acceleration of disease and potentiation of autoimmunity (Havarinasab and Hultman, Citation2006; Hultman et al., Citation2006), leading to the hypothesis that the immunostimulatory effects of exposure to mercury at non-toxic concentrations may be sufficient to function as a trigger of autoimmunity and possibly autoimmune disease especially in a genetically predisposed background. In this study, we asked whether developmental exposure to mercury would lead to activation of autoreactive cells and functions. Specifically, the expansion of IdLNF1 cells, which are strongly correlated with induction of spontaneous and estrogen-induced autoimmunity, would support the hypothesis that exposure to metals such as mercury could pathologically function as triggers of autoimmunity in a background of genetic susceptibility.
Interestingly, we saw that prenatal mercury exposure resulted in a significant expansion of thymic and splenic cells that were reactive for a biomarker (IdLNF1) of autoimmune nephritis in these mice. We saw that mercury exposure induced a significant expansion of potentially pathogenic CD4+IdLNF1+ T-cells in the spleens of mercury-exposed female mice, and a similar trend for increased numbers in male mercury-exposed mice. These results suggested that mercury may modulate IdLNF1-reactive cells, possibly by a similar mechanism to that of estrogen-induced autoimmune disease. Others have noted that exposure to mercuric chloride at subtoxic doses resulted in enhanced activation of autoimmunity and potentiated TH2-type responses (Pollard et al., Citation1999, Citation2001; Hansson et al., Citation2005), which are also associated with a lupus phenotype. In further support for the effects of mercury in potentiating autoimmunity, we also found that IL-4 production by lymphocytes stimulated with the pathogenic peptide IdLNF1 was significantly increased in mercury-exposed mice, similar to the TH2-type bias that is seen in spontaneous lupus-like SNF1 disease. We also noted that ConA-stimulated lymphocytes from mercury-exposed mice produced increased levels of IFNγ, which is believed to mediate prototypic IgG responses in TH2-dominated mercury-induced autoimmunity (Kono et al., Citation1998). Furthermore, cells from mercury-exposed female mice showed significantly increased proliferation in response to idiotypic peptide aa62-73, suggesting that enhanced autoreactivity may be a consequence of mercury exposure.
Thus, our data suggest that immune stimulation due to mercury exposure could potentially lead to the development of autoimmunity. However, since proteinuria was not increased and there was no evidence of glomerulonephritis on histological examination, developmental exposure to mercury in the present study did not result in overt postnatal autoimmune disease despite evidence of enhanced autoreactivity. We speculate that because, in the context of the idiotypic dysregulation model of lupus nephritis, the increase in IdLNF1-reactive CD4+ cells is an early event in the pathogenesis of lupus nephritis that is followed by activation of IdLNF1-reactive B-cells and production of autoantibodies, there may not have been sufficient levels or specific types of nephritogenic autoantibodies produced at 10 weeks. Spontaneous lupus-like disease in SNF1 female mice is typically observed starting at 16 wk-of-age, again supporting that evaluation of clinical disease at 10 weeks may have been too early. In future studies, additional timepoints will be evaluated for the clinical impact of mercury exposure. Alternatively, a more extended period of exposure and/or exposure to possibly higher mercury concentrations might be required before clinical disease is observed.
In summary, we have found that gestational exposure to non-toxic levels of mercuric chloride resulted in altered immunophenotypes and immune functions that persisted in adult offspring. These changes, even in the absence of overt thymic toxicity (e.g., atrophy), could have important consequences in terms of adult immune responses, if they contribute to; modulating immune responses to pathogens; aberrant immune responses such as hypersensitivity; or the induction of autoimmunity in genetically-susceptible individuals. While mercury immunotoxicity has been extensively reviewed in literature, few studies to date have addressed the potential immune consequences that might occur after developmental exposure. In light of recent evidence showing increased risk of mercury exposure among newborns as a result of maternal mercury exposure, our study shows that there are potential long-term health risks associated with intrauterine exposure to mercury. It also underlies the relevance of assessing immunological parameters when determining health risks associated with prenatal xenobiotic exposures.
Acknowledgments
This work was supported by a grant from the Children’s Miracle Network Foundation (to JG) and the James A Pucello, Sr. and Margie Kehoskie Memorial Foundation.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Apostolou, I., Sarukhan, A., Klein, L., and von Boehmer, H. 2002. Origin of regulatory T-cells with known specificity for antigen. Nature Immunol. 3:756–763.
- Axtell, C. D., Cox, C., Myers, G. J., Davidson, P. W., Choi, A. L., Cernichiari, E., Sloane-Reeves, J., Shamlaye, C. F., and Clarkson, T. W. 2000. Association between methylmercury exposure from fish consumption and child development at five and a half years of age in the Seychelles Child Development Study: An evaluation of nonlinear relationships. Environ. Res. 84:71–80.
- Bagenstose, L. M., Salgame, P., and Monestier, M. (1999) Murine mercury-induced autoimmunity: A model of chemically-related autoimmunity in humans. Immunol. Res. 20:67–78.
- Belles-Isles, M., Ayotte, P., Dewailly, E., Weber, J. P., and Roy, R. 2002. Cord blood lymphocyte functions in newborns from a remote maritime population exposed to organochlorines and methylmercury. J. Toxicol. Environ. Health 65:165–182.
- Bunn, T. L., Marsh, J. A., and Dietert, R. R. 2000. Gender differences in developmental immunotoxicity to lead in the chicken: Analysis following a single early low-level exposure in ovo. J. Toxicol. Environ. Health 61:677–693.
- Bunn, T. L., Parsons, P. J., Kao, E., and Dietert, R. R. 2001a. Exposure to lead during critical windows of embryonic development: Differential immunotoxic outcome based on stage of exposure and gender. Toxicol. Sci. 64:57–66.
- Bunn, T. L., Parsons, P. J., Kao, E., and Dietert, R. R. 2001b. Gender-based profiles of developmental immunotoxicity to lead in the rat: Assessment in juveniles and adults. J. Toxicol. Environ. Health 64:223–240.
- Dantas, D., and Quieroz, M. 1997. Immunogobulin E and autoantibodies in mercury-exposed workers. Immunopharmacol. Immunotoxicol. 19:383–392.
- Davidson, P. W., Myers, G. J., Cox, C., Axtell, C., Shamlaye, C., Sloane-Reeves, J., Cernichiari, E., Needham, L., Choi, A., Wang, Y., Berlin, M., and Clarkson, T. W. 1998. Effects of prenatal and postnatal methylmercury exposure from fish consumption on neurodevelopment: Outcomes at 66 months of age in the Seychelles Child Development Study. JAMA 280:701–707.
- Dietert, R. R., and Piepenbrink, M. S. 2006. Perinatal immunotoxicity: Why adult exposure assessment fails to predict risk. Environ. Health Perspect. 114:477–483.
- Dietert, R. R., Lee, J. E., and Bunn, T. L. 2002. Developmental immunotoxicology: Emerging issues. Human Exp. Toxicol. 21:479–485.
- Dudek, K. 1996. Molecular Identification/Characterization of a Pathogenic Idiotype, IdLNF1, from the (NZBxSWR)F1 Model for Systemic Lupus Erythematosus. PhD Dissertation, State University of New York Health Science Center at Syracuse.
- Gehrs, B. C., and Smialowicz, R. J. 1997. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin I. Toxicology 122:219–228.
- Hansson, M., Djerbi, M., Rabbani, H., Mellstedt, H., Gharibdoost, F., Hassan, M., Depierre, J. W., and Abedi-Valugerdi, M. 2005. Exposure to mercuric chloride during the induction phase and after the onset of collagen-induced arthritis enhances immune/autoimmune responses and exacerbates the disease in DBA/1 mice. Immunology 114:428–437.
- Havarinasab, S., and Hultman, P. 2006. Alteration of the spontaneous systemic autoimmune disease in (NZB × NZW)F1 mice by treatment with thimerosal (ethyl mercury). Toxicol. Appl. Pharmacol. 214:43–54.
- Holladay, S. D., and Smialowicz, R. J. 2000. Development of the murine and human immune system: differential effects of immunotoxicants depend on time of exposure. Environ. Health Perspect. 108(S3):463–473.
- Holsapple, M. P. 2003. Developmental immunotoxicity testing: A review. Toxicology 185:193–203.
- Hultman, P., and Enestrom, S. 1992. Dose-response studies in murine mercury-induced autoimmunity and immune-complex disease. Toxicol. Appl. Pharmacol. 113:199–208.
- Hultman, P., and Hansson-Georgiadis, H. 1999. Methyl mercury-induced autoimmunity in mice. Toxicol. Appl. Pharmacol. 154:203–211.
- Hultman, P., and Nielsen, J. B. 2001. The effect of dose, gender, and non-H-2 genes in murine mercury-induced autoimmunity. J. Autoimmun. 17:27–37.
- Hultman, P., Bell, L. J., Enestrom, S., and Pollard, K. M. 1992. Murine susceptibility to mercury. I. Autoantibody profiles and systemic immune deposits in inbred, congenic, and intra-H-2 recombinant strains. Clin. Immunol. Immunopathol. 65:98–109.
- Hultman, P., Bell, L. J., Enestrom, S., and Pollard, K. M. 1993. Murine susceptibility to mercury. II. Autoantibody profiles and renal immune deposits in hybrid, backcross, and H-2d congenic mice. Clin. Immunol. Immunopathol. 68:9–20.
- Hultman, P., Johansson, U., and Dagnaes-Hansen, F. 1995. Murine mercury-induced autoimmunity: The role of T-helper cells. J. Autoimmun. 8:809–823.
- Hultman, P., Taylor, A., Yang, J. M., and Pollard, K. M. 2006. The effect of xenobiotic exposure on spontaneous autoimmunity in (SWR × SJL)F1 hybrid mice. J. Toxicol. Environ. Health 69:505–523.
- Hultman, P., Turley, S. J., Enestrom, S., Lindh, U., and Pollard, K. M. 1996. Murine genotype influences the specificity, magnitude and persistence of murine mercury-induced autoimmunity. J. Autoimmun. 9:139–149.
- Johansson, U., Hansson-Georgiadis, H., and Hultman, P. 1998. The genotype determines the B-cell response in mercury-treated mice. Intl. Arch. Allergy Immunol. 116:295–305.
- Johansson, U., Sander, B., and Hultman, P. 1997. Effects of the murine genotype on T-cell activation and cytokine production in murine mercury-induced autoimmunity. J. Autoimmun. 10:347–355.
- Kim, S. H., Johnson, V. J., and Sharma, R. P. 2003. Oral exposure to inorganic mercury alters T-lymphocyte phenotypes and cytokine expression in BALB/c mice. Arch. Toxicol. 77:613–620.
- Knupp, C. J., Uner, A. H., Korthas, C., and Gavalchin, J. 1993. Characterization of IdLNF1-specific T-cell clones from the (NZB × SWR)F1 murine model for systemic lupus erythematosus. Clin. Immunol. Immunopathol. 68:273–282.
- Knupp, C. J., Uner, A. H., Tatum, A. H., and Gavalchin, J. 1992. The onset of nephritis in the (NZB × SWR)F1 murine model for systemic lupus erythematosus correlates with an increase in the ratio of CD4 to CD8 T-lymphocytes specific for the nephritogenic idiotype (IdLNF1). Clin. Immunol. Immunopathol. 65:167–175.
- Knupp, C. J., Uner, A. H., Tatum, A. H., Kakanar, J. R., and Gavalchin, J. 1995. IdLNF1-Specific T-cell clones accelerate the production of IdLNF1 + IgG and nephritis in SNF1 mice. J. Autoimmun. 8:367–380.
- Kono, D. H., Balomenos, D., Pearson, D. L., Park, M. S., Hildebrandt, B., Hultman, P., and Pollard, K. M. 1998. The prototypic TH2 autoimmunity induced by mercury is dependent on IFNγ and not TH1/TH2 imbalance. J. Immunol. 161:234–240.
- Koos, B. J., and Longo, L. D. 1976. Mercury toxicity in the pregnant woman, fetus, and newborn infant. A review. Am. J. Obstet. Gynecol. 126:390–409.
- Kosuda, L. L., Greiner, D. L., and Bigazzi, P. E. 1993. Mercury-induced renal autoimmunity: Changes in RT6+ T-lymphocytes of susceptible and resistant rats. Environ. Health Perspect. 101:178–185.
- Kosuda, L. L., Hosseinzadeh, H., Greiner, D. L., and Bigazzi, P. E. 1994. Role of RT6+ T-lymphocytes in mercury-induced renal autoimmunity: Experimental manipulations of “susceptible” and “resistant” rats. J. Toxicol. Environ. Health 42:303–321.
- Kosuda, L. L., Wayne, A., Nahounou, M., Greiner, D. L., and Bigazzi, P. E. 1991. Reduction of the RT6.2+ subset of T-lymphocytes in Brown Norway rats with mercury-induced renal autoimmunity. Cell. Immunol. 135:154–167.
- Kosuda, L. L., Whalen, B., Greiner, D. L., and Bigazzi, P. E. 1998. Mercury-induced autoimmunity in Brown Norway rats: Kinetics of changes in RT6+ T-lymphocytes correlated with IgG isotypes of circulating autoantibodies to laminin 1. Toxicology 125:215–231.
- Layland, L. E., Wulferink, M., Dierkes, S., and Gleichmann, E. 2004. Drug-induced autoantibody formation in mice: Triggering by primed CD4+CD25− T-cells, prevention by primed CD4+CD25+ T-cells. Eur. J. Immunol. 34:36–46.
- Leubke, B., Chen, D., Dietert, R., King, M., Yang, Y., and Luster, M. 2006. Increased sensitivity of the developing immune system to xenobiotics: Experimental evidence supporting the concept of developmental immunotoxicity guidelines. J. Toxicol. Environ. Health 69:811–825.
- Martin, M. B., Reiter, R., Pham, T., Avellanet, Y. R., Camara, J., Lahm, M., Pentecost, E., Pratap, K., Gilmore, B. A., Divekar, S., Dagata, R. S., Bull, J. L., and Stoica, A. 2003. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology 144:2425–2436.
- Mayes, M. D. 1999. Epidemiologic studies of environmental agents and systemic autoimmune diseases. Environ. Health Perspect. 107(S5):743–748.
- Mosier, D. 1977. Ontogeny of T-cell function in the neonatal mouse. In: Development of Host Defenses (Cooper, M., and Dayton, D., Eds), Raven Press, New York, pp. 115–127.
- Myers, G. J., Davidson, P. W., Cox, C., Shamlaye, C. F., Palumbo, D., Cernichiari, E., Sloane-Reeves, J., Wilding, G. E., Kost, J., Huang, L.-S., and Clarkson, T. W. 2003. Prenatal methylmercury exposure from ocean fish consumption in the Seychelles child development study.[see comment]. Lancet 361:1686–1692.
- Pilones, K., Lai, Z., and Gavalchin, J. 2007. Prenatal HgCl2 exposure alters fetal cell phenotypes. J. Immunotoxicol. 4(4):295–301.
- Pollard, K. M., and Hultman, P. 1997. Effects of mercury on the immune system. Metal Ions Biol. Syst. 34:421–440.
- Pollard, K. M., and Landberg, G. P. 2001. The in vitro proliferation of murine lymphocytes to mercuric chloride is restricted to mature T-cells and is interleukin 1-dependent. Int. Immunopharmacol. 1:581–593.
- Pollard, K. M., Arnush, M., Hultman, P., and Kono, D. H. 2004. Co-stimulation requirements of induced murine systemic autoimmune disease. J. Immunol. 173:5880–5887.
- Pollard, K. M., Pearson, D. L., Hultman, P., Deane, T. N., Lindh, U., and Kono, D. H. 2001. Xenobiotic acceleration of idiopathic systemic autoimmunity in lupus-prone bxsb mice. Environ. Health Perspect. 109:27–33.
- Pollard, K. M., Pearson, D. L., Hultman, P., Hildebrandt, B., and Kono, D. H. 1999. Lupus-prone mice as models to study xenobiotic-induced acceleration of systemic autoimmunity. Environ. Health Perspect. 107(S5):729–735.
- Risher, J. F., Murray, H. E., and Prince, G. R. 2002. Organic mercury compounds: Human exposure and its relevance to public health. Toxicol. Ind. Health 18:109–160.
- Schober, S. E., Sinks, T. H., Jones, R. L., Bolger, P. M., McDowell, M., Osterloh, J., Garrett, E. S., Canady, R. A., Dillon, C. F., Sun, Y., Joseph, C. B., and Mahaffey, K. R. 2003. Blood mercury levels in U.S. children and women of childbearing age, 1999–2000. JAMA 289:1667–1674.
- Silva, I. A., El Nabawi, M., Hoover, D., and Silbergeld, E. K. 2005. Prenatal HgCl2 exposure in BALB/c mice: Gender-specific effects on the ontogeny of the immune system. Develop. Compar. Immunol. 29:171–183.
- Silva, I. A., Nyland, J. F., Gorman, A., Perisse, A., Ventura, A. M., Santos, E. C. O., Souza, J. M. d., Burek, C. L., Rose, N. R., and Silbergeld, E. K. 2004. Mercury exposure, malaria, and serum antinuclear/antinucleolar antibodies in Amazon populations in Brazil: A cross-sectional study. Environmental Health: A Global Access Science Source 3:11–21.
- Tchounwou, P., Ayensu, W., Ninashvili, N., and Sutton, D. 2003. Environmental exposure to mercury and its toxicopathologic implications for public health. Environ. Toxicol. 18:149–175.
- Uner, A. H., Knupp, C. J., Tatum, A. H., and Gavalchin, J. 1994. Treatment with antibody reactive with the nephritogenic idiotype, IdLNF1, suppresses its production and leads to prolonged survival of (NZB × SWR)F1 mice. J. Autoimmun. 7:27–44.
- Vorderstrasse, B. A., Cundiff, J. A., and Lawrence, B. P. 2006. A dose-response study of the effects of prenatal and lactational exposure to TCDD on the immune response to Influenza A virus. J. Toxicol. Environ. Health 69:445–463.
- Wraith, D., Nicolson, K., and Whitley, N. 2004. Regulatory CD4+ T-cells and the control of autoimmune diseases. Curr. Opin. Immunol. 16:695–701.