Abstract
Activation of aryl hydrocarbon receptor (AhR) by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in T-cells is required for TCDD-induced suppression of the allogeneic CTL response and for induction of CD25hiCD62Llow adaptive regulatory T-cells. Here, the ability of a constitutively-active AhR (CA-AhR) expressed in T-cells alone to replicate the effects of TCDD was examined. The response of CA-AhR-expressing B6 donor T-cells in B6xD2F1 mice was compared to the response of wild-type B6 donor T-cells in B6xD2F1 mice given a single dose of TCDD. Expression of CA-AhR in donor T-cells enhanced the down-regulation of CD62L on Day 2 after injection, similar to a single oral dose of TCDD, but did not induce up-regulation of CD25 on Day 2 or affect CTL activity on Day 10. This suggests that activation of AhR in T-cells alone may not be sufficient to alter T-cell responses in this acute graft-versus-host (GvH) model. Since host APC are responsible for activating the donor T-cells, we examined the influence of the F1 host’s AhR on donor T-cell responses by creating an AhR−/− B6xD2F1 host that had a greatly diminished AhR response to TCDD compared to wild-type F1 mice. As in AhR+/+ B6xD2F1 mice, the CTL response in AhR−/− B6xD2F1 mice was completely suppressed by TCDD. This suggests that either CA-AhR dose not fully replicate the function of TCDD-activated AhR in suppression of the CTL response, or that minimal activation of AhR in host cells is required to combine with activation of AhR in T-cells to elicit the immunosuppressive effects of TCDD.
Introduction
The AhR is an intracellular receptor and transcription factor well known for mediating the immunosuppressive effects of a large group of environmental contaminants, including certain polycyclic aromatic hydrocarbons and polychlorinated dibenzo-furans and -dioxins (Kerkvliet, Citation2002, Citation2003). Human exposure to dioxins and related chemicals occurs primarily through the consumption of food, particularly meat and dairy products, because these chemicals are highly fat-soluble and bioaccumulate (Liem et al., Citation2000; Schecter et al., Citation2001; Startin and Rose, Citation2003). In addition to these man-made ligands, many natural chemicals found in plants as well as products of normal cellular metabolism have been shown to bind and activate AhR (Denison and Nagy, Citation2003; Jeuken et al., Citation2003; Nguyen and Bradfield, Citation2008). Ligand binding leads to nuclear translocation of the AhR and dimerization with its transcriptional partner, ARNT, which in turn binds to the consensus DNA sequence, termed the dioxin response element (DRE). DRE has been found in the promoters of many genes, including one of the most sensitive gene targets, CYP1A1 (Schmidt and Bradfield, Citation1996; Mimura and Fujii-Kuriyama, Citation2003).
Of many chemicals that bind to AhR, TCDD is a well-known ligand having high affinity and eliciting potent suppressive effect on both humoral and cell-mediated immune responses (Holsapple et al., Citation1991; Nohara et al., Citation2002; Kerkvliet, Citation2003; Inouye et al., Citation2003; Lawrence and Kerkvliet, Citation2007). Recent studies have shown that AhR activation by TCDD enhances expansion of CD4+CD25+ cells, particularly CD62LlowCD4+CD25+ cells, and CD4+Foxp3+ regulatory T-(Treg) cells (Funatake et al., Citation2005; Quintana et al., Citation2008). AhR activation also participates in Th17 differentiation (Quintana et al., Citation2008; Kimura et al., Citation2008; Veldhoen et al., Citation2008). Thus, Ahr is involved in a variety of immune responses and suppressions, while the mechanism is not fully understood.
In attempts to clarify the target cells affected by AhR activation, previous studies have shown that T-cells are direct AhR-dependent targets of TCDD-mediated immune suppression, and both CD4+ and CD8+ T-cells must express AhR in order for full suppression of the CTL response to alloantigen (Kerkvliet et al., Citation2002). Subsequent studies showed that activation of AhR in the donor T-cells was also required for TCDD-mediated induction of a subpopulation of T-cells that are CD25hiCD62Llow and exhibit suppressive activity in vitro, for both CD4+ (Funatake et al., Citation2005) and CD8+ T-cells (Funatake et al., Citation2008). However, many other cells in the immune system express AhR and are also known to be directly targeted by TCDD, including dendritic cells and B-cells (Sulentic et al., Citation1998; Vorderstrasse and Kerkvliet, Citation2001; Boverhof et al., Citation2004; Ruby et al., Citation2005). A recent study has shown that activation of AhR within hematopoietic cells, but not within CD8+ T-cells, is necessary for suppression of the CTL response to influenza, suggesting that in the influenza model, the immunosuppressive effects of TCDD are mediated by the activation of AhR in immune cells other than the responding CD8+ T-cells (Lawrence et al., Citation2006). Thus, although studies have shown that activation of AhR within the donor T-cells is required for suppression of the CTL response to alloantigen, it is not known if activation of AhR in T-cells alone is sufficient to mediate this suppression.
In order to address the exact role of the AhR in T-cells, we previously-produced transgenic (Tg) mice expressing a constitutively active AhR (CA-AhR) under the control of the T-cell lineage-specific CD2 promoter (Nohara et al., Citation2005). These mice express the transgene in their thymocytes, CD4 T-cells, and CD8 T-cells, and they express CYP1A1 mRNA in the thymus and spleen, showing that the CA-AhR is transcriptionally active in T-cells. These mice also display thymic atrophy (Nohara et al., Citation2005), which is one of the hallmarks of exposure to TCDD that occurs in an AhR-dependent manner (Laiosa et al., Citation2003). The immune response of these mice against Ovalbumin (OVA)-alum only partially mimicked the immune response of wild-type mice exposed to TCDD (Nohara et al., Citation2005, Citation2009).
For example, splenocytes from CA-AhR Tg mice produced levels of interleukin (IL)-4 and IL-5 and antibody titers upon OVA immunization similar to those of wild-type mice (Nohara et al., Citation2009).
On the other hand, interferon (IFN)-γ production by the splenocytes upon OVA immunization was augmented in the Tg mice similar to that seen in wild-type mice exposed to TCDD (Nohara et al., Citation2009). In the present study, we used the transgenic CA-AhR mice to ask whether activation of AhR in T-cells alone was sufficient to induce the CD25+CD62Llow adaptive Treg-like phenotype and mediate suppression of the CTL response to alloantigen. To do so, we compared the response of CA-AhR-expressing B6 donor T-cells in B6xD2F1 mice and the response of wild-type B6 donor T-cells in B6xD2F1 mice given a single dose of TCDD in the acute graft-versus-host (GVH) model.
Materials and methods
Mice and treatment with TCDD
The generation and genotyping of the transgenic C57Bl/6-CA-AhR (CA-AhR) mice has been described (Nohara et al., Citation2005). The heterozygous Tg mice showed CYP1A1 mRNA expression in the thymus at the level corresponding approximately to the level induced by a single dose of 20 μg TCDD/kg (Nohara et al., Citation2005). The transgenic CA-AhR mice and wild-type (B6-WT) littermates used in the present studies were backcrossed to the B6 background over 10 generations. For some GVH experiments, transgenic CA-AhR mice (CA-AhR+/−) were crossed with B6.PL-Thy1a/CyJ (Thy1.1)mice, purchased from The Jackson Laboratory (Bar Harbor, ME), to generate Thy1.1+ donor T-cells. Thy1.1+ CA-AhR−/− littermates were used as B6-WT controls. B6D2F1/Crlj mice were purchased from Charles River Laboratories (Yokohama, Japan).
B6D2F1/J mice were generated by breeding C57Bl/6J female mice with DBA/2J male mice, originally purchased from CLEA Japan (Tokyo). In one study, B6 mice lacking AhR (AhrΔ2; Schmidt et al., Citation1996) were crossed with DBA/2 mice, (both purchased from The Jackson Laboratory). All mice were used between 2 and 5 months of age. Except where noted, B6D2F1 mice were dosed orally with 20 μg TCDD/kg body weight 1 day before the injection of donor T-cells, as previously described (Kerkvliet et al., Citation2003). TCDD was purchased from Cambridge Isotope Laboratories (Andover, MA) and prepared as a 4% nonane/corn oil solution. A similarly prepared solution of 4% nonane in corn oil was used as a vehicle-control. All mice were handled in a humane manner in accordance with the National Institute for Environmental Studies (NIES, Japan) guidelines for animal experiments.
Preparation of donor T-cells for the GVH model
T-Cells were purified from spleen cells of B6-WT or transgenic CA-AhR mice using the BD IMag™ and a mouse T-lymphocyte enrichment set (BD Biosciences, San Jose, CA), following the manufacturer’s instructions. The percentages of CD4+ and CD8+ cells in the T-cell fraction were checked using flow cytometry (see below for details) and viability was assessed by trypan blue exclusion. T-Cells (i.e., CD4+ and CD8+ cells) were 80-93% pure and viabilities were > 90%. The concentration of purified T-cells was adjusted so that each F1 mouse received 1.5 x 107 T-cells intravenously in 250 μl of 1X Hank's Balanced Salt Solution (HBSS). B6-WT mice were used as syngeneic recipients. In some experiments the purified donor T-cells were labeled with 0.5 μM Carboxyfluorescein Succinimydl Ester (CFSE) (Invitrogen, Carlsbad, CA) before injection into F1 or syngeneic hosts as described previously (Funatake et al., Citation2004).
Flow cytometry
Spleen cells were resuspended in staining buffer containing 0.1% sodium azide and 1% fetal bovine serum in phosphate- buffered saline. The cells were incubated with 200 μg/ml rat IgG (Jackson Immunoresearch, West Grove, PA) to block Fc receptors (FcR), followed by incubation with fluorochrome- or biotin-conjugated monoclonal antibodies. Antibodies for Thy1.1, CD4, CD62L, CD19, and CD25 were purchased from eBiosciences (San Diego, CA). All other antibodies were purchased from BD Biosciences. After incubation with monoclonal antibodies, the cells were washed and incubated with streptavidin conjugated to allophycocyanin, PerCP (BD Biosciences), or PE-Cy5 (eBiosciences). After incubating the cells with streptavidin, the cells were washed and resuspended in staining buffer, and data were collected on 7000-10,000 events of interest using a BD FACSCalibur (BD Biosciences). Data were analyzed using CellQuest software (BD Biosciences).
Cytotoxicity assay
With one noted exception that used the standard 51 Cr-release assay, spleen cells were assayed for CTL activity using the CytoTox 96® non-radioactive cytotoxicity assay kit from Promega (Madison, WI), according to the manufacturer’s instructions. Briefly, P815 cells recovered from the ascites of DBA/2 mice were added to a 96-well round bottom plate at 1 x 104/well. Spleen cells from F1 mice that had been injected with B6 donor T-cells 10 days earlier were then added to the wells in duplicate serial dilutions from 100:1 to 12.5:1. Wells containing only spleen cells were included as background controls, in addition to wells containing only P815 cells with or without lysis buffer used for maximum and spontaneous release controls, respectively. The plates were incubated for 4 hr at 37°C and 5% CO2, after which the CTL activity was measured and the percent cytotoxicity was calculated according to the manufacturer’s instructions.
7-Ethoxyresorufin-O-deethylase (EROD) activity assay
Livers were homogenized and debris was removed by centrifugation at 10,000 x g for 20 min at 4°C. The microsomal fraction was isolated from the supernatants by additional centrifugation at 100,000 x g for 90 min at 4°C. The EROD activity of each microsomal fraction was determined using 2 μg of protein for the WT-F1 samples and 25 μg for the KO-F1 (AhR−/− B6 mice bred with DBA/2 mice to create F1 hosts that would respond poorly to AhR activation) samples. Microsomal protein was aliquoted into microtiter wells containing 200 μM 7-ethoxy-resorufin in 0.1M Tris-HCl, (pH 7.8.) NADPH was added to each well and the fluorescence was measured on a SpectraMAX Gemini plate reader using 535 nm excitation and 585 nm emission filters. Samples were assayed in triplicate and the amount of resorufin produced was calculated using a resorufin standard curve.
Statistics
All experiments were independently conducted twice with n = 3–5 mice per group per experiment. Where appropriate, data from replicate studies were combined, and all data are presented as the mean ± SEM. The differences between means were analyzed by Student’s t-test, with p < 0.05 considered statistically significant.
Results
CA-AhR does not affect cell division or the number of donor T-cells in the spleen on Day 2
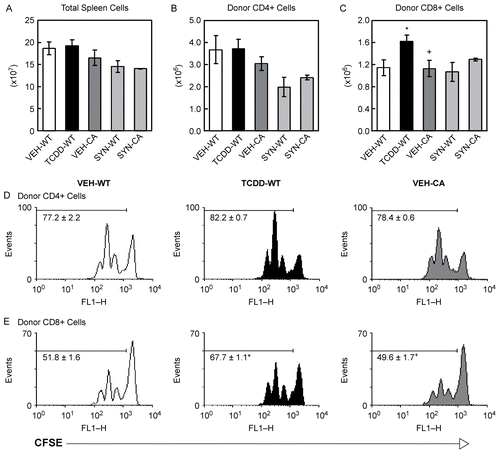
Previous studies have shown that exposure of F1 mice to TCDD has no adverse effect on the proliferation or number of donor T-cells in the spleen on Day 2 of the GVH response (Funatake et al., Citation2005, Citation2008). To determine if expression of CA-AhR in donor T-cells affected cell division or cell number in the spleen, we compared donor T-cells from transgenic CA-AhR mice to donor T-cells from B6-WT mice two days after injection into vehicle- or TCDD-treated F1 hosts. Donor T-cells from CA-AhR mice or wild-type littermates were labeled with CFSE before injection into F1 hosts for evaluation of cell division. As with TCDD, CA-AhR in the donor T-cells had no effect on the total number of spleen cells () or on the number of donor CD4+ T-cells in the spleen on Day 2 (). In addition, neither exposure to TCDD nor CA-AhR had any effect on the cell division of donor CD4+ T-cells (). Exposure to TCDD led to a small but significant increase in the number of donor CD8+ T-cells (), and there was a corresponding increase in the percentage of donor CD8+ T-cells that had undergone one or more cell divisions (, middle panel). CA-AhR in the donor T-cells had no effect on the number of donor CD8+ T-cells in the spleen () or on the percentage of donor CD8+ cells that were dividing (). As expected, in syngeneic B6 recipients, there was no difference in the total number of spleen cells or the number of donor CD4+ or CD8+ T-cells in the spleen two days after injection of donor T-cells from either B6-WT or CA-AhR mice ().
Figure 1. CA-AhR does not alter the number or proliferation of donor T-cells in the spleen on Day 2. Age- and gender-matched F1 mice were dosed with vehicle or 20 μg/kg TCDD one day before the injection of donor T-cells from B6-WT or CA-AhR mice. On Day 2 after injection of donor T-cells, the spleens from F1 mice were collected and analyzed by flow cytometry. After gating on total spleen cells (A), the donor T-cells were identified as CFSE+CD4+ (B) or CFSE+CD8+ (C). For A-C, data are combined from two independent experiments and n = 4-6 mice per group. VEH, vehicle-treated F1 recipient; TCDD, TCDD-treated F1 recipient; SYN, syngeneic B6 recipient; WT, donor T-cells from wild-type mice; CA, donor T-cells from CA-AhR Tg mice. The percentage of donor CD4+ (D) or CD8+ T-cells (E) that were dividing was determined on Day 2. Representative histograms are shown and the numbers on the histograms indicate the mean ± SEM of the percentage of cells in divisions 1-4 for n = 3 mice per group. *Indicates statistically significant difference compared to VEH-WT; +indicates statistically significant difference compared to TCDD-WT.
CA-AhR enhances the down-regulation of CD62L but does not affect the expression of CD25
Next, we compared the effect of the CA-AhR to the effect of TCDD on the expression of CD25 and CD62L on the donor CD4+ and CD8+ T-cells. For donor CD4+ T-cells, exposure to TCDD caused a large increase in the percentage of cells expressing CD25, whereas CA-AhR had a much more modest effect ( ). In contrast, both activation of AhR by TCDD and CA-AhR caused a substantial increase in the percentage of donor CD4+ T-cells that had down-regulated CD62L ( and ). For donor CD8+ T-cells, a similar phenomenon was seen. Exposure to TCDD dramatically increased the frequency of CD25-expressing cells while CA-AhR produced only a slight increase ( ). Additionally, both TCDD and CA-AhR increased the frequency of donor CD8+ T-cells that had down-regulated CD62L ( and ). The decrease in CD62L occurred stepwise as the T-cells divided, as indicated by the decrease in median fluorescence intensity for both TCDD-treated and CA-AhR-expressing donor T-cells as compared to vehicle-treated donor T-cells (). Interestingly, in syngeneic recipients, CA-AhR also caused a small increase in the frequency of donor T-cells that had down-regulated CD62L as compared to B6-WT donors for both CD4+ and CD8+ T-cells (, light grey bars).
Figure 2. CA-AhR enhances the down-regulation of CD62L comparable to TCDD but has only a minimal effect on the up-regulation of CD25. F1 mice were treated with vehicle or TCDD as described for . On Day 2, the donor CD4+ (A and C) and donor CD8+ T-cells (B and D) were analyzed for the expression of CD25 (A and B) and CD62L (C and D). For CD25, data are combined from two independent experiments with n = 4–6 mice per group. For CD62L, data are from a representative experiment with n = 3–4 mice per group. Also on Day 2, the median fluorescence intensity (MedFI) of CD62L was determined for each cell division for donor CD4+ (E) and CD8+ (F) T-cells. *Indicates statistically significant difference compared to VEH-WT; +indicates statistically significant difference compared to TCDD-WT; #indicates statistically significant difference compared to SYN-WT.
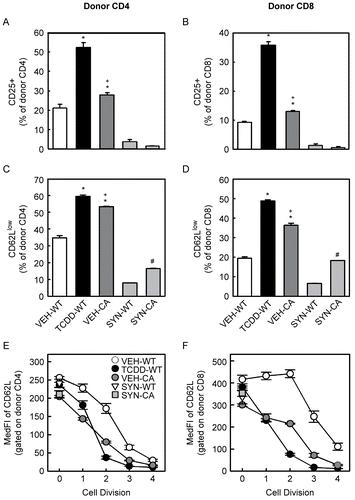
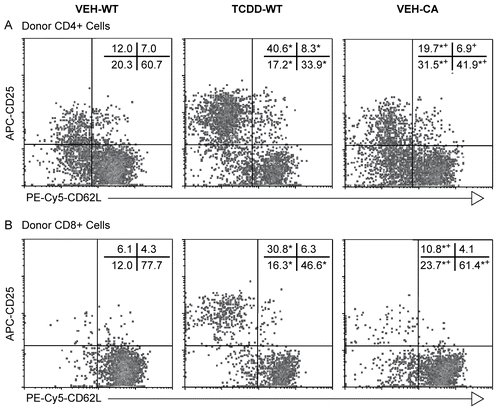
Figure 3. CA-AhR does not expand a subpopulation of CD25hiCD62Llow donor T-cells. F1 mice were treated with vehicle or TCDD as described for . The co-expression of CD25 and CD62L was determined for the donor CD4+ (A) and donor CD8+ T-cells (B) on Day 2. Representative histograms are shown and the numbers on the histograms indicate the mean of the percentage of cells in each quadrant for n = 3 mice per group (SEM is excluded for clarity). *Indicates statistically significant difference compared to VEH-WT; +indicates statistically significant difference compared to TCDD-WT.
Expression of CD25hiCD62Llow Treg phenotype
Recently, we reported that a subpopulation of donor CD4+ T-cells in TCDD-treated mice is CD25hiCD62Llow and that this subpopulation has Treg-like suppressive activity in vitro (Funatake et al., Citation2005). We have also observed the same phenotype and in vitro suppressive activity for donor CD8+ T-cells from TCDD-treated mice (Funatake et al., Citation2008). However, as shown above, for donor T-cells expressing CA-AhR, there were only minor changes in the expression of CD25 while at the same time expression of CD62L was profoundly down-regulated. When we examined the co-expression of these two markers on the donor T-cells two days after injection into F1 hosts, exposure to TCDD greatly increased the percentage of cells that were CD25+CD62Llow for both donor CD4+ and CD8+ T-cells (, middle panel) In contrast, donor T-cells expressing CA-AhR exhibited a significant but much smaller increase in the percentage of donor CD4+ or CD8+ T-cells that were CD25+CD62Llow when compared to vehicle-treated mice (, right panel). Thus, compared to the effect from TCDD treatment, CA-AhR in the donor T-cells alone does not appear to be sufficient to induce a subpopulation of CD25hiCD62Llow donor T-cells.
Development of effector CTL and CTL activity is not impaired by CA-AhR
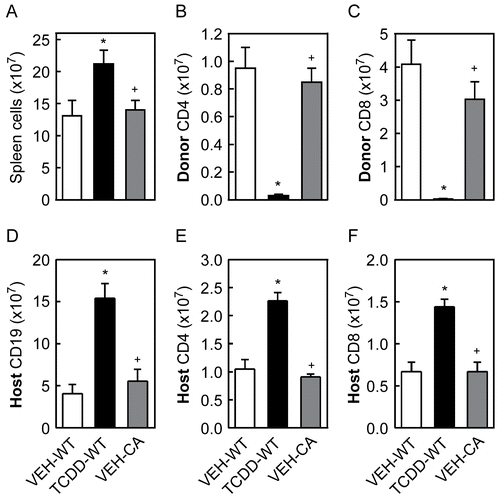
To determine if CA-AhR in T-cells alone was sufficient to suppress the CTL response, we injected F1 mice with donor T-cells from B6-WT or transgenic CA-AhR mice and 10 days later tested the spleen cells from F1 mice for the presence of effector CTL and CTL activity. As shown in , there were significantly more spleen cells in TCDD-treated mice, while CA-AhR in the donor T-cells had no effect on the number of spleen cells on Day 10. The greater number of total spleen cells in TCDD-treated mice on Day 10 was due to greater numbers of host B-cells and host T-cells in the spleen reflecting the absence of an anti-host immune response, as shown in . In addition, the number of donor CD4+ and donor CD8+ T-cells in the spleen was greatly reduced in TCDD-treated mice, while CA-AhR had no effect on the number of donor T-cells compared to vehicle-treated controls ().
Figure 4. Analysis of the distribution of spleen cells on Day 10 after injection of donor T-cells. F1 mice were treated with vehicle or TCDD as described for . On Day 10 after injection of donor T-cells, total spleen cells from F1 mice were counted (A) and analyzed by flow cytometry for enumeration of donor CD4+ (B) and donor CD8+ T-cells (C), host CD19+ cells (D), host CD4+ (E) and host CD8+ T-cells (F). Data from a representative experiment are shown with n = 4-5 mice per group. *Indicates statistically significant difference compared to VEH-WT; +indicates statistically significant difference compared to TCDD-WT.
Of the few donor CD8+ T-cells present in the spleen of TCDD-treated mice on Day 10, significantly fewer expressed an effector CTL phenotype (CD44hiCD62Llow) as compared to donor CD8+ T-cells in vehicle-treated controls (), resulting in essentially no effector CTL present in the spleens of TCDD-treated mice (). In mice receiving CA-AhR donor T-cells, there was no difference in the percentage or number of donor-derived effector CTL as compared to vehicle-treated F1 mice that received WT donor T-cells ().
Figure 5. CA-AhR does not suppress the CTL activity or the percentage and number of CTL effectors in the spleen on Day 10. F1 mice were treated with vehicle or TCDD as described for . Ten days after in the injection of donor T-cells, the spleen cells from F1 mice were tested for CTL activity using a non-radioactive cytotoxicity assay, as described in the Methods (A). The percentage (B) of donor CD8+ cells, identified as Thy1.1+CD8+ cells, expressing a CTL effector (CTLe) phenotype, defined as CD44hiCD62Llow, and the number (C) of CTLe in the spleen were determined by flow cytometry. Data from a representative experiment are shown with n = 4–5 mice per group. *Indicates statistically significant difference compared to VEH-WT; +indicates statistically significant difference compared to TCDD-WT.
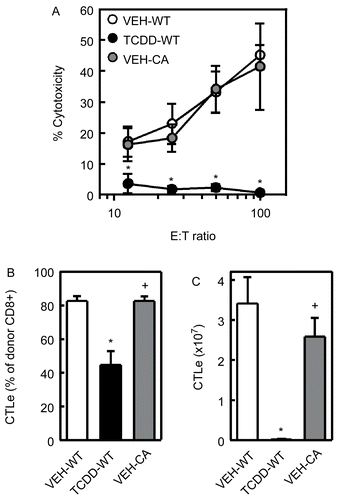
On Day 10, CTL activity in the spleen was measured ex vivo in a 4-hour non-radioactive cytotoxicity assay, as described in the Methods, and the percentage and number of effector CTL was determine by flow cytometry. As shown in , no CTL activity was detected with splenocytes from TCDD-treated mice. In contrast, the CTL activity of spleen cells from mice injected with CA-AhR donor T-cells was indistinguishable from CTL activity in vehicle-treated mice injected with WT donor T-cells (). Together, these data suggest that AhR activation only in T-cells is not sufficient to suppress CTL activity in the GVH model.
Influence of AhR responsiveness of F1 host mice on donor CTL activity
The absence of an expanded population of CD25hiCD62Llow donor T-cells and the lack of suppression of effector CTL when the donor T-cells expressed CA-AhR might suggest that activation of AhR in donor T-cells only is not sufficient to suppress the CTL response, and that activation of AhR in host cells (e.g., dendritic cells that activate the donor T-cells) contribute to the suppression of the GVH response by TCDD. To test this hypothesis, we bred AhR−/− B6 mice with DBA/2 mice to create F1 host mice that would respond poorly to AhR activation (“KO-F1”). If AhR activation in host cells contributes to suppression of the donor CTL response by TCDD, the CTL response should be less affected by TCDD in the KO-F1mice.
To confirm the low responsiveness of these KO-F1 mice, we measured EROD activity of CYP1A1 in the liver after treatment with TCDD. WT-F1 mice reached near-maximal EROD activity with as little as 3 μg TCDD/kg body weight, but little to no EROD activity was detected in KO-F1 mice at this dose (). At 15 μg TCDD/kg body weight, a dose previously shown to completely suppress the GVH CTL response (Kerkvliet et al., Citation2002), the EROD activity in KO-F1 mice was nearly 4 times lower than in WT-F1 mice; even at 30 μg TCDD/kg body weight, the EROD activity of KO-F1 mice was significantly less than in WT-F1 mice ().
Figure 6. Host AhR does not appear to be involved in suppression of the CTL response on Day 10. (A) KO-F1 and WT-F1 mice were dosed with 0, 3, 15, or 30 μg TCDD/kg body weight and, two days later, the livers were collected and EROD activity was determined as described in the Methods. (B) KO- and WT-F1 mice received AhR-WT donor T-cells and treated with 15 μg TCDD/kg body weight. Ten days later, the CTL activity in the spleen was assessed. Data from a representative experiment are shown.
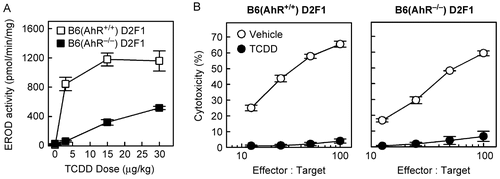
Interestingly, despite the low host response to TCDD, the CTL response of AhR-WT donor T-cells in KO-F1 hosts was suppressed by TCDD to the same extent as in WT-F1 hosts (), suggesting that activation of AhR in host cells plays a minimal role in mediating the suppressive effects of TCDD in this model. These data suggest that CA-AhR does not behave the same as ligand (TCDD)-bound AhR in T-cells that appears to be necessary and sufficient for suppressing the CTL response. However, it remains possible that even low-level activation of AhR within key cells of the KO-F1 host treated with 15 μg/kg TCDD was sufficient to synergize with AhR-mediated signaling in T-cells to suppress the CTL response. Additional studies will be necessary to determine which of these interpretations is correct.
Discussion
In the present study, we used transgenic mice that express CA-AhR only in T-lineage cells (Nohara et al., Citation2005). The CA-AhR mutant has a minimal deletion in the PAS B domain which is required for binding to hsp90 and also contains part of the ligand binding domain. The minimal deletion causes the AhR to localize in the nucleus, heterodimerize with ARNT, and activate transcription by binding DRE sequences in a ligand-independent manner (McGuire et al., Citation2001).
In our recent study (Nohara et al., Citation2009), we found that CA-AhR Tg mice elicit the same level of IL-4 and IL-5 production and antibody production upon OVA immunization as wild-type mice do. We showed in the present study that CA-AhR T-cells normally proliferate and differentiate into CTL effector cells with CTL activity in B6D2F1 hosts as wild-type B6 cells. In CA-AhR Tg mice, CA-AhR is expressed in the T-cells from their fetal stage, which might affect the immune function (Nohara, et al., Citation2005).
Nevertheless, the results obtained so far indicate that the immune system of Tg mice still maintains its normal ability to perform humoral and cellular immune responses. The findings appear to be consistent with the fact that other studies have shown that T-cells must be activated in order to see any AhR-dependent changes in phenotype or function (Shepherd et al., Citation2000; Funatake et al., Citation2004). On the other hand, Tg mice develop thymus atrophy (Nohara et al., Citation2005) and, upon OVA immunization, they showed augmented IFNγ production compared to wild-type mice (Nohara et al., Citation2009). These changes are also observed in wild-type mice given a single dose of TCDD (Nohara et al., Citation2005, Citation2009). The results of the present study show that CA-AhR enhanced the down-regulation of CD62L to the same extent as exposure to a single dose of TCDD (20 μg/kg body weight). Thus, CA-AhR Tg mice show an immune profile, some of which is similar to those of wild-type mice and some of which mimic those observed in wild-type mice given a single dose of TCDD.
In this study, we show that restricting the expression of CA-AhR to T-cells only partially mimics the effects of TCDD in the GVH response. Specifically, CA-AhR enhanced the down-regulation of CD62L, but had no effect on the expression of CD25 and did not suppress CTL activity on Day 10. In previous studies, the down-regulation of CD62L was found to depend strictly on expression of AhR within the affected T-cell population, whereas the up-regulation of CD25, at least for donor CD8+ T-cells, was found to depend indirectly on AhR within the donor CD4+ T-cells (Funatake et al., Citation2008). Sun et al. (Citation2004) performed a genome-wide study of DRE located between -5000 and +2000 bp for over 11,000 mouse genes and identified eight putative DRE in the promoter region of the gene for CD62L. Three of these putative DRE had match scores equivalent to confirmed DRE for CYP1A1; interestingly, no DRE were identified in the Il2ra gene. Together, these findings suggest that CD62L could be a direct target of the AhR-ARNT complex, while CD25 may be an indirect target, dependent on other AhR-ARNT-regulated genes.
It is interesting that although CA-AhR had a profound effect on the down-regulation of CD62L, this did not appear to have any effect on the final outcome of the GVH response, since the CTL activity on Day 10 was unimpaired. CD62L is important for allowing entry of naïve T-cells into the lymph node through the high endothelial venules (Arbones et al., Citation1994; Bradley et al., Citation1994; Warnock et al., Citation1998). Once activated, T-cells down-regulate CD62L; however, this down-regulation is not necessary for the cells to exit out of a lymph node, and is more important for preventing the re-entry of activated T-cells into lymph nodes (Galkina et al., Citation2003). Some studies have suggested that signaling through CD62L induces conformational changes in integrins and alters expression of chemokine receptors that enhance/prolong the interaction between T-cells and antigen-presenting cells (Giblin et al., Citation1997; Ding et al., Citation2003). However, other studies have shown that the lack of CD62L has no adverse effects on the development of autoimmune diabetes (Friedline et al., Citation2002; Mora et al., Citation2004). This latter finding is consistent with our results showing that CTL activity is unimpaired on Day 10 of the GVH response despite the enhanced down-regulation of CD62L on donor T-cells expressing CA-AhR. Thus, although AhR may directly target CD62L and induce its down-regulation, this alone is not sufficient for suppression of effector function.
CA-AhR expressed in T-cells did not suppress the GVH response, suggesting that activation of AhR in other immune cells is involved in the immunosuppressive effects of TCDD. In the GVH response, host cells, in particular dendritic cells, contribute to the T-cell response by acting as antigen presenting cells. Previous studies have shown that dendritic cells, like T-cells, are sensitive, AhR-dependent targets of TCDD (Vorderstrasse and Kerkvliet, Citation2001; Vorderstrasse et al., Citation2003; Ruby et al., Citation2005).
In order to assess the contribution of host-expressed AhR, AhR−/- B6 mice were crossed with D2 mice to generate KO-F1 hosts with low responsiveness to AhR. If the host AhR contributes significantly to the suppression of the CTL response, we would expect that the CTL activity in these KO-F1 mice to be less sensitive to suppression by TCDD. However, we found that the CTL activity was suppressed to the same extent as in wild-type F1 mice. This would imply that activation of AhR in dendritic cells or other host cells does not have a major influence on the outcome of the T-cell response in this GVH model. One caveat to this interpretation is that there was still activation of AhR, albeit at a much lower level, in the KO-F1 hosts at the dose of TCDD used. This leaves open the possibility that low-level activation of AhR within the host is sufficient to synergize with the activation of AhR within the donor T-cells, leading to the full suppression of the CTL response. To assess this possibility, generation of AhR−/- B6 x AhR−/- D2 F1 would be useful.
Another possible interpretation of the results of the present study is that CA-AhR does not behave the same as ligand-bound AhR in the suppression of the GVH response. AhR-dependent changes in expression of genes, particularly those involved in the activation and differentiation of T-cells, and also non-genomic effects of AhR are reported to depend on the interaction of AhR with other proteins, such as transcription factors, co-activators, and co-repressors (reviewed in Hankinson, Citation2005; Harper et al., Citation2006). The deletion in a part of the ligand/Hsp90–binding site in the PAS B domain of AhR may affect the interaction of the CA-AhR with these other proteins and their function. In addition, it is possible that different ligands induce different conformational states, allowing for different protein interactions (Henry and Gasiewicz, Citation2003; Hestermann and Brown, Citation2003; Zhang et al., Citation2008). Recent studies have reported that activation of AhR by TCDD induces Treg, while activation of AhR by 6-formylindolo-[3,2-b]carbazole (FICZ) induces Th17 cells (Quintana et al., Citation2008; Veldhoen et al., Citation2008). These findings may indicate ligand-dependent regulation of AhR function. In contrast, the single-conformation available for the CA-AhR may not allow for interaction with other proteins.
In summary, our findings suggest that either activation of AhR must occur in other cells in addition to T-cells in order to cause immunosuppression or that CA-AhR does not behave exactly the same as ligand (TCDD)-bound AhR in the GvH model. Additional studies in progress of functions of CA-AhR will provide insights into the genomic and non-genomic mechanisms of AhR function.
Acknowledgements
We would like to thank Linda Steppan for her excellent technical advice, Dr. David Shepherd for his critical review of the manuscript, and Michiyo Matsumoto for her outstanding secretarial assistance.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Arbones, M. L., Ord, D. C., Ley, K., Ratech, H., Maynard-Curry, C., Otten, G., Capon, D. J., and Tedder, T. F. 1994. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity 1:247–260.
- Boverhof, D. R., Tam, E., Harney, A. S., Crawford, R. B., Kaminski, N. E., and Zacharewski, T. R. 2004. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces suppressor of cytokine signaling 2 in murine B-cells. Mol. Pharmacol. 66:1662–1670.
- Bradley, L. M., Watson, S. R., and Swain, S. L. 1994. Entry of naive CD4 T-cells into peripheral lymph nodes requires L-selectin. J. Exp. Med. 180:2401–2406.
- Denison, M. S., and Nagy, S. R. 2003. Activation of the aryl hydrocarbon receptor by structurally-diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 43:309–334.
- Ding, Z., Issekutz, T. B., Downey, G. P., and Waddell, T. K. 2003. L-Selectin stimulation enhances functional expression of surface CXCR4 in lymphocytes: Implications for cellular activation during adhesion and migration. Blood 101:4245–4252.
- Friedline, R. H., Wong, C. P., Steeber, D. A., Tedder, T. F., and Tisch, R. 2002. L-Selectin is not required for T-cell-mediated autoimmune diabetes. J. Immunol. 168:2659–2666.
- Funatake, C. J., Dearstyne, E. A., Steppan, L. B., Shepherd, D. M., Spanjaard, E. S., Marshak-Rothstein, A., and Kerkvliet, N. I. 2004. Early consequences of 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure on the activation and survival of antigen-specific T-cells. Toxicol. Sci. 82:129–142.
- Funatake, C. J., Marshall, N. B., Steppan, L. B., Mourich, D. V., and Kerkvliet, N. I. 2005. Cutting edge: activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+CD25+ cells with characteristics of regulatory T-cells. J. Immunol. 175:4184–4188.
- Funatake, C. J., Marshall, N. B., and Kerkvliet, N. I. 2008. 2,3,7,8-Tetrachlorodibenzo-p-dioxin alters the differentiation of alloreactive CD8+ T-cells toward a regulatory T-cell phenotype by a mechanism that is dependent on aryl hydrocarbon receptor in CD4+ T-cells. J. Immunotoxicol. 5:81–91.
- Galkina, E., Tanousis, K, Preece, G., Tolaini, M., Kioussis, D., Florey, O., Haskard, D. O., Tedder, T. F., and Ager, A. 2003. L-Selectin shedding does not regulate constitutive T-cell trafficking but controls the migration pathways of antigen-activated T-lymphocytes. J. Exp. Med. 198:1323–1335.
- Giblin, P. A., Hwang, S. T., Katsumoto, T. R., and Rosen, S. D. 1997. Ligation of L-selectin on T-lymphocytes activates β1 integrins and promotes adhesion to fibronectin. J. Immunol. 159:3498–3507.
- Hankinson, O. 2005. Role of co-activators in transcriptional activation by the aryl hydrocarbon receptor. Arch. Biochem. Biophys. 433:379–386.
- Harper, P. A., Riddick, D. S., and Okey, A. B. 2006. Regulating the regulator: Factors that control levels and activity of the aryl hydrocarbon receptor. Biochem. Pharmacol. 72:267–279.
- Henry, E. C., and Gasiewicz, T. A. 2003. Agonist but not antagonist ligands induce conformational change in the mouse aryl hydrocarbon receptor as detected by partial proteolysis. Mol. Pharmacol. 63:392–400.
- Hestermann, E. V., and Brown, M. 2003. Agonist and chemopreventative ligands induce differential transcriptional cofactor recruitment by aryl hydrocarbon receptor. Mol. Cell. Biol. 23:7920–7925.
- Holsapple, M. P., Snyder, N. K., Wood, S. C., and Morris, D. L. 1991. A review of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced changes in immunocompetence: 1991 update. Toxicology 69:219–255.
- Inouye, K., Ito, T., Fujimaki, H., Takahashi, Y., Takemori, T., Pan, X., Tohyama, C., and Nohara, K. 2003. Suppressive effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the high-affinity antibody response in C57BL/6 mice. Toxicol. Sci. 74:315–324.
- Jeuken, A., Keser, B. J., Khan, E., Brouwer, A., Koeman, J., and Denison, M. S. 2003. Activation of the Ah receptor by extracts of dietary herbal supplements, vegetables, and fruits. J. Agric. Food Chem. 51:5478–5487.
- Kerkvliet, N. I. 2002. Recent advances in understanding the mechanisms of TCDD immunotoxicity. Int. Immunopharmacol. 2:277–291.
- Kerkvliet, N. I. 2003. Immunotoxicology of dioxins and related chemicals. In: Dioxins and Health, 2nd Edition (Schecter, A., and Gasiewicz, T. A., Eds.), New York: John Wiley & Sons, Inc., pp. 299–328.
- Kerkvliet, N. I., Shepherd, D. M., and Baecher-Steppan, L. 2002. T-Lymphocytes are direct, aryl hydrocarbon receptor (AhR)-dependent targets of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): AhR expression in both CD4+ and CD8+ T-cells is necessary for full suppression of a cytotoxic T-lymphocyte response by TCDD. Toxicol. Appl. Pharmacol. 185:146–152.
- Kimura, A., Naka, T., Nohara, K., Fujii-Kuriyama, Y., and T., Kishimoto. 2008. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc. Natl. Acad. Sci. USA 105:9721–9726.
- Laiosa, M. D., Wyman, A., Murante, F. G., Fiore, N. C., Staples, J. E., Gasiewicz, T. A., and Silverstone, A. E. 2003. Cell proliferation arrest within intrathymic lymphocyte progenitor cells causes thymic atrophy mediated by the aryl hydrocarbon receptor. J. Immunol. 171:4582–4591.
- Lawrence, B. P., and Kerkvliet, N. I. 2007. Immune modulation by TCDD and related polyhalogenated aromatic hydrocarbons. In: Immunotoxicology and Immunopharmacology, 3rd Edition (Luebke, R., House, R., and Kimber, I., Eds.), Boca Raton, FL: Taylor & Francis, pp. 239–258.
- Lawrence, B. P., Roberts, A. D., Neumiller, J. J., Cundiff, J. A., and Woodland, D. L. 2006. Aryl hydrocarbon receptor activation impairs the priming but not the recall of influenza virus-specific CD8+ T-cells in the lung. J. Immunol. 177:5819–5828.
- Liem, A. K., Furst. P., and Rappe. C. 2000. Exposure of populations to dioxins and related compounds. Food Addit. Contam. 17:241–259.
- McGuire, J., Okamoto, K., Whitelaw, M. L., Tanaka, H., and Poellinger, L. 2001. Definition of a dioxin receptor mutant that is a constitutive activator of transcription: Delineation of overlapping repression and ligand binding functions within the PAS domain. J. Biol. Chem. 276:41841–41849.
- Mimura, J., and Fujii-Kuriyama, Y. 2003. Functional role of AhR in the expression of toxic effects by TCDD. Biochem. Biophys. Acta 1619:263–268.
- Moennikes, O., Loeppen, S., Buchmann, A., Andersson, P., Ittrich, C., Poellinger, L., and Schwarz, M. 2004. A constitutively-active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 64:4707–4710.
- Mora, C., Grewal, I. S., Wong, F. S., and Flavell, R. A. 2004. Role of L-selectin in the development of autoimmune diabetes in non-obese diabetic mice. Int. Immunol. 16:257–264.
- Nguyen, L. P., and Bradfield, C. A. 2008. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 21:102–116.
- Nohara, K., Fujimaki, H., Tsukumo, S., Inouye, K., Sone, H., and Tohyama, C. 2002. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on T-cell-derived cytokine production in ovalbumin (OVA)-immunized C57Bl/6 mice. Toxicology 172:49–58.
- Nohara, K., Pan, X., Tsukumo, S., Hida, A., Ito, T., Nagai, H., Inouye, K., Motohashi, H., Yamamoto, M., Fujii-Kuriyama, Y., and Tohyama, C. 2005. Constitutively active aryl hydrocarbon receptor expressed specifically in T-lineage cells causes thymus involution and suppresses the immunization-induced increase in splenocytes. J. Immunol. 174:2770–2777.
- Nohara, K., Suzuki, T., Ao, K., Murai, H., Miyamoto, Y., Inouye, K., Pan, X., Motohashi, H., Fujii-Kuriyama, Y., Yamamoto, M., and Tohyama, C. 2009. Constitutively active aryl hydrocarbon receptor expressed in T-cells increases immunization-induced IFNγ production in mice but does not suppress TH2-cytokine production or antibody production Int. Immunology. 21:769–777.
- Quintana, F. J., Basso, A. S., Iglesias, A. H., Korn, T., Farez, M. F., Bettelli, E., Caccamo, M., Oukka, M., and Weiner, H. L. 2008. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 453:65–71.
- Ruby, C. E., Funatake, C. J., and Kerkvliet, N. I. 2005. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) directly enhances the maturation and apoptosis of dendritic cells in vitro. J. Immunotoxicol. 1:159–166.
- Schecter, A., Cramer, P., Boggess, K., Stanley, J., Papke, O., Olson, J., Silver, A., and Schmitz, M. 2001. Intake of dioxins and related compounds from food in the U.S. population. J. Toxicol. Environ. Health A 63:1–18.
- Schmidt, J. V., and Bradfield, C. A. 1996. Ah receptor signaling pathways. Annu. Rev. Cell Dev. Biol. 12:55–89.
- Schmidt, J. V., Su, G. H., Reddy, J. K., Simon, M. C., and Bradfield, C. A. 1996. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 93:6731–6736.
- Shepherd, D. M., Dearstyne, E. A., and Kerkvliet, N. I. 2000. The effects of TCDD on the activation of ovalbumin (OVA)-specific DO11.10 transgenic CD4+ T-cells in adoptively transferred mice. Toxicol. Sci. 56:340–350.
- Startin, J. R., and Rose, M. D. 2003. Dioxins and dioxin-like PCBs in food. In: Dioxins and Health, 2nd Edition (Schecter, A., and Gasiewicz, T. A., Eds.), New York: John Wiley & Sons, Inc., pp. 89–136.
- Sulentic, C. E. W., Holsapple, M. P., and Kaminski, N. E. 1998. Aryl hydrocarbon receptor-dependent suppression by 2,3,7,8-tetrachlorodibenzo-p-dioxin of IgM secretion in activated B-cells. Mol. Pharmacol. 53:623–629.
- Sun, Y. V., Boverhof, D. R., Burgoon, L. D., Fielden, M. R., and Zacharewski, T. R. 2004. Comparative analysis of dioxin response elements in human, mouse and rat genomic sequences. Nucl. Acids Res. 32:4512–4523.
- Veldhoen, M., Hirota, K., Westendorf, A. M., Buer, J., Dumoutier, L., Renauld, J. C., and Stockinger, B. 2008. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453:106–109.
- Vorderstrasse, B. A., and Kerkvliet, N. I. 2001. 2,3,7, 8-Tetrachlorodibenzo-p-dioxin affects the number and function of murine splenic dendritic cells and their expression of accessory molecules. Toxicol. Appl. Pharmacol. 171:117–125.
- Vorderstrasse, B. A., Dearstyne, E. A., and Kerkvliet, N. I. 2003. Influence of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the antigen-presenting activity of dendritic cells. Toxicol. Sci. 72:103–112.
- Warnock, R. A., Askari, S., Butcher, E. C., and von Andrian, U. H. 1998. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 187:205–216.
- Zhang, S., Rowlands, C., and Safe, S. 2008. Ligand-dependent interactions of the Ah receptor with co-activators in a mammalian two-hybrid assay. Toxicol. Appl. Pharmacol. 227:196–206.