
Abstract
Accumulation and aggregation of misfolded proteins is a hallmark of several diseases collectively known as proteinopathies. Autophagy has a cytoprotective role in diseases associated with protein aggregates. Age-related macular degeneration (AMD) is the most common neurodegenerative eye disease that evokes blindness in elderly. AMD is characterized by degeneration of retinal pigment epithelial (RPE) cells and leads to loss of photoreceptor cells and central vision. The initial phase associates with accumulation of intracellular lipofuscin and extracellular deposits called drusen. Epidemiological studies have suggested an inverse correlation between dietary intake of marine n-3 polyunsaturated fatty acids (PUFAs) and the risk of developing neurodegenerative diseases, including AMD. However, the disease-preventive mechanism(s) mobilized by n-3 PUFAs is not completely understood. In human retinal pigment epithelial cells we find that physiologically relevant doses of the n-3 PUFA docosahexaenoic acid (DHA) induce a transient increase in cellular reactive oxygen species (ROS) levels that activates the oxidative stress response regulator NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2). Simultaneously, there is a transient increase in intracellular protein aggregates containing SQSTM1/p62 (sequestosome 1) and an increase in autophagy. Pretreatment with DHA rescues the cells from cell cycle arrest induced by misfolded proteins or oxidative stress. Cells with a downregulated oxidative stress response, or autophagy, respond with reduced cell growth and survival after DHA supplementation. These results suggest that DHA both induces endogenous antioxidants and mobilizes selective autophagy of misfolded proteins. Both mechanisms could be relevant to reduce the risk of developing aggregate-associate diseases such as AMD.
Abbreviations
| AA | = | arachidonic acid |
| AMD | = | age-related macular degeneration |
| AREDS | = | Age-Related Eye Disease Study |
| ATF4 | = | activating transcription factor 4 |
| ATG4 | = | autophagy-related 4 |
| BafA1 | = | bafilomycin A1 |
| CHX | = | cycloheximide |
| CREB | = | cAMP responsive element binding protein |
| DCF | = | 5-(and-6)-carboxy-2',7'-dichlorodihydrofluorescein diacetate |
| DHA | = | docosahexaenoic acid |
| ER | = | endoplasmatic reticulum |
| HMOX1 | = | heme oxygenase 1 |
| KEAP1 | = | kelch-like ECH-associated protein 1 |
| MAP1LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MEF | = | mouse embryonic fibroblast |
| NAC | = | N-acetyl cysteine |
| NQO1 | = | NAD(P)H dehydrogenase, quinone 1 |
| NFE2L2 | = | nuclear factor, erythroid derived 2, like 2 |
| OA | = | Oleic acid |
| POS | = | photoreceptor outer segment |
| PPARA | = | peroxisome proliferator-activated receptor α |
| PUFA | = | polyunsaturated fatty acid |
| qRT-PCR | = | quantitative real-time polymerase chain reaction |
| ROS | = | reactive oxygen species |
| RPE | = | retinal pigment epithelial |
| SLC7A11 | = | solute carrier family 7 (anionic amino acid transporter light chain, xc- system), member 11 |
| SRXN1 | = | sulfiredoxin 1 |
| SQSTM1 | = | sequestosome 1 |
| TFEB | = | transcription factor EB |
| UBA | = | ubiquitin associated domain |
| WT | = | wild type. |
Introduction
Damaged proteins may have deleterious effects on cellular functions, and accumulation of misfolded proteins is the hallmark of several neurodegenerative diseases such as Huntington, Parkinson, and Alzheimer diseases and other age-related disorders.Citation1-6 Age-related macular degeneration (AMD) is a neurodegenerative disease of the eye and the leading cause of central blindness in western countries.Citation7,8 Currently, for 80% to 85% of the 30 to 50 million AMD patients worldwide there are no effective treatment alternatives.Citation9 Therefore, one major public health challenge is to devise an effective primary prevention of AMD and to improve current treatments.
The pathogenesis of AMD is initiated by degeneration and death of retinal pigment epithelial (RPE) cells followed by loss of the overlying photoreceptor neurons rod and cones.Citation10 Increased accumulation of intracellular auto-oxidative and autofluorescent lipofuscin in the lysosomes of RPE cells, as well as drusen formation in the extracellular space between the RPE and the Bruch membrane are hallmarks of AMD.Citation8,11-13 Lipofuscin is a brown-yellow, electron-dense, age-related pigment composed of a complex heterogeneous mixture of lipid–protein aggregates.Citation14 In drusens various acute phase inflammatory markers and oxidative stress-related proteins have been characterized.Citation15-17 Oxidative processes have been proposed to play a contributing role in AMD. RPE cells are exposed to chronic oxidative stress due to constant exposure to sunlight and relatively high oxygen tension, and high concentration of lipid peroxidation products from the ingested photoreceptor outer segments (POS).Citation10,18,19 Oxidatively damaged proteins post-translationally modified e.g. with carboxyethylpyrrole, malondialdehyde, 4-hydroxynonenal, and advanced glycation end products, accumulate in the macular area and serve to further elevate oxidative stress.Citation20 It is unclear whether accumulation and aggregation is the cause or the consequence of the disease. However, high amounts of deposits predict AMD progression and severity and it is thought that this aggregation of misfolded proteins occurs after failure of the cellular protein quality control mechanisms of the cells.Citation21 However, the role of aggregates in pathologies is controversial and may even protect the cells by sequestering putatively harmful soluble misfolded proteins. In Huntington disease it has been suggested that formation of aggregates may serve a cytoprotective role.Citation22 In addition, aggregates may also facilitate the clearance of the toxic materials.Citation21
Two major proteolytic systems are responsible for maintaining the cellular function: the proteosomal and lysosomal system. Both systems remove irreversibly damaged proteins and recycle amino acids for protein synthesis. The activity of these systems decline upon aging.Citation23 Macroautophagy (hereafter referred to as autophagy) is a catabolic process that removes damaged and foreign intracellular components by lysosomal degradation.Citation24 During autophagy, targeted cytoplasmic proteins and organelles are sequestered by a growing double membrane that forms an autophagosomal vesicle where the content is degraded after fusion with a lysosome. The rate of cellular turnover by autophagy increases in response to cellular stresses like starvation, endoplasmatic reticulum (ER) stress, and elevated levels of reactive oxygen species (ROS) for maintenance of cellular homeostasis.Citation25-27 The SQSTM1/p62 protein (hereafter referred to as SQSTM1) binds both to ubiquitinated cargos, such as misfolded proteins and protein aggregates via its ubiquitin-associated (UBA) domain and also to the mammalian orthologs of yeast Atg8, located on the phagophore membrane.Citation28 In this way, SQSTM1 selectively targets ubiquitinated protein aggregates to lysosomal degradation.Citation28,29 A specific binding of SQSTM1 to misfolded and ubiquitinated proteins, and its presence in cytoplasmic inclusions in diverse human diseases have suggested a general role of SQSTM1 in diseases associated with protein aggregates.Citation5
Tissue-specific knockout of autophagy genes in neurons or hepatocytes results in early onset neurodegeneration and liver failure, respectively, accompanied by accumulation of misfolded protein aggregates and ubiquitinated proteins.Citation30,31 Inducible knockout of autophagy in mice limits survival to 2 to 3 mo due to development of severe neurodegeneration.Citation32 These findings suggest that autophagy is an important cytoprotective mechanism that counteracts the development of several age-related diseases, especially proteinopathies, by clearence of damaged proteins and organelles.Citation33 Lysosomal-mediated clearance is also important in RPE cellsCitation34 supporting the idea that also in RPE cells, autophagy is critical to maintain cellular homeostasis. Whether autophagy could be induced as a disease preventive mechanism in these cells is still uncertain. However, caloric restriction and compounds like resveratrol can extend life span in different model organisms possibly due to increased autophagy.Citation35,36
Epidemiological studies indicate an inverse correlation between dietary intake of fish and the risk of developing AMD.Citation37-42 The disease preventive effects of increased fish intake have been associated with the content of marine omega-3 polyunsaturated fatty acids (n-3 PUFAs).Citation43 Deficiency of n-3 PUFAs in photoreceptors is associated with the development of AMD.Citation44 Increased intake of marine n-3 PUFAs has also been suggested to reduce the risk of other age-related disorders such as neurodegenerative diseases, different types of cancer, and heart and circulatory diseases.Citation45-47 However, the disease preventive mechanism(s) mobilized by dietary n-3 PUFAs are not completely understood. Previously, several reports have suggested a change in autophagy in cancer cell lines that are sensitive and display cytotoxic and/or cytostatic responses to physiological doses of PUFAs.Citation48-53 We explore whether increased autophagy is a part of the cellular response to PUFAs also in spontaneously arising ARPE-19 cells. If so, we hypothesize a correlation between the disease preventive effects of n-3 PUFAs and stimulation of autophagy in retinal pigment epithelial cells where the initial phases of AMD occurs.
Here we report that the n-3 PUFA docosahexaenoic acid (DHA, 22:6, n-3), in contrast to the n-6 PUFA arachidonic acid (AA, 20:4, n-6) and the n-9 fatty acid oleic acid (OA, 18:1, n-9), induces a transient increase in reactive oxygen species (ROS) in spontaneously arising retinal pigment epithelial cells. This mild, subtoxic stress is counteracted by activation of the antioxidant stress response transcription factor NFE2L2/NRF2 (nuclear factor, erythroid derived 2, like 2) that controls the transcription of a number of genes encoding endogenous enzymatic and nonenzymatic antioxidants. DHA also causes a selective rise in SQSTM1 mRNA and protein levels in an NFE2L2-dependent manner. Further we observe a transient increase in sequestration of misfolded proteins into aggregates after DHA that coincides with an increase in autophagy that could facilitate clearance of the protein aggregates. In line with a mobilization of a protective response, we find that DHA increases the tolerance for oxidative stress and misfolded proteins in retinal pigment epithelial cells.
Results
The n-3 PUFA DHA induces protein aggregation and autophagy in ARPE-19 cells
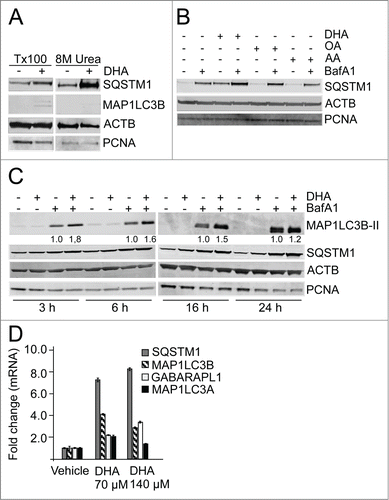
n-3 PUFAs have been suggested to mobilize disease preventive effects for several age-related diseases. Since insufficient autophagy has been proposed to contribute in the development of several of the same diseases, we asked if n-3 PUFA supplementation could induce autophagy. Cellular responses to lipids were determined in the presence of serum to mimic the in vivo situation. The n-3 PUFA DHA (22:6, n-3) was used in final concentrations of 70 µM and 140 µM in the cell culture experiments which is well within physiological relevant levels found in serum of healthy individuals.Citation54 The diploid, spontaneously derived ARPE-19 human retinal pigment epithelial cellsCitation55 were used as a model system since these cells are relevant for the development of AMD. To determine the basal autophagy flux in these cells, cell extracts were analyzed by immunoblot for accumulation of lipidated microtubule-associated protein 1 light chain 3 β (MAP1LC3B-II/LC3B-II) and SQSTM1 in the absence or presence of the autophagy/lysosomal inhibitor bafilomycin A1 (BafA1) for different time points. In exponentially growing ARPE-19 cells, both SQSTM1 and MAP1LC3B-II protein levels displayed a linear increase with time throughout the experiment. The doubling time for SQSTM1 and MAP1LC3B-II was determined to be approximately 7 h and 5 h, respectively (Fig. S1). Since SQSTM1 is associated with protein aggregates that might be resistant to detergents, the pellets that remain after Triton X-100 protein extraction and centrifugation of the lysates, were resolved in a buffer containing 8 M urea to also solubilize also detergent-resistant proteins. A very clear increase in SQSTM1 protein level was observed in response to DHA in the detergent-resistant pellet while very little MAP1LC3B could be detected (). During the rest of the study, cells were lysed directly in a buffer containing 8 M urea to avoid losing part of the cellular pool of SQSTM1. We then tested whether other lipids also could induce the level of SQSTM1. Interestingly, whereas stimulation with DHA clearly increased the protein level of SQSTM1, that was further elevated when combining DHA and BafA1 (), no increase was observed after treatment with OA nor AA (). The SQSTM1 gene is induced in response to different types of cellular stresses and the protein is continously turned over by autophagy. Since combining the DHA stimuli and the lysosomal inhibitor caused an additive effect, this suggests an increased autophagic turnover of SQSTM1 in response to DHA. Consistently, supplementation with DHA also increased the level of MAP1LC3B-II when combined with lysosomal inhibition (). This observation indicates an autophagy-inducing activity of DHA in the ARPE-19 cells. To determine the time needed for DHA to induce autophagy and increase the level of SQSTM1, cell extracts were prepared after 3, 6, 16, and 24 h supplementation with DHA and BafA1. Already after 3 h with DHA supplementation, the turnover of both SQSTM1 and MAP1LC3B-II was induced, and the effect lasted for the duration of the experiment (). The additive effect of DHA supplementation and inhibition of autophagic degradation by BafA1 suggests lipid-induced synthesis of the 2 proteins. In line with this notion, quantitative real-time PCR (qRT-PCR) analyses revealed a more than 7-fold increase in SQSTM1 mRNA and more than 4-fold increase in MAP1LC3B mRNA levels in response to 16 h DHA treatment (). Interestingly, among the mammalian orthologs of yeast Atg8, the induction of MAP1LC3B seems selective since only minor changes could be detected in mRNA levels of MAP1LC3A and GABARAPL1. Together, these data suggest that the synthesis of SQSTM1 and MAP1LC3B is induced and autophagy increased in response to DHA in a lipid-specific manner.
Figure 1. The n-3 PUFA DHA increases protein level of SQSTM1 and induces autophagy in ARPE-19 cells. (A) Cells were treated with DHA (70 µM) for 24 h and lysed in Triton X-100 (Tx100) buffer. Equal amounts of protein (20 µg) from T × 100 fraction were centrifugated at 10,000 x g and the pellet was dissolved in the same volume of 8 M urea buffer before loading on the gel. The membrane was immunoblotted for SQSTM1 and MAP1LC3B. β-actin (ACTB) and PCNA are used as loading controls. (B) The cells were treated with DHA, OA or AA (70 µM) with or without BafA1 (100 nM) for 16 h. Total cell extracts were immunoblotted for SQSTM1. ACTB and PCNA are used as loading controls. (C) Protein levels of SQSTM1 and MAP1LC3B determined by immunoblotting of cells treated with DHA (70 µM), BafA1 (100 nM) or a combination of DHA and BafA1 for the indicated time points. The numbers below the MAP1LC3B-II bands represent fold change relative to BafA1 for each time point normalized to PCNA intensity. ACTB and PCNA are used as loading controls. (D) The mRNA levels of SQSTM1, MAP1LC3B, MAP1LC3A, and GABARAPL1 relative to ACTB after DHA (70 and 140 µM) supplementation for 16 h determined by quantitative real-time PCR. qRT-PCR data displayed are representative for 2 independent experiments. Mean fold change from triplicate wells ± SD is displayed. Data shown are representative of 3 or more independent experiments, unless otherwise stated.
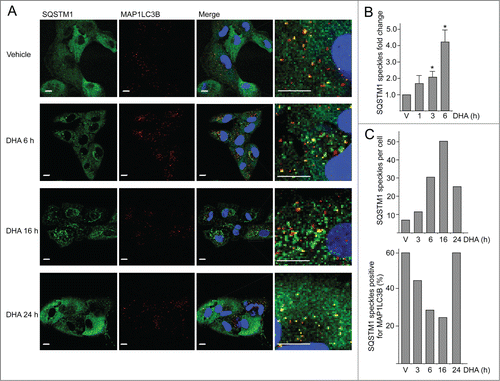
Since SQSTM1 was found in the detergent-resistant fraction after DHA supplementation, the cells were immunostained for SQSTM1 and MAP1LC3B. In response to DHA, a transient increase in number and size of SQSTM1-positive punctate cytosolic structures was observed (). The number of SQSTM1-positive structures increased with time up to 16 h. A partial colocalization with MAP1LC3B was observed, which might represent autophagosomes. To quantify the number of punctate SQSTM1-positive structures per cell, more than 500 cells per condition were analyzed using automated imaging. Consistent with the manual inspection, automated image analyses demonstrated that the average number of SQSTM1 punctate structures increased with time after DHA supplementation (). The average number of SQSTM1-positive speckles increased from less than 10 per cell in untreated cells to approximately 50 per cell in cells treated with DHA for 16 h. Interestingly, the number of SQSTM1 speckles that colocalized with MAP1LC3B decreased from approximately 60% in the untreated cells to less than 30% in the cells treated with DHA for 16 h. By extending the treatment time to 24 h, the number of punctate SQSTM1 structures was reduced, and the frequency of colocalization with MAP1LC3B increased (). Together, these data indicate that cells respond to DHA by inducing a transient increase in SQSTM1-positive speckles. The reduction in the number of these speckles coincides with an increased turnover of MAP1LC3B-II and elevated colocalization between SQSTM1 and MAP1LC3B.
Figure 2. The number of SQSTM1-positive protein speckles in ARPE-19 cells increases after DHA supplementation. (A) Immunostaining for SQSTM1 and MAP1LC3B after DHA (70 µM) treatment for indicated time points. Nuclear DNA was stained using Draq5 (5 µM). Scale bar: 10 µm. (B) Cells were treated with vehicle (V) or DHA (70 µM) for 1, 3, and 6 h. The SQSTM1-positive speckles were automatically quantified using ScanR automated image acquisition. The quantification displayed are representative for 3 independent experiments from where 2 are automatically quantified for more than 1,000 cells per condition and one is manually counted. *) indicates significantly different from control, Student t test P < 0.05. (C) The number of SQSTM1-positive speckles per cell (upper panel) and SQSTM1 speckles positive for MAP1LC3B (lower panel) in ARPE-19 cells supplemented with vehicle (V) or DHA (70 µM) for the indicated time points. The quantification displayed was performed manually for more than 100 cells per condition from one representative experiment. This quantification is representative for 3 independent experiments.
DHA induces a transient increase in ROS and activation of NFE2L2 in ARPE-19 cells
PUFA supplementation causes a rise in the level of reactive oxygen species (ROS) in different cell types,Citation56 and to induce oxidative stress response genes in colon cancer cells.Citation57 In response to DHA (70 µM and 140 µM) there was a significant increase in ROS levels at 3 h and then the level was reduced with time (). Interestingly, 24 h after adding DHA (140 µM ) the level of ROS was lower compared to control cells. The DHA-induced increase in ROS levels could be counteracted by pretreating the cells with the exogeneous antioxidants N-acetyl-cysteine (NAC) or vitamin E (). DHA treatment for 3 h resulted in significantly higher levels of ROS compared to treatment with AA or OA for the same time-period (). Also, no further increase in ROS levels was observed after 6 h and 24 h supplementation with OA or AA (data not shown). Increased levels of ROS represent a stress situation that is counteracted by numerous cellular responses including changes in gene expression coordinated by the transcription factor NFE2L2. In response to ROS, NFE2L2 is stabilized and translocated to the nucleus.Citation58 Consistent with the ROS measurements, immunofluorescent analyses demonstrated a clear increase in nuclear localization of NFE2L2 in response to DHA while AA and OA did not affect the level of nuclear NFE2L2 significantly (). The immunostaining approach for evaluating NFE2L2 activation was specific since NFE2L2 siRNA caused a loss in intensity (data not shown). In line with a ROS-mediated activation of NFE2L2 after DHA supplementation, pretreatment with NAC counteracted nuclear translocation of NFE2L2 () and induction of HMOX1 (heme oxygenase 1) (), representing one of the typical NFE2L2 targets induced by oxidative stress.Citation59 Immunoblot analyses also demonstrated a clear increase in NFE2L2 protein level in response to DHA consistent with a stabilization of the protein (). Induction of HMOX1 was further analyzed after adding different lipids and found to be selective for DHA (). Further, induction of NFE2L2 was validated by microarray gene expression analyses Using MetaCore for pathway analyses, it was revealed that NFE2L2-associated stress responses were significantly activated after 12 h DHA treatment (P < 10−6). The identified, upregulated NFE2L2 target transcripts included HMOX1, NQO1 (NAD[P]H dehydrogenase, quinone 1), SRXN1 (sulfiredoxin 1), ATF4 (activating transcription factor 4), and SLC7A11 (solute carrier family 7 [anionic amino acid transporter light chain, xc- system], member 11). In line with these findings, the mRNA level of SQSTM1, another NFE2L2 target gene, was highly increased after 16 h DHA supplementation determined by qRT-PCR (). In summary, these data are consistent with a lipid selective induction of ROS that results in elevated transcription of NFE2L2 controlled genes in response to DHA in ARPE-19 cells.
Figure 3. DHA induces a transient increase in ROS and induce NFE2L2 cytoprotective genes. (A) Changes in ROS levels measured at different time points after DHA (70 and 140 µM) using a fluorescent ROS DCF probe. The data represent the mean fold change ± SD for 6 independent experiments for 3 h and 3 independent experiments for 6 h and 24 h. Each experiment was performed in duplicates where the mean intensity of 10,000 cells per well ± SD was measured. *) indicates significantly different from control, Student t test P < 0.05 and **) P < 0.01. (B) Where indicated the cells were pretreated with antioxidants (5 mM N-acetyl-cysteine (NAC) or 150 µM vitamin E) for 16 h before further stimulations with 140 µM DHA for 3 h. The data represent the average of 3 independent experiments for the DHA and NAC treatments and the average of 2 independent experiments for the Vitamin E treatment. Each experiment was performed in duplicates where the mean intensity of 10,000 cells per well ±SD was measured. **) indicates significantly different from DHA, Student t test P < 0.01. (C) Changes in ROS levels measured 3 h after DHA, OA or AA (140 µM) supplementation using a DCF fluorescent probe. The data represent the average of 3 independent experiments ±SD for DHA treated samples and 2 independent experiments ±SD for AA and OA treated samples. Each experiment was performed in duplicates where the mean intensity of 10,000 cells ±SD per well was measured. (D) Immunostaining of NFE2L2 after 70 µM DHA, OA, AA for 6 h. Nuclear DNA was stained using Draq5 (5 µM). Scale bar: 10 µm. The results are representative for 3 independent experiments. Nuclear NFE2L2 staining from one representative experiment was automatically quantified using ScanR automated image acquisition of more than 3,000 cells. Each experiment was performed in duplicates and the data are presented as average percentage number of cells with NFE2L2 nuclear staining ± SD. (E) The cells were pretreated with NAC (5 mM) for 1 h prior to further stimulation with DHA (70 µM) in combination with NAC for 6 h. After fixation, the cells were immunostained for NFE2L2. Data are representative for 2 independent experiments. The percentage of cells with NFE2L2 nuclear staining from one representative experiment was automatically quantified using ScanR automated image acquisition. Each experiment was performed in duplicate and the data displayed represent the average percentage number of cells with NFE2L2 nuclear staining ± SD. (F) ARPE-19 cells were pretreated with NAC (5 mM) for 1 h following stimulation with DHA (70 µM) for 6 h and BafA1 (100 nM) the last 2 h. Levels of HMOX1 was determined by immunoblotting. COX4I1 was used as loading control. (G) The ARPE-19 cells were treated with DHA, OA or AA (70 µM) with or without BafA1 (100 nM) for 16 h before immunoblotting for HMOX1 (100 µg protein loaded). ACTB/β-actin and PCNA were used as loading controls. (H) Immunoblot of NFE2L2, HMOX1, KEAP1 (100 µg protein loaded), SQSTM1, ACTB and PCNA (loaded 20 µg protein) after DHA (70 µM) with or without BafA1 (100 nM) for 16 h. Arrows represent NFE2L2 and KEAP1 bands while *) represents a nonspecific NFE2L2 band. ACTB and PCNA were used as loading controls. (I) Cells were treated as in (G) and immunoblotted for phosphorylated SQSTM1 (Ser351) and total SQSTM1 (100 µg protein loaded). ACTB and PCNA were used as loading controls. Data shown are representative of 3 or more independent experiments, unless otherwise stated.
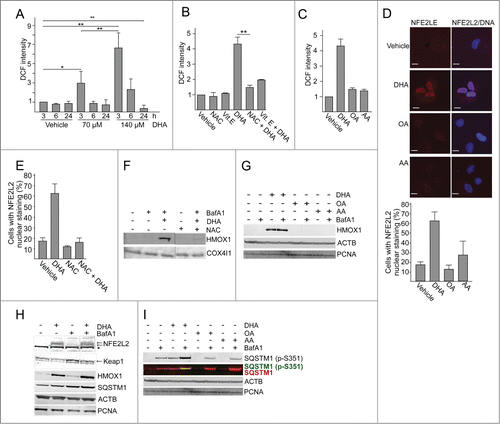
The DHA-induced increase in the protein level of NFE2L2 corresponded with a slight reduction in the protein level of one of its negative regulators KEAP1 (kelch-like ECH-associated protein 1) (). This reduction was blocked by BafA1, supporting the notion that KEAP1 is degraded by selective autophagy.Citation60 Under resting conditions, KEAP1 sequesters NFE2L2 and targets it for proteosomal degradation.Citation61 Elevated cellular levels of ROS cause KEAP1 to dissociate from NFE2L2.Citation62 In addition, SQSTM1 can sequester KEAP1 and the affinity increases upon phosphorylation of SQSTM1 at serine 351 (Ser351) and this mechanism represents an alternative route to activate NFE2L2.Citation63-66 Interestinly, using an antibody specific for SQSTM1 phosphorylated at Ser351, a clear raise was observed in response to DHA, but not to AA or OA (). Inhibition of lysosmal degradation also caused an increase in the cellular level of this modified form of SQSTM1 that was further enhanced by cotreatment with DHA. Together, these data is consistent with DHA-induced ROS and a resulting stabilization and nuclear translocation of NFE2L2. However, we cannot exclude that phosphorylation of SQSTM1 at Ser351 also contributes to the observed activation of NFE2L2. Activation of NFE2L2 results in elevated mRNA and protein levels of SQSTM1 and HMOX1. Prolonged exposure to DHA results in cellular ROS levels that are lower compared with control cells indicating an induction of endogenous antioxidants by DHA. Interestingly, pretreating the cells with exogenous antioxidants counteracted the DHA-induced ROS levels as well as nuclear translocation and activation of NFE2L2, indicating that ROS is clearly involved in the DHA-dependent activation of NFE2L2.
NFE2L2, SQSTM1, and ATG5 are important in the cellular responses to DHA by limiting oxidative stress and mediating cell survival
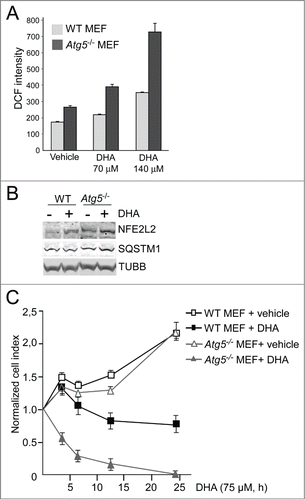
To evaluate the importance of NFE2L2, SQSTM1 and autophagy in the cellular responses toward DHA, the cells were transfected with targeted siRNAs. More than 60% reduction in ATG5 protein level was observed in cells transfected with ATG5 siRNA. However, the cells with reduced ATG5 protein levels did not display any reduced ability to form MAP1LC3B-II or degrade SQSTM1 (), indicating that the remaining ATG5 protein provides sufficient catalytic activity to maintain autophagy largely unchanged. This is in line with previous findings reporting that very low levels of ATG12–ATG5 might be sufficient for maintaining autophagy.Citation67 Even though no clear reduction in autophagy could be observed in the ATG5 siRNA tranfected cells, we could still observe a tendency of potentiation of ROS levels in response to DHA compared to the control siRNA-transfected cells (). In addition, downregulation of ATG5 protein levels affected the growth of the cells treated with DHA compared to the control-transfected cells (). These relative differences in sensitivity toward DHA were also observed by counting the number of viable cells 48 h after adding DHA (data not shown). However, since we were unable to establish a clear reduction in autophagy in the ARPE-19 cells after downregulation of ATG5, we are uncertain how these effects relate to autophagy. We therefore analyzed ROS levels and cell survival after DHA supplementation in wild-type (WT) and atg5-deficient mouse embryonic fibroblasts (MEFs). Compared to the WT MEFs, the atg5−/− MEFs displayed an elevated basal level of ROS () and NFE2L2 () in line with previous notions.Citation26 In response to DHA, the level of both ROS and NFE2L2 increased to higher levels in atg5−/− MEFs () and the autophagy-deficient cells were found more sensitive to DHA (). Together, these findings indicate a cytoprotective role of autophagy in the cellular responses toward DHA.
Figure 4. ATG5 is important in the cellular responses to DHA. (A) ARPE-19 cells were transfected with control siRNA or ATG5 siRNA (100 nM) and left for 24 h before reseeding. Following incubation for 24 h, the cells were added DHA (70 μM) or BafA1 (100 nM) for 24 h and immunoblotted for ATG5, SQSTM1, and MAP1LC3B. ACTB and PCNA were used as loading controls. (B) The cells were siRNA-transfected as in (A). After DHA (140 µM) treatment for 3 h changes in ROS levels were measured using a fluorescent ROS DCF probe. The results are representative for 2 independent experiments. Each experiment was performed in duplicates where the mean intensity ±SD of 10,000 cells per well was measured. The control is normalized to one and the relative fold changes are shown. (C) Relative cell index after transfection with control or ATG5 siRNA (100 nM) after vehicle or DHA (140 μM) based on real-time monitoring using the xCELLigence instrument. The cell index for each treatment was normalized to one at the start of the experiment. For each time point the cell index of control samples (Control siRNA + vehicle and ATG5 siRNA + vehicle) was normalized to 1. The effect of DHA treatment after transfection with either Control siRNA or ATG5 siRNA is shown relative to the corresponding controls. Mean normalized cell index with standard deviation of triplicate wells of vehicle or DHA treated cells is displayed. Data shown are representative for 2 independent experiments.
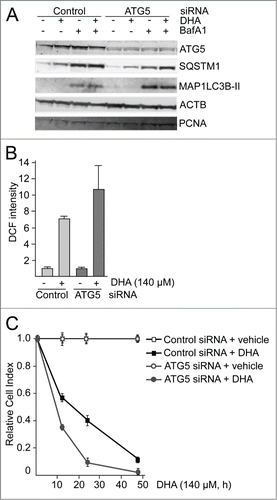
Figure 5. The atg5 knockout MEFs are more sensitive to DHA compared to wild-type MEFs. (A) The levels of ROS were measured in wild-type (WT) and atg5−/− MEFs after 3 h DHA treatment (70 and 140 µM) using the fluorescent DCF probe. The data from one representative experiment of 3 independent experiments are displayed. Each experiment was performed in triplicate wells where the mean intensity ±SD of 10,000 cells per well was measured. (B) The levels of NFE2L2 and SQSTM1 after vehicle or DHA (70 μM, 16 h) treatment in wild-type and atg5−/− MEFs (85 μg protein loaded). TUBB/β-tubulin was used as loading control. The immunoblot is representative for 3 independent experiments. (C) Wild-type and atg5−/− MEFs were exposed to DHA (75 μM) and cellular responses observed over time using the xCELLigence real-time monitoring system. The cell index was normalized to one at the start of the experiment. Mean normalized cell index ±SD of triplicate wells of vehicle or DHA treated cells are displayed. The results are representative for 5 independent growth experiments scored by cell index using xCELLigence.
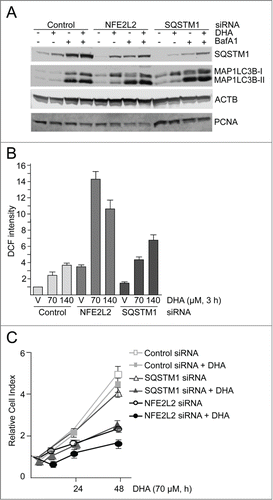
Using well-established siRNA probes targeting NFE2L2,Citation28,64 we estimated NFE2L2 to be approximately 70% downregulated by immunostaining (data not shown) and SQSTM1 was reduced more than 50% after cotreatment with DHA and BafA1 (). Also, the DHA-induced level of HMOX1 was clearly reduced in the NFE2L2 siRNA-transfected cells compared to the cells transfected with control siRNA (Fig. S2). These results are consistent with a important role of NFE2L2 in the regulation of both HMOX1 and SQSTM1 in response to DHA. The clear reduction in SQSTM1 and HMOX1 indicates an efficient and functional downregulation of NFE2L2. Targeting SQSTM1 with siRNA caused a more than 90% reduction in the protein level of SQSTM1 (). Interestingly, the level of MAP1LC3B-I was elevated in response to both NFE2L2 and SQSTM1 siRNA transfection compared to control siRNA-transfected cells. The induced level of MAP1LC3B-I was evident in the untreated control and further increased in response to DHA, indicating that the cells compensate for reduced levels of NFE2L2 and SQSTM1 by inducing the synthesis of MAP1LC3B protein and activation of autophagy. Consistent with an important role of NFE2L2 in the cellular responses to DHA, a more than 3-fold increase in basal ROS levels was observed that was further strongly increased by DHA in the NFE2L2 siRNA-transfected cells (). Downregulation of SQSTM1 also resulted in a slightly increased basal ROS level and a further elevation after DHA supplementation compared to control siRNA-transfected cells. Since SQSTM1 is only one of a number of different cytoprotective genes controlled by NFE2L2, the data demonstrate that downregulation of NFE2L2 is more severe in terms of basal and induced levels of ROS compared to siRNA-mediated downregulation of SQSTM1. Accordingly, real-time monitoring of cells transfected with siRNA against NFE2L2 and SQSTM1 displayed a reduced cell growth in the presence of 70 μM DHA, consistent with a cytostatic effect. Again, siRNA mediated downregulation of NFE2L2 was found more severe than downregulation of SQSTM1 after DHA supplementation (). By increasing DHA concentration (140 μM), the sensitivity was further increased (data not shown). Together, these results indicate that NFE2L2, SQSTM1 and autophagy cooperate in regulating the cellular responses toward DHA; interfering with any of these processes turns a transient, mild increase in ROS into a cytotoxic stress condition.
Figure 6. NFE2L2 and SQSTM1 are important in the cellular responses to DHA in ARPE-19 cells. (A) Cells were transfected with control, NFE2L2 and SQSTM1 siRNA (25 nM) and left for 24 h before reseeding. Following incubation for 24 h, the cells were added DHA (70 μM) or BafA1 (100 nM) for 24 h. Immunoblot for SQSTM1 and MAP1LC3B. ACTB/β-actin and PCNA were used as loading controls. (B) The cells were siRNA-transfected as in (A). After vehicle (V) and DHA (70 and 140 µM) treatment for 3 h changes in ROS levels were measured using a fluorescent ROS DCF probe. The data are representative for 2 independent experiments both performed in duplicates. The data represent the mean intensity ±SD of 10,000 cells per well and is displayed as relative DCF intensity. (C) Relative cell index after transfection with control, NFE2L2 or SQSTM1 siRNA (25 nM) after vehicle and DHA treatment (70 μM) based on real-time monitoring using the xCELLigence instrument. The cell index was normalized to one at the start of the experiment. Mean normalized cell index with standard deviation of triplicate wells of vehicle and DHA treated cells is displayed. Data shown are representative for 3 independent experiments.
DHA mobilizes cytoprotection toward protein aggregates and oxidative stress
Protein aggregates are involved in aging, diseases, and cell death, and autophagic degradation of these aggregates is important for cell survival. We observed that DHA stabilizes and activates NFE2L2, and causes a subsequent increase in SQSTM1 mRNA and protein levels. SQSTM1 are involved in sequestration of misfolded, ubiquitinated proteins into protein aggregates, and ensures selective degradation of these by autophagy.Citation29,68 Therefore, we wanted to investigate if DHA makes the cells more resistant to accumulation of protein aggregates or subsequent oxidative stress. Treating the cells with puromycin, a protein synthesis inhibitor that causes release of premature and misfolded proteins during translation,Citation28,69 led to a clear increase in mono- and polyubiquitinated protein aggregates (). In contrast, no such increase could be observed in response to the translational inhibitor cycloheximide (CHX) for the same period of time.
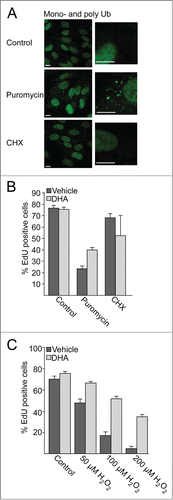
After pretreating the cells with DHA, the cells were further treated with puromycin or cycloheximide for 4 h. The drugs were then removed and the cells were incubated for a further 16 h in the presence of the tymidine analog 5-ethynyl-2′-deoxyuridine (EdU) to evaluate the frequency of cells performing S phase during this time.Citation70 By automated imaging and image analyses of more than 3,500 cells, it was found that more than 75% of the untreated cells displayed nuclei positive for EdU (). Pretreatment with DHA did not affect the frequency of cells that performed S phase. In response to a 4 h treatment with puromycin, but not cycloheximide, the number of nuclei positive for EdU was clearly reduced compared to control. Interestingly, pretreating the cells with DHA partially rescued the cell proliferation following a puromycin challenge. These results indicate that under conditions where SQSTM1 is induced, there is also an improved tolerance for misfolded proteins formed in response to puromycin.
To test whether activation of NFE2L2 observed after DHA stimulation makes the cells more resistant to subsequent exposure to oxidative stress, ARPE-19 cells were pretreated with DHA for 16 h following a 30 min challenge with hydrogen peroxide (H2O2). After washing, cells were incubated for 16 h in the presence of EdU. Again, pretreatment with DHA partially rescued proliferation of the cells (). Together, these results indicate that DHA induces cellular responses that mobilize resistance to misfolded proteins and oxidative stress.
Discussion
We here demonstrate that the n-3 PUFA DHA selectively induces a transient increase in cellular ROS levels that is counteracted by activation of NFE2L2 and induction of oxidative stress response genes and proteins in ARPE-19 cells. In addition, DHA stimulates the synthesis of SQSTM1 and elevates autophagy. Pretreatment with DHA counteracted cell cycle arrest induced by misfolded proteins or oxidatives stress. Together our data indicate that DHA induces an interplay between NFE2L2 activation and autophagy in retinal pigment epithelial cells that makes the cells more tolerant to misfolded proteins and oxidative stress. These responses could represent putative disease preventive mechanisms mobilized after a mild, transient oxidative stress induced by DHA in a lipid selective manner.
Elevated levels of ROS are implicated in the pathogenesis of a number of neurodegenerative diseases, including AMD.Citation18,71 NFE2L2 is the main regulator of the expression of genes encoding proteins that controls cellular redox status.Citation58,72 Upon aging, nfe2l2-knockout mice have an increased risk of developing AMD-like phenotype,Citation73 emphasizing the important cytoprotective role of the cell's endogenous antioxidative system. Physiologically relevant doses of DHA caused a transient increase in ROS followed by increased protein levels and nuclear translocation of NFE2L2. Subsequently, increased expression of NFE2L2-modulated genes was detected by gene-expression arrays, and as increased protein levels of the NFE2L2 regulated proteins HMOX1 and SQSTM1. The n-6 PUFA arachidonic acid and the monounsaturated oleic acid did not cause a similar increase in ROS or activation of NFE2L2. Currently, the mechanisms underlying this lipid selectivity is incompletely understood. However, these lipids serve as precursors for different types of lipid-derived signaling compounds resulting from both nonenzymatic and enzymatic reactions.Citation74 The ARPE-19 cells did not display any changes in cell growth or survival after adding any of the lipids (70 µM). Nevertheless, in cells where NFE2L2 was downregulated, both the basal and DHA-induced levels of ROS were increased consistent with a central role of NFE2L2 in redox balance. Interestingly, in cells depleted for NFE2L2 the growth rate was clearly affected even under normal growth conditions. After DHA supplementation the cytostatic response was further potentiated. These data are consistent with previous reports demonstrating that DHA directly or indirectly induces NFE2L2.Citation74,75 Also, we found that DHA pretreatment protected the ARPE-19 cells from a cytostatic effect upon a subsequent challenge with hydrogen peroxide. Together, these results indicate that DHA induces activation of NFE2L2 and an increased buffer capacity for oxidative stress in retinal pigment epithelial cells.
Autophagy has emerged as a cellular process for selective clearance of damaged proteins and organells, and is crucial to avoid the development of several age-related diseases, including different types of neurodegeneration.Citation76-78 Mice genetically modified to lack autophagy in a tissue-specific manner display early onset neurodegeneration accompanied by accumulation of misfolded protein aggregates and ubiquitinated proteins.Citation30,31 Inducible, systemic knockout of autophagy restricts lifespan in mice to 2 to 3 mo due to the development of severe neurodegeneration.Citation32 Interestingly, deletion of ATG5 in the lens has been reported to result in age-related cataract accompanied by accumulation of polyubiquitinated and oxidized proteins and SQSTM1.Citation79 ATG5 is also required for lysosomal fusion of phagosomes containing photoreceptor outer segments (POS) important in renewal of photoreceptors in RPE cells and thus optimal vision.Citation80 Also, it was recently reported that prolonged use of the autophagy inhibitor chloroquine in treatment of malaria, could cause chloroquine-associated visual loss due to degeneration of RPE cells.Citation81 AMD develops with age and a marked reduction in autophagy activity in the retina of aged mice have been suggested to be involved in age-associated visual loss and retinal dystrophy.Citation82 Together, these findings suggest that elevation of autophagy could protect from development of AMD as well as other neurodegenerative diseases. In this context it is interesting that physiologically relevant concentrations of DHA increases autophagy in the ARPE-19 cells. In addition to increased autophagy, we also observed an increased level of SQSTM1 mRNA and SQSTM1 protein level. SQSTM1 selectively targets misfolded, ubiquitinated proteins for lysosomal degradation by binding both to ubiquitinated cargos, via its UBA domain, and to mammalian orthologs of yeast Atg8 on the growing phagophore membrane.Citation28 In this way, the elevated levels of SQSTM1 observed after DHA might enhance the cell's capacity to sequester damaged and ubiquitinated proteins into aggregates. In accordance, a transient increase in the number and size of cytosolic protein aggregates positive for SQSTM1 was observed after DHA. The reduction in the number of these speckles coincides with an increased turnover of MAP1LC3B-II and elevated colocalization between SQSTM1 and MAP1LC3B, which may indicate selective removal of these structures by increased autophagy.
Intriguingly, we observe DHA pretreatment to protect ARPE-19 cells from cell cycle arrest induced by misfolded proteins released during protein translation in the presence of puromycin. We speculate that this may be due to DHA-induced increase in SQSTM1 and subsequent aggregation and autophagic clearance. Such a model would be consistent with the protective role of ubiquitination and aggregation of mutant aggregate-prone huntingtin as a mechanism for cell survival,Citation22,83 and autophagy as the mechanism responsible for the clearance of these huntingtin aggregates.Citation29,84 Also, others have reported that DHA-derived ROS is involved in protein quality control by regulating aggregation and further autophagic degradation of misfolded apolipoprotein B in hepatocytes.Citation85
Interestingly, both the basal and DHA-induced level of MAP1LC3B-I were elevated in cells depleted for NFE2L2 and SQSTM1. This increase in MAP1LC3B-I could indicate a compensatory role of autophagy under conditions of increased ROS levels. Downregulation of SQSTM1 did not influence cell growth under normal conditions but resulted in a prominant cytostatic response when combined with DHA treatment. As expected, the cytostatic responses toward DHA after downregulating SQSTM1 were not as clear compared to cells with downregulated NFE2L2. These differential effects likely reflect that NFE2L2 controls a number of mechanisms that counteract oxidative stress where SQSTM1-mediated targeting of misfolded proteins for autophagic degradation is only one of these. The complex interplay between oxidative stress and autophagy is also illustrated by the finding that ROS levels are increased in cancer cells lacking autophagy.Citation86 In addition, underscoring the complexity of this interplay, NFE2L2 controls the expression of the autophagy cargo receptor SQSTM1, but at the same time, phosphorylated SQSTM1 partly controls the activity of NFE2L2 by sequestering its negative regulator KEAP1.Citation58,64-66 NFE2L2 transcriptionally cooperates with several cellular stress response pathways, including ATF4-regulated stress responses,Citation87 that further regulates SQSTM1,Citation88 and autophagy.Citation89 Also, in addition to TFEB (transcription factor EB),Citation90 recent studies have pointed to CREB (cAMP responsive element binding protein) and PPARA (peroxisome proliferator-activated receptor α) important for the transcriptional regulation of autophagy.Citation91,92
The pathogenesis of AMD is strongly associated with oxidative stress.Citation10,18,20,93,94 Currently, there is no established prevention or therapy for early AMD. The Age-Related Eye Disease Study (AREDS) is among the largest and most robust randomized clinical trials designed to investigate the role of daily oral supplementation of antioxidant vitamins and minerals.Citation13 The AREDS study demonstrated that daily supplementation with antioxidant reduced the risk of developing advanced AMD by 25% at 5 y. Epidemiological studies have suggested a protective role of n-3 PUFAs for developing AMD.Citation37-42,95-97 Also, subgroup analysis from the Nutritional AMD Treatment 2 Study from 2013, revealed that high levels of n-3 PUFAs in red blood cells can prevent AMD progression.Citation98 To assess if antioxidants and n-3 PUFAs could induce additive effects, a second AREDS2 study was designed to evaluate if inclusion of n-3 PUFAs to the AREDS formulation further reduced the risk of progression to advanced AMD. However, no additional effect was observed in preventing AMD progression.Citation99 Our results demonstrate that DHA potently induces the endogenous oxidative stress defense coordinated by NFE2L2 in ARPE-19 cells. The DHA-induced activation of NFE2L2 was abolished by cotreatment with exogenous antioxidants. Thus, it is possible that the 2 approaches to prevent AMD, either by reducing ROS via exogenous antioxidants, or by n-3 PUFAs to mobilize the endogenous ROS scavenging systems, neutralize each other. Based on our results, it would be interesting to determine if additional effects of the 2 approaches could be present if antioxidants and n-3 PUFAs are sequentially supplemented. However, further research is needed in order to determine the kinetics of the 2 responses in relevant cell types.
Further studies are needed to determine if aggregation, autophagy and NFE2L2 activation are part of the physiological responses to DHA also in vivo. Our data indicate that activation of NFE2L2 and elevated autophagy could represent markers for the disease preventive effects of DHA supplementation. Interestingly, our results emphasize that exogenous antioxidants may interfere with and counteract some of the putative positive effects of DHA, including the activation of NFE2L2. The current study shows that DHA induces the cellular antioxidant responses controlled by NFE2L2 and stimulate protein quality control and autophagy. These cellular mechanisms harbor a number of putative biomarkers that may be utilized in the future to defined and improve disease preventive effects of marine n-3 PUFAs.
Materials and Methods
Cell lines and reagents
ARPE-19 were obtained from ATCC (CRL-2302) and cultured in DMEM:F12 medium (Sigma, D8437), supplemented with fetal bovine serum (10%) (Gibco, 10270-106) and gentamicin (0.05 mg/mL; Gibco, 15710049). Immortalized wild-type (WT) and atg5−/− MEFs were a kind gift from Noboru Mizushima and were grown in DMEM (Sigma, D5796) supplemented with fetal bovine serum (10%) (Gibco, 10270-106) and gentamicin (0.05 mg/mL; Gibco, 15710049). All cell lines were maintained in a humidified atmosphere of 5% CO2; 95% air at 37°C. All experiments were performed in subconfluent, exponentially growing cells that never exceeded passage number 25.
Docosahexaenoic acid (DHA; Cayman, 90310), oleic acid (OA; Cayman, 90260) and arachidonic acid (AA; Cayman, 90010) were added to prewarmed complete medium to the final desired concentration and vortexed at full speed before added to the cells. Vehicle-treated samples were added to the same volumes of absolute ethanol and was used as control throughout all experiments.
Other reagents used: bafilomycin A1 (BafA1; Sigma, B1793), puromycin (Sigma, P9620), cycloheximide (CHX; Sigma, C4859), and hydrogen peroxide (H2O2; Merck Millipore, 108600).
The following antibodies were used: anti-SQSTM1/p62 (Progen, GP62-C); anti- NFE2L2/NRF2 (Santa Cruz Biotechnology, sc-13032); anti-MAP1LC3B/LC3B (Cell Signaling Technology, D11); anti-HMOX1 (Enzo, ADI-OSA-110), anti-ATG5 (Novus Biologicals, NB110-53818), anti-ACTB/β-actin (Abcam, ab6276), anti-KEAP1 (Santa Cruz Biotechnology, E20), anti-phospho-SQSTM1/p62 (Ser351; MBL, PM074), anti–mono- and polyubiquitininated conjugates (clone FK2; Biomol, PW8810), anti-TUBB/β-tubulin (Abcam, ab6046), anti-PCNA (Santa Cruz Biotechnology, sc-7907), anti-COX4I1/COX IV (Abcam, ab33985). All secondary antibodies were from Invitrogen (Alexa conjugates, catalog numbers A-11073, A-21428 and A-11001) or Li-Cor Biotechnology (NIR dye conjugates, catalog numbers 926-32211, 926-32214, 926-32411926-68077, 926-68071, 926-68070).
Microarray gene expression profiling
ARPE-19 cells were treated with vehicle (ethanol) or 70 µM DHA for 1, 3, 6, 12, and 24 h before RNA isolation using High Pure RNA isolation kit (Roche, 11828665001). Microarray gene expression profiling was performed in independent triplicates for all time points using Illumina HumanHT-12 v4 Expression BeadChip according to the manufacturer's protocol (Illumina). The statistical analysis was based on summary expression measures using the raw data (CEL) files performed by the robust multiarray average method. The statistical analysis was performed in R (http://www.r-project.org), using packages Limma from Bioconductor.Citation100 Differentially expressed genes were selected based on a threshold of 0.05 on the adjusted P values. Enrichment analyses was performed in MetaCore™ (Thomson Reuters, UK) a data-mining and pathway analysis tool. All data have been submitted to ArrayExpress with the accession number E-MTAB-3016.
Quantitative real-time PCR
Total RNA was extracted from DHA treated cells using the High Pure RNA isolation kit. Purity and quantity were measured by Nanodrop. 1 µg total RNA was used for cDNA synthesis using the iScript Select cDNA synthesis kit (Bio-Rad, 170-8896). The cDNA was diluted 1:10 before real-time PCR was performed in parallel 25 µl reactions containing 12.5 µl 2X QuantiTect SYBR Green PCR master mix (Qiagen, 204141) and 2.5 µl 10X QuantiTect Primer Assay (Qiagen, catalog numbers Hs_SQSTM1_1_SG, Hs_MAP1LC3B_2_SG, Hs_GABARAPL1_1_SG, Hs_MAP1LC3A_1_SG, Hs_GAPDH_2_SG, Hs_ACTB_2_SG ). The cycling conditions for the StepOne plus system (Applied Biosystems, Foster City, CA, USA) were 95°C for 15 min, 40 cycles of 94°C for 15 sec, 55°C for 30 sec and 72°C for 30 sec. Relative RNA transcription levels were transformed into linear form by 2 (−deltadeltaCt). Transcripts were normalized to the quantity of ACTB for each condition.
siRNA-mediated knockdown
For NFE2L2 and SQSTM1 downregulation cells were transfected using 25 nM siRNA oligo (final concentration) by DharmaFECT transfection reagent 1 (Dharmacon, T-2001-03) and compared to 25 nM control nontargeting siRNA. For ATG5 downregulation cells were transfected using 100 nM siRNA oligos (final concentration) and compared to the same final concentration of nontargeting siRNA. Following 24 h, the cells were collected by trypsinization and seeded for real-time cell monitoring, ROS measurements and immunoblot analysis. The following smartpool siRNA oligonucleotides were obtained from Dharmacon; control nontargeting siRNA (D-001210-01); NRF2/NFE2L2 siRNA (D-003755-02), target sequence: 5′- CCAAAGAGCAGUUCAAUGA; SQSTM1 siRNA (J-010230-06) and ATG5 siRNA (L-004374-00)
Real-time cell monitoring
Real-time growth curves were obtained using an xCELLigence system (Roche) according to the supplier's recommendations in the presence or absence of DHA (70 µM and 140 µM). Where indicated, the cells were pretreated with siRNA (nontargeting, NFE2L2, SQSTM1, or ATG5) 24 h before monitoring in real time with or without DHA (70 µM and 140 µM).
ScanR automated image acquisition
The microscope-based imaging platform ScanˆR (Olympus, Hamburg, Germany) were used to image SQSTM1-positive structures in the presence and absence of DHA (70 µM). Images were taken with a 20 × objective, using the excitation filters (wavelength [nm]/width [nm]): FITC (485/20) and Draq5 (650/13). For emission, a combination filter (440,521,607 and 700 nm) was used for all fluorophores (Chroma Technology Corp, Bellows Falls, VT). For each well, approximately 2000 cells were counted. The images were analyzed by the ScanR Analysis software (Olympus). Using the ScanR analysis software (Olympus) the number of cells was counted (based on the nuclear-stain) and the number of SQSTM1 dots within the cells (nucleus and surrounding cytosol) or Click EdU-positive nuclei.
Cell proliferation assay
Cell proliferation was monitored using Click-iT® EdU Alexa Fluor® 488 Imaging Kit (Invitrogen, C10337) according to the manufacturer's protocol. For EdU incorporation experiments, cells were pretreated with vehicle or DHA (70 µM) for 16 h, washed 2X PBS before further stimulation with puromycin (10 µM), cycloheximide (10 µg/ml) for 4h or hydrogen peroxide (50, 100 or 200 µM) for 30 min. After 2X washing in PBS the cells were added 5-ethynyl-2′-deoxyuridine (EdU) (5 µM) for 16 h and fixated using 4% paraformaldehyde. For the click reaction the cells were washed in 3% BSA (Sigma, A7906) and permabilized using 0.5% Triton X-100 (Sigma, T8787) for 20 min. After additional washing the cells were incubated with Click-It reaction cocktail containing Alexa Fluor 488 azide (Invitrogen, C10337) for 30 min. DNA was stained using 5 µg/ml Hoechst 33342 included in the kit (Invitrogen, C10337). EdU-positive cells were automatically quantified using ScanR automated image acquisition.
Detection of reactive oxygen species
ROS levels were determined using the Image-iT ™ LIVE Green Reactive Oxygen Species Detection Kit (which utilizes 5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate [DCF]; Invitrogen, C6827) and a BD FACS Canto flow cytometer (BD Biosciences, San Jose, CA, USA). The cells were treated with 70 µM or 140 µM of designated lipids for the indicated time points. When indicated, the cells were pretreated for 16 h with 150 µM vitamin E (Sigma, T3251) and 5 mM N-acetyl cysteine (NAC; Sigma, A9165) before DHA treatment (140 µM) for 3 h. The cells were incubated at 37°C and 5% CO2 with 0.3 µM DCF for 30 min before intracellular ROS was determined. The experiments were performed in duplicates and the data represent mean intensity of 10,000 cells per well ± SD. The results represent the average of 6 independent experiments for 3 h and 3 independent experiments for 6 and 24 h. For the antioxidant treatments, the results represent the average of 3 and 2 independent experiments for NAC and vitamin E, respectively. For WT and atg5 knockout MEFs the experiments were performed in triplicates and the data represent the average of 3 independent experiments. P values were calculated using the Student t test.
Immunostaining
The cell cultures were treated as specified for indicated time points and fixed in 4% paraformaldehyde before immunostaining using indicated antibodies and visualization by fluorescently labeled secondary antibodies. Nuclear DNA was stained using Draq5 (5 µM; Biostatus, DR50200) or Hoechst 33342. Immunostaining was imaged with an Axiovert200 microscope equipped with a 63 × 1.2W objective and the confocal module LSM510 META (Carl Zeiss, Jena, Germany). Images were processed using the LSM software and mounted using Canvas 11 (Deneba). Images are representative of more than 200 randomly selected cells in each condition and of 2 or more independent experiments. All images to be compared were taken with the same settings.
Immunoblotting
After the indicated treatment the cells were harvested by trypsinization and lysed in a urea buffer containing 8 M urea (Merck Millipore, 1084870500), 0.5% (v/v) Triton X-100 (Sigma, T8787), 100 mM DTT (Sigma, 646563), Complete® protease inhibitor (Roche, 1187350001) and phosphatase inhibitor cocktail II (Sigma, P5726) and III (Sigma, P0044). When indicated, cells were lysed in a buffer containing 0.25% Triton X-100 (Sigma, T8787), 1 % NP40 (Sigma, NP40S), 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA pH 8, Complete® protease inhibitor, and phosphatase inhibitor cocktail II (Sigma, P5726) and III (Sigma, P0044). For wild-type and atg5−/− MEFs, the extracts for NFE2L2 detection were prepared using a nuclear extract kit (Active Motif, 11447358). Total protein concentration in the lysates was determined by BioRad protein assay (Bio-Rad, 500-0006). Equal amounts of proteins (20 µg if nothing else stated) were separated using NuPAGE® Novex® 12% or 4-12% Bis-Tris Gels (Invitrogen). Bound antibodies were imaged by near infrared fluorescence using appropriate fluorescent dye labeled secondary antibodies and an Odyssey Near Infrared scanner (Li-Cor Biosciences, Lincoln, Nebraska, USA). PCNA and COX4I1 were used for normalization purposes. In addition, loading was also detected using immunoblotting for ACTB/β-actin and TUBB/β-tubulin. Images were processed using Li-Cor Odyssey software and mounted using the Canvas 11 software (Deneba).
Statistics
Values were expressed as mean ± standard deviation (SD). Statistical analyses were performed by the 2-tailed Student t test, 2-sample assuming equal variances. P values < 0.05 was considered statistically significant and is labeled with *) and P < 0.01 is labeled with **). Error bars for qRT-PCR represent the standard deviation of triplicate wells.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1061170_Fig_S2.pdf
Download PDF (499.3 KB)1061170_Fig_S1.pdf
Download PDF (260 KB)Acknowledgments
We thank Noboru Mizushima for wild-type and atg5 knockout MEFs. Arnar Flatberg and Vidar Beisvåg at the Genomics Core Facility (GCF), Faculty of Medicine, NTNU are acknowledged for the support on gene expression profiling. The Cellular and Molecular Imaging Core Facility (CMIC), Faculity of Medicine, NTNU is acknowledged for access to instruments and support.
Funding
This work was supported by grants from the Norwegian Cancer Society and from the Research Council of Norway through its Centres of Excellence funding program, project number 223255/F50.
References
- Martinez A, Portero-Otin M, Pamplona R, Ferrer I. Protein targets of oxidative damage in human neurodegenerative diseases with abnormal protein aggregates. Brain Pathol 2010; 20:281-97; PMID:19725834; http://dx.doi.org/10.1111/j.1750-3639.2009.00326.x.
- Sorolla MA, Rodriguez-Colman MJ, Vall-llaura N, Tamarit J, Ros J, Cabiscol E. Protein oxidation in Huntington disease. BioFactors 2012; 38:173-85; PMID:22473822; http://dx.doi.org/10.1002/biof.1013.
- Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A 1991; 88:10540-3; PMID:1683703; http://dx.doi.org/10.1073/pnas.88.23.10540.
- Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer's disease: possible role in tangle formation. Neuropathol Appl Neurobiol 2002; 28:228-37; PMID:12060347; http://dx.doi.org/10.1046/j.1365-2990.2002.00394.x.
- Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, Kleinert R, Prinz M, Aguzzi A, Denk H. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol 2002; 160:255-63; PMID:11786419; http://dx.doi.org/10.1016/S0002-9440(10)64369-6.
- Nagaoka U, Kim K, Jana NR, Doi H, Maruyama M, Mitsui K, Oyama F, Nukina N. Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J Neurochem 2004; 91:57-68; PMID:15379887; http://dx.doi.org/10.1111/j.1471-4159.2004.02692.x.
- Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD): Alzheimer's disease in the eye? J Alzheimers Dis 2011; 24:615-31; PMID:21297256.
- Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, Boulton ME, Petrovski G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013; 9:973-84; PMID:23590900; http://dx.doi.org/10.4161/auto.24546.
- Gehrs KM, Anderson DH, Johnson LV, Hageman GS. Age-related macular degeneration–emerging pathogenetic and therapeutic concepts. Ann Med 2006; 38:450-71; PMID:17101537; http://dx.doi.org/10.1080/07853890600946724.
- Jarrett SG, Boulton ME. Consequences of oxidative stress in age-related macular degeneration. Mol Aspects Med 2012; 33:399-417; PMID:22510306; http://dx.doi.org/10.1016/j.mam.2012.03.009.
- Dorey CK, Wu G, Ebenstein D, Garsd A, Weiter JJ. Cell loss in the aging retina. Relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989; 30:1691-9; PMID:2759786.
- Holz FG, Bellmann C, Margaritidis M, Schutt F, Otto TP, Volcker HE. Patterns of increased in vivo fundus autofluorescence in the junctional zone of geographic atrophy of the retinal pigment epithelium associated with age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 1999; 237:145-52; http://dx.doi.org/10.1007/s004170050209.
- Age-Related Eye Disease Study Research G. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, β carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol 2001; 119:1417-36; PMID:11594942; http://dx.doi.org/10.1001/archopht.119.10.1417.
- Murdaugh LS, Mandal S, Dill AE, Dillon J, Simon JD, Gaillard ER. Compositional studies of human RPE lipofuscin: mechanisms of molecular modifications. J Mass Spectrom 2011; 46:90-5; PMID:21182214; http://dx.doi.org/10.1002/jms.1865.
- Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J 2000; 14:835-46; PMID:10783137.
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, et al. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 2002; 99:14682-7; PMID:12391305; http://dx.doi.org/10.1073/pnas.222551899.
- Crabb JW. The proteomics of drusen. Cold Spring Harb Perspect Med 2014; 4:a017194; PMID:24799364; http://dx.doi.org/10.1101/cshperspect.a017194.
- Winkler BS, Boulton ME, Gottsch JD, Sternberg P. Oxidative damage and age-related macular degeneration. Mol Vis 1999; 5:32; PMID:10562656.
- Kaarniranta K, Salminen A, Eskelinen EL, Kopitz J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD). Ageing Res Rev 2009; 8:128-39; PMID:19274853; http://dx.doi.org/10.1016/j.arr.2009.01.001.
- Handa JT. How does the macula protect itself from oxidative stress? Mol Aspects Med 2012; 33:418-35; PMID:22503691; http://dx.doi.org/10.1016/j.mam.2012.03.006.
- Kaganovich D, Kopito R, Frydman J. Misfolded proteins partition between two distinct quality control compartments. Nature 2008; 454:1088-95; PMID:18756251; http://dx.doi.org/10.1038/nature07195.
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431:805-10; PMID:15483602; http://dx.doi.org/10.1038/nature02998.
- Höhn A, König J, Grune T. Protein oxidation in aging and the removal of oxidized proteins. J Proteomics 2013; 92:132-59; PMID:23333925; http://dx.doi.org/10.1016/j.jprot.2013.01.004.
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/10.1038/ncb1007-1102.
- Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl−2. Mol Cell 2007; 25:193-205; PMID:17244528; http://dx.doi.org/10.1016/j.molcel.2006.12.009.
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/10.1038/sj.emboj.7601623.
- Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Ann Rev Biochem 2011; 80:125-56; PMID:21548784; http://dx.doi.org/10.1146/annurev-biochem-052709-094552.
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; http://dx.doi.org/10.1074/jbc.M702824200.
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002.
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724.
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-i, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723.
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014; 4:914-27; PMID:24875857; http://dx.doi.org/10.1158/2159-8290.CD-14-0363.
- Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol 2010; 12:842-6; PMID:20811357; http://dx.doi.org/10.1038/ncb0910-842.
- Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, et al. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy 2014; 10:480-96; PMID:24468901; http://dx.doi.org/10.4161/auto.27292.
- Weindruch R. The retardation of aging by caloric restriction: studies in rodents and primates. Toxicol Pathol 1996; 24:742-5; PMID:8994305; http://dx.doi.org/10.1177/019262339602400618.
- Rascon B, Hubbard BP, Sinclair DA, Amdam GV. The lifespan extension effects of resveratrol are conserved in the honey bee and may be driven by a mechanism related to caloric restriction. Aging 2012; 4:499-508; PMID:22868943.
- SanGiovanni JP, Chew EY, Agron E, Clemons TE, Ferris FL, 3rd, Gensler G, Lindblad AS, Milton RC, Seddon JM, Klein R, et al. The relationship of dietary omega-3 long-chain polyunsaturated fatty acid intake with incident age-related macular degeneration: AREDS report no. 23. Arch Ophthalmol 2008; 126:1274-9; PMID:18779490; http://dx.doi.org/10.1001/archopht.126.9.1274.
- Seddon JM, Cote J, Rosner B. Progression of age-related macular degeneration: association with dietary fat, transunsaturated fat, nuts, and fish intake. Arch Ophthalmol 2003; 121:1728-37; PMID:14662593; http://dx.doi.org/10.1001/archopht.121.12.1728.
- Seddon JM, George S, Rosner B. Cigarette smoking, fish consumption, omega-3 fatty acid intake, and associations with age-related macular degeneration: the US Twin Study of Age-Related Macular Degeneration. Arch Ophthalmol 2006; 124:995-1001; PMID:16832023; http://dx.doi.org/10.1001/archopht.124.7.995.
- Chong EW, Kreis AJ, Wong TY, Simpson JA, Guymer RH. Dietary omega-3 fatty acid and fish intake in the primary prevention of age-related macular degeneration: a systematic review and meta-analysis. Archives of ophthalmology 2008; 126:826-33; PMID:18541848; http://dx.doi.org/10.1001/archopht.126.6.826.
- Merle BM, Delyfer MN, Korobelnik JF, Rougier MB, Malet F, Feart C, Le Goff M, Peuchant E, Letenneur L, Dartigues JF, et al. High concentrations of plasma n3 fatty acids are associated with decreased risk for late age-related macular degeneration. J Nutr 2013; 143:505-11; PMID:23406618; http://dx.doi.org/10.3945/jn.112.171033.
- Merle BM, Benlian P, Puche N, Bassols A, Delcourt C, Souied EH, Nutritional AMDTSG. Circulating omega-3 Fatty acids and neovascular age-related macular degeneration. Invest Ophthalmol Visual Sci 2014; 55:2010-9; PMID:24557349; http://dx.doi.org/10.1167/iovs.14-13916.
- Serini S, Fasano E, Piccioni E, Cittadini AR, Calviello G. Differential anti-cancer effects of purified EPA and DHA and possible mechanisms involved. Curr Med Chem 2011; 18:4065-75; PMID:21824086; http://dx.doi.org/10.2174/092986711796957310.
- Bazan NG. Cellular and molecular events mediated by docosahexaenoic acid-derived neuroprotectin D1 signaling in photoreceptor cell survival and brain protection. Prostaglandins Leukot Essent Fatty Acids 2009; 81:205-11; PMID:19520558; http://dx.doi.org/10.1016/j.plefa.2009.05.024.
- Dyerberg J. Coronary heart disease in Greenland Inuit: a paradox. Implications for western diet patterns. Arctic Med Res 1989; 48:47-54; PMID:2736000.
- Friedland RP. Fish consumption and the risk of Alzheimer disease: is it time to make dietary recommendations? Arch Neurol 2003; 60:923-4; PMID:12873846; http://dx.doi.org/10.1001/archneur.60.7.923.
- Caygill CP, Hill MJ. Fish, n-3 fatty acids and human colorectal and breast cancer mortality. Eur J Cancer Prev 1995; 4:329-32; PMID:7549825; http://dx.doi.org/10.1097/00008469-199508000-00008.
- Jing K, Song KS, Shin S, Kim N, Jeong S, Oh HR, Park JH, Seo KS, Heo JY, Han J, et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011; 7:1348-58; PMID:21811093; http://dx.doi.org/10.4161/auto.7.11.16658.
- Rovito D, Giordano C, Vizza D, Plastina P, Barone I, Casaburi I, Lanzino M, De Amicis F, Sisci D, Mauro L, et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARgamma activation in MCF-7 breast cancer cells. J Cell Physiol 2013; 228:1314-22; PMID:23168911; http://dx.doi.org/10.1002/jcp.24288.
- Shin S, Jing K, Jeong S, Kim N, Song KS, Heo JY, Park JH, Seo KS, Han J, Park JI, et al. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed Res Int 2013; 2013:568671; PMID:23841076; http://dx.doi.org/10.1155/2013/568671.
- Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-kappaB activation and enhancing autophagy. PloS One 2014; 9:e97957; PMID:24911523; http://dx.doi.org/10.1371/journal.pone.0097957.
- Hsu HC, Chen CY, Chiang CH, Chen MF. Eicosapentaenoic acid attenuated oxidative stress-induced cardiomyoblast apoptosis by activating adaptive autophagy. Eur J Nutr 2014; 53:541-7; PMID:23887854; http://dx.doi.org/10.1007/s00394-013-0562-2.
- Fukui M, Kang KS, Okada K, Zhu BT. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: role of ROS accumulation, caspase-8 activation, and autophagy induction. J Cell Biochem 2013; 114:192-203; PMID:22903547; http://dx.doi.org/10.1002/jcb.24354.
- Elvevoll EO, Barstad H, Breimo ES, Brox J, Eilertsen KE, Lund T, Olsen JO, Osterud B. Enhanced incorporation of n-3 fatty acids from fish compared with fish oils. Lipids 2006; 41:1109-14; PMID:17269556; http://dx.doi.org/10.1007/s11745-006-5060-3.
- Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM. ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp Eye Res 1996; 62:155-69; PMID:8698076; http://dx.doi.org/10.1006/exer.1996.0020.
- Dai J, Shen J, Pan W, Shen S, Das UN. Effects of polyunsaturated fatty acids on the growth of gastric cancer cells in vitro. Lipids Health Dis 2013; 12:71; PMID:23663688; http://dx.doi.org/10.1186/1476-511X-12-71.
- Jakobsen CH, Storvold GL, Bremseth H, Follestad T, Sand K, Mack M, Olsen KS, Lundemo AG, Iversen JG, Krokan HE, et al. DHA induces ER stress and growth arrest in human colon cancer cells: associations with cholesterol and calcium homeostasis. J Lipid Res 2008; 49:2089-100; PMID:18566476; http://dx.doi.org/10.1194/jlr.M700389-JLR200.
- Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 2014; 39:199-218; PMID:24647116; http://dx.doi.org/10.1016/j.tibs.2014.02.002.
- Anwar AA, Li FY, Leake DS, Ishii T, Mann GE, Siow RC. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: role of mitogen-activated protein kinases and Nrf2. Free Radic Biol Med 2005; 39:227-36; PMID:15964514; http://dx.doi.org/10.1016/j.freeradbiomed.2005.03.012.
- Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, Motohashi H, Yamamoto M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A 2012; 109:13561-6; PMID:22872865; http://dx.doi.org/10.1073/pnas.1121572109.
- Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 2004; 24:7130-9; PMID:15282312; http://dx.doi.org/10.1128/MCB.24.16.7130-7139.2004.
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 1999; 13:76-86; PMID:9887101; http://dx.doi.org/10.1101/gad.13.1.76.
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010; 30:3275-85; PMID:20421418; http://dx.doi.org/10.1128/MCB.00248-10.
- Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 2010; 285:22576-91; PMID:20452972; http://dx.doi.org/10.1074/jbc.M110.118976.
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742.
- Ichimura Y, Waguri S, Sou Y-s, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol Cell 2013; 51:618-31; PMID:24011591; http://dx.doi.org/10.1016/j.molcel.2013.08.003.
- Hosokawa N, Hara Y, Mizushima N. Generation of cell lines with tetracycline-regulated autophagy and a role for autophagy in controlling cell size. FEBS Lett 2006; 580:2623-9; PMID:16647067; http://dx.doi.org/10.1016/j.febslet.2006.04.008.
- Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell 2014; 53:167-78; PMID:24462201; http://dx.doi.org/10.1016/j.molcel.2013.12.014.
- Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2006; 2:189-99; PMID:16874109; http://dx.doi.org/10.4161/auto.2731.
- Asano M, Yamamoto T, Tsuruta T, Nishimura N, Sonoyama K. Dual labeling with 5-bromo-2′-deoxyuridine and 5-ethynyl-2′-deoxyuridine for estimation of cell migration rate in the small intestinal epithelium. Dev Growth Differ 2014; 57(1):68-73.
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009; 7:65-74; PMID:19721819; http://dx.doi.org/10.2174/157015909787602823.
- Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer 2012; 12:564-71; PMID:22810811; http://dx.doi.org/10.1038/nrc3278.
- Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J. Age-related retinopathy in NRF2-deficient mice. PloS One 2011; 6:e19456; PMID:21559389; http://dx.doi.org/10.1371/journal.pone.0019456.
- Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem 2007; 282:2529-37; PMID:17127771; http://dx.doi.org/10.1074/jbc.M607622200.
- Cipollina C, Salvatore SR, Muldoon MF, Freeman BA, Schopfer FJ. Generation and dietary modulation of anti-inflammatory electrophilic omega-3 fatty acid derivatives. PloS One 2014; 9:e94836; PMID:24736647; http://dx.doi.org/10.1371/journal.pone.0094836.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018.
- Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ 2009; 16:1-2; PMID:19079285; http://dx.doi.org/10.1038/cdd.2008.139.
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443:780-6; PMID:17051204; http://dx.doi.org/10.1038/nature05291.
- Morishita H, Eguchi S, Kimura H, Sasaki J, Sakamaki Y, Robinson ML, Sasaki T, Mizushima N. Deletion of autophagy-related 5 (Atg5) and Pik3c3 genes in the lens causes cataract independent of programmed organelle degradation. J Biol Chem 2013; 288:11436-47; PMID:23479732; http://dx.doi.org/10.1074/jbc.M112.437103.
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, et al. Noncanonical autophagy promotes the visual cycle. Cell 2013; 154:365-76; PMID:23870125; http://dx.doi.org/10.1016/j.cell.2013.06.012.
- Bae EJ, Kim KR, Tsang SH, Park SP, Chang S. Retinal damage in chloroquine maculopathy, revealed by high resolution imaging: a case report utilizing adaptive optics scanning laser ophthalmoscopy. Korean J Ophthalmol 2014; 28:100-7; PMID:24505207; http://dx.doi.org/10.3341/kjo.2014.28.1.100.
- Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P. Balance between autophagic pathways preserves retinal homeostasis. Aging cell 2013; 12:478-88; PMID:23521856; http://dx.doi.org/10.1111/acel.12072.
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, et al. SUMO modification of Huntingtin and Huntington's disease pathology. Science 2004; 304:100-4; PMID:15064418; http://dx.doi.org/10.1126/science.1092194.
- Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002; 11:1107-17; PMID:11978769; http://dx.doi.org/10.1093/hmg/11.9.1107.
- Pan M, Maitin V, Parathath S, Andreo U, Lin SX, St Germain C, Yao Z, Maxfield FR, Williams KJ, Fisher EA. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc Natl Acad Sci U S A 2008; 105:5862-7; PMID:18391222; http://dx.doi.org/10.1073/pnas.0707460104.
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/10.1016/j.cell.2009.03.048.
- Miyamoto N, Izumi H, Miyamoto R, Bin H, Kondo H, Tawara A, Sasaguri Y, Kohno K. Transcriptional regulation of activating transcription factor 4 under oxidative stress in retinal pigment epithelial ARPE-19/HPV-16 cells. Invest Ophthalmol Vis Sci 2011; 52:1226-34; PMID:21087962; http://dx.doi.org/10.1167/iovs.10-5775.
- B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683-99; PMID:23804767; http://dx.doi.org/10.1093/nar/gkt563.
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29:4424-35; PMID:20514020; http://dx.doi.org/10.1038/onc.2010.191.
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592.
- Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, Sun X, Yoon G, Kang Y, Zhong W, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014; 516:108-11; PMID:25383523.
- Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, Moore DD. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014; 516:112-5; PMID:25383539.
- Kinnunen K, Petrovski G, Moe MC, Berta A, Kaarniranta K. Molecular mechanisms of retinal pigment epithelium damage and development of age-related macular degeneration. Acta Ophthalmol 2012; 90:299-309; PMID:22112056; http://dx.doi.org/10.1111/j.1755-3768.2011.02179.x.
- Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol Aspects Med 2012; 33:295-317; PMID:22542780; http://dx.doi.org/10.1016/j.mam.2012.04.005.
- Chong EW, Robman LD, Simpson JA, Hodge AM, Aung KZ, Dolphin TK, English DR, Giles GG, Guymer RH. Fat consumption and its association with age-related macular degeneration. Arch Ophthalmol 2009; 127:674-80; PMID:19433719; http://dx.doi.org/10.1001/archophthalmol.2009.60.
- Christen WG, Schaumberg DA, Glynn RJ, Buring JE. Dietary omega-3 fatty acid and fish intake and incident age-related macular degeneration in women. Arch Ophthalmol 2011; 129:921-9; PMID:21402976; http://dx.doi.org/10.1001/archophthalmol.2011.34.
- Tan JS, Wang JJ, Flood V, Mitchell P. Dietary fatty acids and the 10-year incidence of age-related macular degeneration: the Blue Mountains Eye Study. Arch Ophthalmol 2009; 127:656-65; PMID:19433717; http://dx.doi.org/10.1001/archophthalmol.2009.76.
- Souied EH, Delcourt C, Querques G, Bassols A, Merle B, Zourdani A, Smith T, Benlian P, Nutritional AMDTSG. Oral docosahexaenoic acid in the prevention of exudative age-related macular degeneration: the Nutritional AMD Treatment 2 study. Ophthalmology 2013; 120:1619-31; PMID:23395546; http://dx.doi.org/10.1016/j.ophtha.2013.01.005.
- Age-Related Eye Disease Study 2 Research G. Lutein + zeaxanthin and omega-3 fatty acids for age-related macular degeneration: the Age-Related Eye Disease Study 2 (AREDS2) randomized clinical trial. Jama 2013; 309:2005-15; PMID:23644932; http://dx.doi.org/10.1001/jama.2013.4997.
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80; PMID:15461798; http://dx.doi.org/10.1186/gb-2004-5-10-r80.