
ABSTRACT
Polycystic ovary syndrome (PCOS) is a unification of endocrine and metabolic disorders and has become immensely prevalent among women of fertile age. The prime organ affected in PCOS is the ovary and its distressed functioning elicits disturbed reproductive outcomes. In the ovary, macroautophagy/autophagy performs a pivotal role in directing the chain of events starting from oocytes origin until its fertilization. Recent discoveries demonstrate a significant role of autophagy in the pathogenesis of PCOS. Defective autophagy in the follicular cells during different stages of follicles is observed in the PCOS ovary. Exploring different autophagy pathways provides a platform for predicting the possible cause of altered ovarian physiology in PCOS. In this review, we have emphasized autophagy’s role in governing follicular development under normal circumstances and in PCOS, including significant abnormalities associated with PCOS such as anovulation, hyperandrogenemia, metabolic disturbances, and related abnormality. So far, few studies have linked autophagy and PCOS and propose its essential role in PCOS progression. However, detailed knowledge in this area is lacking. Here we have summarized the latest knowledge related to autophagy associated with PCOS. This review’s main objective is to provide a background of autophagy in the ovary, its possible connection with PCOS and suggested a novel proposal for future studies to aid a better understanding of PCOS pathogenesis.
Abbreviations: AE: androgen excess; AF: antral follicle; AKT/PKB: AKT serine/threonine kinase; AMH: anti-Mullerian hormone; AMPK: AMP-activated protein kinase; ATG: autophagy-related; BCL2: BCL2 apoptosis regulator; BECN1: beclin 1; BMP: bone morphogenetic protein; CASP3: caspase 3; CL: corpus luteum; CYP17A1/P450C17: cytochrome P450 family 17 subfamily A member 1; CYP19A1: cytochrome P450 family 19 subfamily A member 1; DHEA: dehydroepiandrosterone; EH: endometrial hyperplasia; FF: follicular fluid; FOXO: forkhead box O; FSH: follicle stimulating hormone; GC: granulosa cell; GDF: growth differentiation factor; HA: hyperandrogenemia; HMGB1: high mobility group box 1; IGF1: insulin like growth factor 1; INS: insulin; IR: insulin resistance; LHCGR/LHR: luteinizing hormone/choriogonadotropin receptor; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MAPK/ERK: mitogen-activated protein kinase; MAPK8/JNK: mitogen-activated protein kinase 8; MTOR: mechanistic target of rapamycin kinase; MTORC: mechanistic target of rapamycin complex; NAFLD: nonalcoholic fatty liver disease; NFKB: nuclear factor kappa B; OLR1/LOX-1: oxidized low density lipoprotein receptor 1; oxLDL: oxidized low-density lipoproteins; PA: palmitic acid; PCOS: polycystic ovary syndrome; PF: primary follicle; PGC: primordial germ cell; PI3K: phosphoinositide 3-kinase; PMF: primordial follicle; ROS: reactive oxygen species; RP: resting pool; SIRT1: sirtuin 1; SQSTM1/p62: sequestosome 1; T2DM: type 2 diabetes mellitus; TC: theca cell; TUG1: taurine up-regulated 1
Introduction
Polycystic ovary syndrome (PCOS) is acknowledged as a diversified disorder in women of fertile age [Citation1]. Stein and Leventhal were the first to delineate PCOS in 1935 as a heterogeneous disorder with an amalgam of various signs and symptoms including (i) androgen excess (AE) (clinically manifested as hirsutism and/or acne), (ii) polycystic ovarian morphology, (iii) ovulatory dysfunction (oligo-ovulation/anovulation) [Citation2,Citation3]. PCOS is the most widespread metabolic and endocrine disorder with a global prevalence of 6–20% in reproductive-aged women [Citation4–6]. However, the conflict arises in determining the prevalence rate of PCOS is due to different criteria for PCOS diagnosis. Combining all the available epidemiological data, in India, the prevalence of PCOS is turning out to be 3.7% – 22.5% [Citation7]. Presently, three criteria are given for the diagnosis of PCOS with different combinations of clinical manifestations: (i) National Institutes of Health 1990 criteria, (ii) Rotterdam criteria 2003, (iii) Androgen Excess-PCOS Society criteria 2006. As a consequence of different criteria for determining PCOS, the diagnosis of PCOS is still disputable. Accordant to the current guidelines given by International PCOS Network, the Rotterdam criterion is preferred over others in adolescence which requires oligo-anovulation and hyperandrogenism for a confirmed diagnosis of PCOS [Citation8]. The pervasiveness of PCOS was determined by considering all diagnostic criteria that is 49% fulfilling all the three diagnostic categories, whereas 51%, 83%, 70.6% of cases are as per National Institute of Health criteria, Rotterdam criteria, and Androgen Excess-PCOS Society criteria, respectively [Citation9,Citation10]. The PCOS leads to other complications such as women diagnosed with PCOS are more prone to develop diseases associated with metabolic and reproductive deformities. Consequently, they have more probability of developing type 2 diabetes mellitus (T2DM), obesity, endometrial carcinoma, infertility, mood and eating disorders, cardiovascular disease [Citation11].
Autophagy, a process of self-eating of cellular materials, requisite to reprocess the damaged cell organelles to procure new building blocks for maintaining cellular homeostasis [Citation12]. Autophagy is obligatory for cell survival during stress conditions (nutrient deprivation) by regulating cellular bioenergetics and eliminating toxic agglomerates of protein [Citation13]. Autophagy is ordinally accompanied by cell death, possibly via remodeling autophagic constituents for cell death or by inducing apoptotic cell death [Citation14]. Kim et al. [Citation15] divulged the impact of autophagy in governing cellular metabolism by performing an autophagy-related (Atg) gene knockout study in mice. Features associated with metabolic disorders (obesity and T2DM) like dyslipidemia, oxidative stress, hyperglycemia, hyperinsulinemia are contributing to interrupted and compromised autophagy leading to cell death, cardiomyopathy, and cardiac malfunction [Citation16,Citation17]. Obesity-related complications are associated with autophagy, as autophagy supervises the cellular bio-power during starvation, demonstrated the essential role of autophagy in obesogenesis [Citation18]. In T2DM, autophagy might have a part in preventing beta-cell death in the pancreas [Citation19].
In the ovary, starting from the origin of oocytes to the development of follicles as well as their degeneration requires proper functional autophagy [Citation20]. Autophagy is necessary for follicular cells in maintaining the development of oocytes, follicular growth and differentiation, follicular atresia, and reproductive cycle under normal circumstances [Citation21–23]. Poor quality oocytes lead to female infertility as a result of defective autophagy in ovarian follicular cells [Citation24]. Thereupon, the role of autophagy in PCOS pathogenesis has gained insight nowadays. Till now, only a few studies have focused on linking autophagy with PCOS, but data from these studies suggest a prominent role played by autophagy in key affected organs in PCOS. But still, there is a need to explore this area in detail, as the exact underlying mechanism is unfamiliar. In this review, we have provided a comprehensive knowledge of autophagy in ovarian follicular development and summarized autophagic adaptation that arises due to altered physiology in PCOS, and illustrated the role of autophagy associated with different conditions recognized in PCOS, including insulin resistance (IR), oocyte development, follicular cysts formation, metabolic abnormalities, anovulation, AE, alteration in ovarian follicular cells and conditions associated with PCOS, and also suggested an approach for future investigation.
Ovarian follicular development and role of autophagy
Origin of oocytes, formation of ovarian reserve, follicular recruitment, and autophagy role
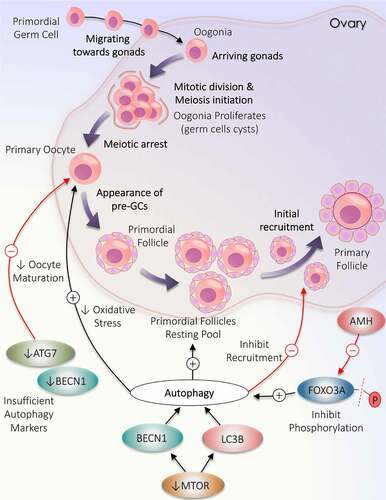
Ovary is the reproductive organ in females, produces ovum and sex hormones (androgens, estrogen, and progesterone) on stimulation by gonadotropins [Citation25]. Female gamete is produced from primordial germ cell (PGC); once these PGCs arrive into developing gonads, these PGCs are now referred as oogonia. This oogonia holds mitotic activity and is subject to extensive mitotic division, reaches about 6–7 million by the 20th week in the mammalian ovary. Termination of oogonial mitosis occurs during the 28th week of pregnancy [Citation26,Citation27]. The survival of oogonia becomes difficult without undergoing meiosis, and because of this, neonatal ovaries lacking oogonia [Citation28]. Conversion of oogonia into oocytes occurs once meiosis is initiated followed by the appearance of single squamous cells layer around the oocyte and meiotic arrest, which defend its degeneration and is now called primordial follicles (PMF). These PMFs are non-growing follicles that represent the ovarian reserve or the resting pool (RP) [Citation29–31]. However, the population of PMFs declines prenatally, after birth ovary of a neonate carries up to 1–2 million population of PMFs, out of which only 300,000–500,000 survive at puberty [Citation27,Citation30,Citation32,Citation33]. The population of resting follicles in ovarian reserve is approximately similar in both the ovaries of humans. The recruitment of PMFs is initiated before the birth and keeps going after the birth until the RP is exhausted: from >250,000 PMFs at the menarche, only ~1000 PMFs remain in the ovary at menopause [Citation34]. The majority of ovarian follicles die due to follicular atresia; only a small proportion (less than 1%) of follicles got matured and finally undergo ovulation in mammalian ovaries. Taken together, follicles facing mitosis, meiosis, and follicular atresia play a critical part in forming a final ovarian reserve.
The PMFs will remain in the dormant state until they are not recruited to enter the growth phase. Recruitment refers to the growth of the follicles to such an extent that allows follicles to surpass the atresia and remain in the developmental stage. Signals received at the time of recruitment enable them to proceed as a bunch of follicles rather than a single follicle, which permits them to keep up the growing phase [Citation35,Citation36]. Depending upon the time and stage of the follicle, recruitment is of two types: (i) initial recruitment – it starts shortly after the formation of the follicle in the ovary, which mainly recruits PMFs into a growth phase in a continuous fashion for the entire reproductive life and is gonadotropin independent. However, remaining non-recruited follicles stay in dormant phase only, (ii) cyclic recruitment – once the follicles achieve antral stage, majority of follicles undergo atresia whereas cohort of antral follicles (AF) escapes from atresia by recruiting toward the pre-ovulatory stage, this process is gonadotropin-dependent that is FSH (follicle stimulating hormone) which occurs after the onset of puberty cyclically (every 28 d in human) [Citation37]. Recent studies suggest different ways of cyclic recruitment; earlier, it was proposed that only a single wave for recruitment, but now it is believed that multiple waves are required in human ovaries [Citation38].
Ovary takes various measures to preserve RP of PMFs in a quiescent state, including factors secreted by oocytes, intra-ovarian factors, and mesenchymal-epithelial cell interactions. Intra-ovarian factors can either promote the follicle to progress in the developmental phase (GDF [growth differentiation factor], and BMPs [bone morphogenetic proteins]) or can downgrade this process (AMH [anti-Mullerian hormone]). GDF is produced by somatic cells in the ovary; specifically, GDF9 present in oocytes promotes folliculogenesis. Both GDF9 and BMP15 engage in transforming the PMF to the primary follicle (PF) and helping in expressing LHCGR/LHR (luteinizing hormone/choriogonadotropin receptor) in the later stage of follicle development [Citation39]. AMH is a hormone that belongs to tissue growth factor β family generated by granulosa cells (GC) of early developing follicles. It interferes with the process of follicular recruitment and FSH action. During the initial follicular growth phase, a rise in AMH level stops the recruitment of PMFs to the growth phase and prevents the activity of CYP19A1/aromatase enzyme (cytochrome P450 family 19 subfamily A member 1); hence, inhibits the transition of primordial to the PF [Citation40]. Control over AMH production is maintained by negative inhibition of AMH production via estradiol, restricting the AMH to follicles up to 8 mm in size. Therefore, prior to the selection of dominant follicles, only a small amount of AMH is secreted by small developing follicles [Citation41,Citation42]. AMH impedes the progression of PMFs in a paracrine manner. In a study by Durlinger et al. [Citation40], amh−/- accelerates the exhaustion of PMFs, hence reduces the resting follicle population.
In the intra-ovarian environment, an increase in age-related oxidative stress has been linked to a decline in female fertility. Oocytes are extremely vulnerable to the oxidizing environment, which instigate an imbalance in the protein level required to maintain oocyte health and maturation. To overcome this stressful situation, autophagy is essential to regulate balanced and functional proteome, which protects oocytes and favors embryonic development. Autophagy reduces the oxidative burden in oocytes and promotes their health [Citation43]. In addition to oocytes, autophagy is also seen during the pre-implantation of murine embryos in response to stress conditions [Citation44].
In mammalian ovaries, it is well known that ovaries experience a significant loss of immature follicles (PMFs) during birth. Although the exact mechanism behind the significant decline in the PMFs population is not known. Recent investigations uncover the role of autophagy in the ovary while monitoring the RP inside the ovary. AMH produced by GCs of early developing ovarian follicles also takes part in protecting PMFs reservoir via inducing autophagy in ovaries by inhibiting FOXO3/FOXO3A (forkhead box O3) phosphorylation (causes PMF activation) in the case of follicle depletion [Citation45]. Besides this, it is well known that MTOR (mechanistic target of rapamycin kinase) and autophagy both have an inverse relationship. Obstructing the MTOR pathway by giving rapamycin (MTOR inhibitor) induces autophagy by enhancing MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 beta) and BECN1 (beclin 1) expression, which further encourages the gathering of immature PMFs and the existence of oocytes [Citation46]. Moreover, in ovaries of pre-pubertal rats, apoptosis and autophagy are working simultaneously to remove germ cells from rat ovaries. However, rat oocytes show more autophagic markers proposing that autophagy might have a dominant role during the destruction of germ cells [Citation47].
Studies have confirmed the impact of autophagy in female reproductive functions starting from oocyte development to postpartum remodeling, including implantation, carrying out the pregnancy, and maintaining the placenta’s physiology. Autophagy plays a significant part in developing oocytes and germ cell survival before initiation of PMFs pools. It is reported in an animal study that the probability of oocyte maturation defect and germ cell deformity is related to the insufficiency of key autophagic molecules (), particularly BECN1 and ATG7 [Citation48]. Therefore, a clear understanding of autophagy’s role and its corresponding gene is needed in female reproduction. A steady level of an autophagy gene, ATG7, is essential throughout oocytogenesis and is involved in reserving primordial ovarian follicles. Song et al. [Citation49] suggested that ATG7 can probably have pathogenic potential in causing primary ovarian insufficiency, as germ-cell specific atg7−/- in mice results in depletion of autophagy causes significant decline in oocytes and germ cells results in poor ovarian reserve of PMFs along with producing litters with small size and makes mice infertile eventually. Similarly, Becn1± results in considerable loss of oocytes up to 50–60% on the 1st day of birth in contrast to control [Citation50]. During the neonatal transition, autophagy could prevent over-loss of germ cells by masking apoptosis in neonatal ovaries under the deprivation of food. Compromised autophagy occurs due to the loss of Atg7 or Becn1 within the prenatal ovary. Hence, as a result, the majority of germ cells lost prematurely [Citation50]. Finally, all the discoveries signify that any alteration in the process of autophagy disrupts the ovarian reserve of the follicles and oocyte development.
Figure 1. Deviation in autophagy during initial follicular growth phase: from the origin of oocytes to initial recruitment. Folliculogenesis begins when the precursor primordial germ cells arrive in the ovary. The resultant oogonia increase its population by mitotic division, subsequently induces meiosis. The formation of oocytes is marked with the arrest of meiotic division. Primordial follicle production constitutes the resting pool of follicles. Follicles are continuously recruited from the primordial resting pool to enter the growth phase to become primary follicles. Autophagy supports the growth of oocytes and the formation of the resting follicles pool but opposes the initial recruitment of the primordial follicles. A decrease in the level of autophagy markers (ATG7 and BECN1) directly inhibits oocytes maturation. MTOR inactivation causes an increase in LC3B and BECN1 to enhance autophagy. AMH decreases FOXO3A activation and supports autophagy
Follicular development, selection of dominant follicle, and autophagy role
Ovary is the key reproductive organ in females where all the necessary steps are taken to develop the female gametocyte required for fertilization. In a full-grown ovary, the dormant follicles quit the ovarian resting follicles pool to participate in the developmental phase and go through proliferation and differentiation of follicular cells, consequently favor folliculogenesis. For complete maturation, follicles have to follow various developmental stages, starting from the primordial stage to primary, secondary, pre-antral, early-antral, antral, Graafian follicle, and finally ovulate. Division of follicles in different stages is based on the morphological as well as functional changes acquired with progression, including the difference in oocyte size, follicles size, number of follicular cells surrounding the oocyte, the formation of antrum cavity, appearance of layers around the oocyte () [Citation23].
Figure 2. Participation of autophagy in the development of follicles. The developmental process begins with the formation of primordial follicles which is comprising of immature oocyte encircled by a single layer of pre-granulosa cells. The primary follicle is defined by granulosa cells with the appearance of zona pellucida. The proliferation of granulosa cells to multiple layers and the formation of theca cells layer results in the development of secondary follicle. Follicle with multiple small cavities is referred to as early antral follicles. A follicle with a single large cavity filled with antral fluid is called the antral follicle. Cyclic recruitment of antral follicles leads to the formation of a fully matured Graafian follicle. Estradiol and INH (inhibin) are secreted by the Graafian follicle, which stops the recruitment process. Activation of AKT and MTOR in granulosa cells prevents autophagy and favors follicular development. After the selection of dominant follicles, the remaining subordinate follicles are degenerated by autophagy-induced apoptosis. Increased expression of OLR1 receptor in granulosa cells of regressing follicles supports autophagy via inducing ROS production, resulting in granulosa cells death by apoptosis. NOX: NADPH oxidase
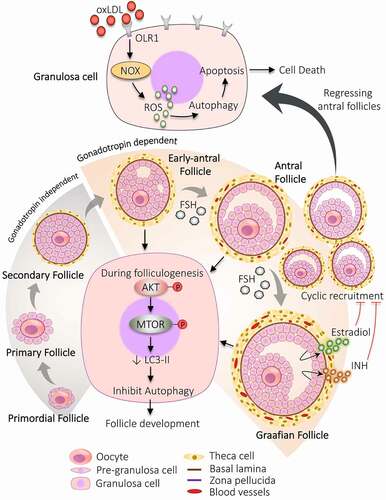
After the transition of oogonia to oocytes, the oocyte undergoes meiotic arrest in the diplotene stage of prophase I. Firstly, the PMF forms by surrounding the primary oocyte with a single squamous layer of follicular somatic cells (flattened shape), namely pre-GCs. When the shape of these surrounding cells changes from flattened to cuboidal shape (now called GCs) along with an enhanced diameter of oocyte and appearance of zona pellucida, then it is called PF [Citation51]. The proliferation of cuboidal GCs into multiple layers, the formation of the theca cells (TC) layer, enlarged oocyte, basal lamina, and zona pellucida are the characteristic features of the secondary follicle. When follicles begin to enlarge, these surrounding layers of connective tissue differentiate into the outer part (theca externa) and inner part (theca interna) [Citation52,Citation53].
Furthermore, to proceed the development process to AF requires the formation of an antral cavity, which is initiated by the formation of small separate fluid-filled sacs between GCs (called early-AF) and that combine to form a single large antral cavity (called AF). The space generated between the GCs during the formation of the antral cavity divides the GCs into two parts: (i) cumulus cells – remain attached to oocytes, (ii) mural cells – facing the follicular wall [Citation54]. Apart from the antral cavity, the AF also contains multiple GCs layers and TCs layer, increased blood supply in the TCs layer [Citation55]. Sometimes AFs are also referred to as tertiary follicles. The fully-grown AF is commonly called the Graafian follicle and is the primary site of estrogen production. Although, just before the ovulation step (pre-ovulatory stage), these follicles are often called pre-ovulatory follicles [Citation54].
At every step of follicular development, some follicles culminate in the follicular atresia; hence, it is necessary to recruit more follicles. After the development of AFs, generally, a cohort of AFs is recruited from which the dominant follicle is assorted. Some of the follicles continue to grow more and achieve the required ovulatory size, out of which a single follicle is chosen over others for further growth and termed as “dominant follicle”. To select the dominant follicle, specific characteristics have to be optimized in the follicle such as the prohibition of premature luteinization, high sensitivity toward gonadotropins, and the steroidogenic property [Citation56]. In humans, one ace follicle qualifies to participate in the late phase of follicular development during each ovarian cycle. The rest of the follicles (subordinate follicles) are eliminated by the time [Citation57] because their growth is halted via negative feedback inhibition by estradiol and INH (inhibin) secretion by the dominant follicle. Additionally, a decline in FSH opposes the growth of subordinate follicles. Although, the development of dominant follicle is not compromised even in the low level of FSH due to increasing the expression of FSH receptors which makes them more susceptible toward FSH [Citation58], also by achieving sufficient stage of differentiation, which supports its growth [Citation59] by acquiring more LHR on GCs of the dominant follicle and enhanced blood supply in follicles [Citation60]. As a result, plenty of follicles undergoing the recruitment process assure that if anything goes wrong with the prime follicle, another follicle will be ready to take its place and allow the continuation of the cycle [Citation60].
Primarily, the follicle gradation from the resting stage (PMFs) to the growing stage (PFs) is an entirely autonomous process and does not require gonadotropins. The initial stages of follicle development up to secondary follicle are independent of gonadotropins; after entering into the early antral stage, the gonadotropins become necessary for their growth [Citation56]. Further development and existence of follicles after the antral stage is mainly governed by FSH as their GCs become hyper-responsive toward FSH. Therefore, a minute change in FSH level immediately recruits them to the next stage of development. FSH causes a change in the morphology of the follicles by increasing GCs proliferation [Citation61]. Growth of pre-ovulatory follicles is supported by higher estradiol production, mandatory for GCs differentiation [Citation62]. LH surge during the mid-menstrual cycle leads to higher expression of LHR and is found to be increased by 10-fold in pre-ovulatory follicles. Expression of LHR is induced primarily by FSH, enhancing the secretion of steroidal hormone (progesterone and estrogen) [Citation63].
Increasing evidence supports the essential role of autophagy in regulating the fate of ovarian follicular cells [Citation21]. Not surprisingly, with all the stages follicle undergoes during their life cycle and as autophagy plays a critical role in maintaining cellular bioenergetics – pointed out the crucial role of autophagy in deciding follicular fate. In the human ovary, autophagy is associated with ovarian functioning and regulating follicular development, such as managing GCs life span [Citation64]. The downregulation of autophagy in GCs within sight of gonadotropins has been recently exhibited in vivo [Citation22]. Choi et al. [Citation65] reported that AKT/PKB (AKT serine/threonine kinase)-mediated MTOR activation halts the autophagy of GCs at the time of follicular development in gonadotropin primed model in immature rats. In rat GCs, expression of LC3-II is turned down along with increased AKT and RPS6KB1 (ribosomal protein S6 kinase B1) phosphorylation followed by gonadotropin injection. Past researches support that apoptosis can be provoked by autophagy in the presence of gonadotropins in follicular cells like GCs and luteal cells. Therefore, activation of the MTOR pathway in FSH exposed GCs prohibits cell death by apoptosis by hampering the autophagy, hence favors follicular development [Citation22,Citation65–67].
Both types of programmed cell death have been noticed in GCs of follicles of distinct sizes, i.e., undergoing different follicular stages. In particular, autophagy was predominantly observed in medium-sized follicles, whereas; apoptosis was more common in large follicles [Citation68]. Information derived from these studies states the important role of autophagy in carrying normal ovarian folliculogenesis as it can favor follicular survival; conversely, it can result in follicular atresia when needed [Citation22].
Additionally, the level of reactive oxygen species (ROS) in specific tissue favors different program cell death to proceed. For instance, in young, healthy women with normal weight, cells choose to repair themselves by inducing autophagy rather than going for apoptosis in response to low ROS levels; whereas, with an increase in age, cells opt for apoptosis. In GCs, a decrease in the level of autophagy marker (LC3-II) is reported with an increase in the level of ROS in old women, results in apoptosis of GCs. It has been found that autophagy can be induced by smoking, which leads to a reduction in the number of GCs [Citation64]. Thus, autophagy can be the reason for the controlling of fertility in women as it is involved in maintaining follicular cell population and subsequent folliculogenesis.
Autophagy and ATG play an influential part in follicular turn of events. In humans, exposure of GCs to oxidized low-density lipoproteins (oxLDL) gives rise to autophagy, which further leads to cell death by apoptosis. Activation of autophagy by oxLDL occurred via lectin-like OLR1/LOX-1 (oxidized low density lipoprotein receptor 1), which stimulates oxidative stress by increasing ROS which can be via NADPH oxidase [Citation69]. According to the studies, obese women have excessive oxLDL causes enhanced autophagy in GCs. Regressing AFs are found to be rich in OLR1 in the human ovary [Citation64,Citation70]. Therefore, the oxLDL-OLR1-ROS pathway induces autophagy in GCs. Altogether these discoveries propose that autophagy is involved in regulating the growth of follicles at different developmental stages.
Ovulation and post-ovulatory events: formation of corpus luteum and autophagy role
During the mid-follicular phase, the pre-ovulatory follicle produces a high amount of estradiol. This triggers the burst release of LH which causes the ovulation of the dominant follicle by dissociating GCs from the cumulus cells [Citation71,Citation72]. Before ovulation, the dominant follicle increases its size by escalating cell mass and depositing follicular fluid (FF) in the antrum cavity [Citation73].
Corpus luteum (CL) is the important component of the reproductive cycle formed after the ovulation of the matured follicle. Pre-ovulatory LH surge activates the LHR on follicular cells induces ovulation, which further promotes terminal differentiation of ovulated follicular cells into luteal cells by a process termed luteinization. The luteal cells derived from terminal differentiation of GCs and TCs are now referred to as granulosa-lutein and theca-lutein, respectively and both types of cells express LHR. CL comprises not only luteinized follicular cells but also endothelial and immune cells; hence, CL is considered a temporary organ. CL is the major source of progesterone hormone secretion; regression in CL results in a decline in progesterone secretion. Therefore, CL’s follicular cells are termed as non-dividing progesterone-producing luteal cells [Citation74,Citation75].
Significance of autophagy is discovered in the cumulus cell covering the ovulated oocyte (). After ovulation, the primary function of cumulus cells is to protect the oocyte from the exterior oviduct environment to achieve successful fertilization by repressing the autophagy-provoking stimulus. Kang et al. [Citation76] suggested that defected cumulus cells in humans favor disruption in the autophagy pathway as they contain disturbing levels of autophagic proteins (high LC3, low ATG7, and low lysosome number). Therefore, defected autophagy causes the transport of cytotoxic substances between cumulus cells and oocytes, which harms oocyte quality and may result in infertility. Interestingly, a higher amount of autophagosome has been noticed shortly after fertilization, indicating the necessity of autophagy post-fertilization where it can probably involve removing the unneeded cellular proteins and provide the derived amino acids for embryo development. In post-fertilization, inside the oocyte, autophagy is induced, which is mechanistic target of rapamycin complex 1 (MTORC1)-independent. Therefore, pointing to a new regulatory mechanism in which autophagy plays a unique role in controlling intracellular turnover in the early embryo [Citation77]. A previous study demonstrated that oocytes generated after oocyte-specific atg5−/- leads to developmental failure after eight-celled stage of the mouse embryo. Thus, it signifies the role of autophagy during the pre-implantation process [Citation78].
Figure 3. Autophagy implication during ovulatory and post-ovulatory events. Autophagy regulates the functioning of follicular cells during and after ovulation. (A) Ovulation: Disturbed level of autophagy markers in cumulus cells attached with oocytes damages the oocyte, which hampers the oocyte quality resulting in infertility. (B) Corpus luteum: becn1−/- causes a defect in lipid droplet formation and affects steroidogenesis decreases progesterone production. (C) Regression of corpus luteum: Enhanced expression of autophagy markers along with autophagic gene expression, inhibition of MTOR pathway, and activation of MAPK1-MAPK3 pathway triggers the autophagy and autophagy induced apoptosis
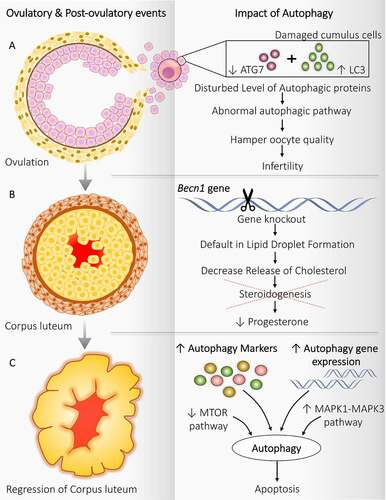
Females without key autophagic gene Becn1 in progesterone-producing ovarian GCs exhibit a defect in progesterone production during pregnancy contributes to preterm phenotype. Progesterone is a mandatory hormone throughout pregnancy, which makes the uterus ready for conceiving. Production of progesterone is a prime function of CL after ovulation, which requires cholesterol; BECN1 plays an essential role in regulating autophagy for the appropriate functioning of CL (). This cholesterol is derived from the lipid droplets present in CL, which initiates steroidogenesis. In the murine granulosa-lutein cell, becn1−/- causes default in the formation of lipid droplets further reduces progesterone secretion. Thus, the manifest role of autophagy in governing steroidogenesis in CL as well as supports pregnancy [Citation79].
During the regression of CL and late luteal phase, the expression of genes involved in autophagy is found to be increased, such as BECN1 suggests the importance of autophagy during luteolysis. Autophagy performs a different role in CL depending upon its stage, in particular: (i) In the early phase – it fosters luteal generation, (ii) In the late phase – it gets involved in luteal deterioration. This is supported by a higher level of autophagy proteins, namely BECN1 and LAMP1 (lysosomal associated membrane protein 1), during CL regression () [Citation80]. In cattle, an elevated level of autophagy marker MAP1LC3-II is present in late-stage CL compared to the earliest one. Besides this, the expression of ATG genes, including ATG3, ATG7, MAP1LC3A, MAP1LC3B also raises significantly in late CL [Citation81]. Increased ATG3 and ATG7 support the formation of the phagophore membrane via making LC3 and phosphatidylethanolamine complex [Citation82].
Additionally, MTOR expression is also observed to be decreased during late CL, proposing the essential part of autophagy in the regression of CL [Citation81]. A study in porcine ovary showed an incidence of autophagy in CL throughout the life cycle of CL proposes its role in governing the life span of CL. Previous studies suggest a crucial role of BECN1 in controlling the lifespan of CL through promoting autophagic survival of luteal cells [Citation83,Citation84]. Another study demonstrates the involvement of the MAPK/ERK (mitogen-activated protein kinase) pathway specifically increased MAPK3/ERK1 and MAPK1/ERK2 in inducing autophagy in luteal cells during CL regression, which is MTOR independent [Citation85]. Evidence from electron microscope cytochemistry proves the participation of autophagy in the regression of luteal cells following childbirth via increasing lysosomes population in CL and occurrence of autophagic vacuoles [Citation86].
Collectively, compiling the observations from previous studies indicates that numerous factors supervise the regression of CL in various ways; in one way, they eliminate the cells constituting the CL, while in another way destroying CL structurally. However, both the types of program cell death, apoptosis and autophagy, works simultaneously to destroy the CL completely. Luteal cell autophagy is supported by the gathering of autophagosomes, which in turn stimulate apoptosis in luteal cells, confirmed by a fall in the ratio of BCL2 (BCL2 apoptosis regulator):BAX (BCL2 associated X apoptosis regulator) along with caspase activation. Moreover, expression of the autophagic protein (LC3-II) goes parallel with apoptotic protein (CASP3) indicates the involvement of both types of programmed cell death during CL regression [Citation66]. Autophagy protein BECN1 interacts with anti-apoptotic protein BCL2 in CL regression. Apart from GCs, the presence of BECN1 is confirmed by immunostaining in TCs. Moreover, both theca-lutein and granulosa-lutein express BECN1 following ovulation, where it is involved in regulating the survival of CL by promoting autophagy rather than apoptosis. Taken together, both types of programmed cell death are noticed in CL [Citation83].
Steroidogenesis by theca cell and granulosa cell and autophagy association
Ovary expresses all the necessary enzymes required for the formation of sex steroids. Not all ovarian cells can produce all types of steroidal hormones; specific cells in the ovary carry specific enzymes for making a particular hormone. TCs produces progesterone and androgens, whereas GCs produce estrogen. Both follicular cells require stimulation from specific gonadotropins to induce hormone production; TCs require LH stimulation, whereas GCs require FSH [Citation87].
Steroidogenesis is the process of steroidal hormone formation, which requires cholesterol as a precursor. LH is the primary hormone responsible for inducing steroidogenesis in ovarian cells. Initiation of steroidogenesis requires STAR (steroidogenic acute regulatory protein)-mediated translocation of cholesterol inside the mitochondria, which further get converted into pregnenolone by another enzyme called cholesterol side-chain cleavage enzyme [Citation88]. The entire steroidogenesis pathway demands a variety of transport proteins, enzymes, cofactors; especially cytochrome P450 enzymes and hydroxysteroid dehydrogenases. Expression of CYP17A1/P450C17 (cytochrome P450 family 17 subfamily A member 1) is found in the adrenal and ovary, where its expression is governed by LH stimulation in the ovary and adrenocorticotropic hormone in the adrenal. CYP17A1 is the only enzyme which holds two different activity and catalyzes the different reactions, considered as key androgen producing enzyme: (i) 17-hydroxylase activity – convert pregnenolone to 17-hydroxypregnenolone, and (ii) 17,20 lyase activity – convert 17-hydroxypregnenolone to dehydroepiandrosterone (DHEA), further sex steroids and androstenedione is made from DHEA by HSD3B2 (hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2). Additionally, both the activities of CYP17A1 are required to convert progesterone into androstenedione. In TCs, androstenedione is converted to testosterone by AKR1C3/HSD17B5 (aldo-keto reductase family 1 member C3). TC is the only site for androgen production in the ovary as no other follicular cell expresses CYP17A1 [Citation41].
Once the androgen is formed successfully by TCs, it is permeated to GCs, where they act as a precursor for estrogen formation as GCs only express the CYP19A1 required for estrogen production (estrone form), which is crucial for GCs differentiation. Further, estrone is converted to estradiol by HSD17B1 (hydroxysteroid 17-beta dehydrogenase 1). In GCs, androstenedione and testosterone both act as a substrate for the CYP19A1. Still, androstenedione is converted more prominently as it is present in higher quantities as compared to testosterone. In contrast, testosterone is majorly converted into more potent dihydrotestosterone by the SRD5A1 (steroid 5-alpha reductase) enzyme [Citation87]. Enhanced capacity of androgen biosynthesis is observed after LH-mediated luteinization. Besides androgens, estradiol production also required increased LH level as androgens are the precursor of estradiol [Citation89,Citation90]. As estradiol is made from androgens only, the lower level of androgens in FF of healthy dominant follicles possibly states that their substantial amount is consumed in estradiol production [Citation91,Citation92].
Signals from different hormones command the synthesis of steroidal hormones in endocrine cells along with a monitored supply of precursors required for steroidogenesis, i.e., cholesterol. The cells in which steroidogenesis is active show a higher autophagy rate, whereas reduced turnover of steroidal hormones is linked to a decline in autophagy. Still, the relationship between autophagy and steroidogenesis is unclear. In mice, atg5−/- in steroid-producing cells is associated with a decrease in the production rate of steroid hormones. This reduction was further found to be due to cholesterol deficiency resulting from reduced autophagy. This study states the regulation of steroid hormone production via autophagy [Citation93].
Various growth factors are known to manage the activity of estrogen-producing enzymes. IGF1 (insulin like growth factor 1) is found to augment the activity of the CYP19A1, although the exact mechanism involved is unfamiliar. Metabolically, IGF1 prevents the degeneration of CYP19A1 protein. Zhang et al. [Citation94] reported rapamycin, an autophagy inducer, completely opposes the increase in CYP19A1 activity by IGF1. Thus, it suggests that the inhibition of autophagy by IGF1 is responsible for promoting CYP19A1 activity. Therefore, defining the role of autophagy in controlling estrogen production. Additionally, IGF1 is reported to have a mutagenic property that elevates the proliferation of ovarian cells. Besides mutagenicity, it also promotes the productivity of progesterone and inhibits apoptosis [Citation95]. In conclusion, IGF1 is involved in regulating the turnover of estrogen and progesterone but not androgen.
Low-density lipoprotein, considered as bad cholesterol, induces the synthesis of steroid hormones in the ovary. Degradation of low-density lipoprotein through lysosome activation in GCs is required for the synthesis of progesterone. In ovarian endometriosis, autophagy is found to be enhanced in the GCs evidenced by higher expression of BECN1. In vitro study on BECN1 knockdown GCs, reduced expression of steroidogenic enzymes alters the steroidogenesis pathway; hence, it reduces progesterone production. Therefore, these findings suggested upregulation of autophagy is involved in increased progesterone secretion in the late follicular stage in endometriosis [Citation96].
Autophagy implication in follicular atresia
Ovarian follicles being the functional unit of ovaries, experience different stages throughout their developmental phases [Citation97]. If follicles do not ovulate at the end of the ovarian cycle, ultimately, they encounter follicular degeneration by a process defined as follicular atresia [Citation98]. A minor percentile of the follicles gets away from follicular death by atresia and ends up in ovulation. In primates, after bypassing the RP, follicles compete together to achieve maturation. Among all, only a single follicle hits the pre-ovulatory phage in each ovarian cycle, remaining all confront demise by follicular atresia [Citation99]. Atresia can occur at any stage during folliculogenesis. The probability of undergoing follicular atresia is different and depends upon the stage of follicular development. However, the maximum possibility of facing atresia is at the antral phase [Citation59] ().
Figure 4. Significance of autophagy during the follicular atresia. Atresia is noticed during all the developmental stages of follicles; however, maximum atresia is observed at the antral stage. Autophagy contributes to follicular atresia by induction of apoptosis and/or autophagic cell death of granulosa cells. Activation of MAPK8 triggers autophagy by phosphorylation of apoptotic protein BCL2. Phosphorylated BCL2 is detached from the autophagy protein BECN1 and promotes autophagy. FSH hormone prevents oxidative stress damage by inhibiting autophagy via PI3K-AKT-MTOR pathway activation and mitophagy
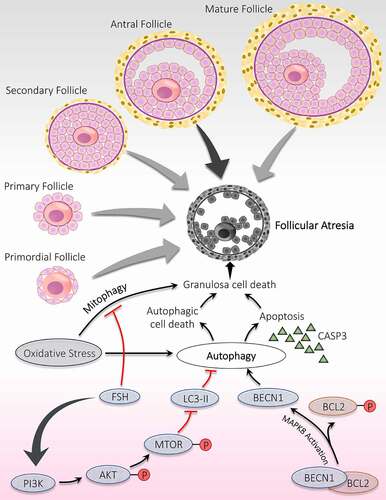
Role of autophagy and ATG has been established in controlling follicular atresia in ovaries. Choi et al. [Citation22] have demonstrated an inverse relationship between autophagy and gonadotropins in vivo. In GCs, autophagy is responsible for inducing follicular atresia through influencing cell death by apoptosis. MTOR has a detrimental effect on autophagy, is under the control of the upstream phosphoinositide 3-kinase (PI3K)-AKT pathway. Inhibition of PI3K-AKT-MTOR pathway stimulates the autophagy accompanied by apoptosis leads to massive degeneration of GCs, which subsequently trigger follicular atresia, proved by obstruction of AKT and MTOR activation by AKT and FSH inhibitor. These findings indicated a significant role of AKT in GCs autophagy and autophagy induced apoptosis during follicular atresia [Citation65]. Duerrschmidt et al. [Citation70] reported the presence of autophagosomes in the GCs of atretic follicles in humans. Furthermore, GCs of atretic follicles exhibit an extreme level of LC3-II and cleaved CASP3 and later found to be gonadotropin-dependent; hence, states autophagy and its involvement in apoptotic induction in rat GCs [Citation22]. Therefore, in rat ovaries, a unique process of cell death is reported in atretic follicles, which involves the combination of type I (apoptosis) and type II cell death (autophagy) [Citation100].
Pieces of evidence from previous studies have confirmed that autophagy is explicitly pronounced in mammalian cells facing oxidative stress. In ovarian tissue, enhanced autophagy resulting from oxidative stress damages the GCs, which provokes follicular atresia. Research conducted on melatonin reflects its protective role in follicular health by preventing oxidative damage in GCs. This protective effect of melatonin is because of blocking MAPK8/JNK (mitogen-activated protein kinase 8) directed autophagy, in which increased interaction between BCL2 and BECN1 is responsible for repressing autophagy. Therefore, blocking autophagy via the melatonin-MAPK8-BCL2-BECN1 axis might support GCs from oxidative stress, although it does not participate in scavenging ROS neither restores GCs viability [Citation101]. FSH involvement has been disclosed in defending GCs from oxidative stress via suppressing autophagy, although the exact mechanism is not known. This study shows that mitophagy induced by oxidative stress is mainly responsible for GCs death, which is found to be reduced after FSH treatment, hence, supports the involvement of FSH in protecting GCs death via inhibiting mitophagy [Citation102]. Additionally, Shen et al. [Citation103] reported that activation of the PI3K-AKT-MTOR pathway is requisite for the action of FSH in subduing oxidative stress instigated autophagy in GCs.
Generally, under stress conditions, cells promote autophagy, making cells adapted to the unfavorable environment and favors cell survival. Sometimes, it is also accountable for cell death (autophagic cell death); hence, autophagy is referred to as an ambiguous nature [Citation21]. Apoptosis does not have complete control over follicular atresia; In fact, recent studies emphasize the death of GCs in ovarian tissue via induction of autophagy [Citation104]. Aggregation of autophagosomes involved enhancing the death of GCs and fostering apoptosis by diminishing BCL2 level accompanied by activation of caspase. Various studies have reported a close link between both types of programmed cell death, induction of apoptosis via autophagy [Citation105].
Crosstalk between PCOS and autophagy
Hyperandrogenism in PCOS and autophagy involvement
Hyperandrogenism is marked by an increased testosterone level in females leading to clinical manifestations like acne, hirsutism, female-pattern alopecia, etc. The root cause behind hyperandrogenism is many, including adrenal/ovary tumor, congenital/acquired adrenal hyperplasia, acromegaly, hyperprolactinemia, and side effects of various drugs. Androgens are mainly produced from three locations: ovary, adrenal, and peripheral (skin, liver, etc.). The term “functional hyperandrogenism” is specially coined when hyperandrogenism is recognized with clinical symptoms without noticeable etiology, mainly used when the main contributor to AE is the ovary. When AE results from follicular TCs, then the ovary generates multiple cysts and shows polycystic morphology, whereas when a non-follicular stromal source causes AE, then called stromal hyperthecosis. For those cases in which the prime cause is nonidentified, these particular cases are mostly diagnosed with PCOS. However, in PCOS, the primary source of hyperandrogenemia (HA) is the ovary (major androgens secreted: androstenedione and testosterone) [Citation106].
Under the ordinary situation, the adrenal gland and ovaries contribute equally to testosterone hormone production depending upon adrenocorticotropic hormone and LH secretion, respectively [Citation41]. In functional ovarian hyperandrogenism, the hypothalamic-pituitary axis plays a vital role as the pituitary becomes more responsive toward gonadotropin-releasing hormone induces LH release [Citation106]. Normally the androgens produced are metabolized by the liver, but in HA, the androgens derived from either source get metabolized into another potent androgen called dihydrotestosterone by the skin. In addition to metabolism, the production of SHBG (sex hormone-binding globulin), a transport protein for androgens synthesized by the liver, is also inhibited in ovarian hyperandrogenism [Citation106]. In PCOS, it is observed that elevated LH:FSH ratio, increased oxidative stress, and the heightened level of unbound testosterone all are interconnected with each other and present in PCOS women with functional ovarian hyperandrogenism [Citation41,Citation107].
Apart from testosterone, the production of intermediate steroids during steroidogenesis is also enhanced, indicating the overall upregulation of enzymes involved in the steroid biosynthesis pathway. This abnormality suggests a disturbed endocrine environment such as higher LH and INS (insulin) levels. Excessive stimulation by LH induces androgen production in ovarian TCs by upregulating the key androgen-producing enzyme that is CYP17A1 [Citation108]. To maintain the balanced level of hormones, androgen produced by TCs is harmonized with estrogen produced by GCs. Several steps in the steroid synthesis pathway are done by the CYP17A1 enzyme, which is dependent on LH action; the action of CYP17A1 is compensated by decreased responsiveness toward LH activity. Moreover, other intra-ovarian secretions also play a crucial part in regulating the activity of CYP17A1, including testosterone and estradiol act as inhibitory factors, whereas INH, INS, and IGF1 an excitatory factor. Disequilibrium between inhibitory and excitatory factors leads to ovarian HA regardless of LH discharge [Citation109].
Compared to normal TCs, TCs of PCOS patients convert precursors of androgens with greater efficacy [Citation110]. Evidence from the previous investigation highlights the intra-ovarian abnormality specifically associated with steroidogenesis in TCs is the root cause of hyperandrogenism linked with PCOS, as TC is the only place where androgens are synthesized in the ovaries. To prove this hypothesis, the activity of androgen producing enzymes, and their mRNA expression were examined in the long-run culture of TCs from PCOS and normal ovaries. Synthesis of progesterone, 17a-hydroxyprogesterone, and testosterone are enhanced in the culture of PCOS TCs. Additionally, mRNA expression of cholesterol side-chain cleavage enzyme and CYP17A1 is also magnified in PCOS TCs culture, as well as activity of HSD3B and HSD17B were also enhanced, whereas no significant change in mRNA expression of the steroidogenic acute regulatory protein was noticed. Therefore, this study confirmed that the enhanced production of androgens in the TCs is the intrinsic property of TCs [Citation111].
IR associated with genetic variance plays a significant part in inducing HA in direct and indirect ways: directly by acting on the adrenal or ovary, and indirectly by inducing hyperinsulinemia. For instance, TNF (tumor necrosis factor) interferes with INS signaling by interacting with IRS1 (insulin receptor substrate 1), alters reproductive functions, which closely resemble PCOS and hyperandrogenism [Citation112]. Growth factors secreted from oocytes and follicular cells mainly regulate follicle development in the early phase by acting in a paracrine manner. However, the androgens produced from TCs play a crucial part during the development of AF; complementarily, they stimulate the FSH receptors expression on GCs. Furthermore, FSH induces LHR expression on GCs, thereby luteinizing the GCs; this process is also augmented by INS. Excess of androgenic hormones provoke proliferation of small AF, subsequently increase in AMH prevent the entry of PMFs into the growth phase [Citation41].
HA being the characteristic feature of PCOS, previous studies disclose that regulation of autophagy in response to androgen might play a noteworthy role in PCOS pathogenesis (). Deterioration in autophagy is associated with protein aggregates accompanying an increased SQSTM1/p62 (sequestosome 1; an autophagosome cargo protein) and ubiquitin [Citation113]. It has been reported that PCOS affected ovary shows the accumulation of SQSTM1 and ubiquitin in the TCs layer using immunohistochemistry. Suppression of autophagy has been noticed in TCs of PCOS ovaries, which can induce ROS production. Palmitic acid (PA) inhibits late-stage autophagy and promotes the elevation of CYP17A1 and SERPINE1/PAI1 (serpin family E member 1) mRNA level as well as increases the production of androgen through ROS-MAPK/p38 and MAPK8 signaling [Citation114]. Consequently, a raised level of PA has been noticed in the serum and FF of PCOS patients and prompt androgen formation [Citation115,Citation116]. Therefore, PA has been hypothesized to be involved in PCOS pathogenesis. Similar expression of LC3-II:LC3-I but increased SQSTM1 was observed on TCs exposed to PA in vitro. Inclusively, PA supports the accumulation of phagosomes and alters autophagic degradation in bovine TCs () [Citation117,Citation118].
Figure 5. Link between autophagy and hyperandrogenism in PCOS. Different autophagic responses are observed at various locations due to an increased level of androgen in PCOS. (A) Theca cells: Increase in palmitic acid augments SQSTM1 and ubiquitin, which inhibits autophagy. Declined autophagy induces testosterone production by increasing ROS and enhanced mRNA expression of CYP17A1 and SERPINE1 via MAPK/p38 and MAPK8 activations. (B) Skeletal Muscles: Testosterone causes activation of MTORC1, which inhibits the autophagy proteins and suppresses autophagy. (C) Uterus: Altered gene expression in response to hyperandrogenemia inhibits autophagy. (D) Granulosa Cells: Testosterone induces expression of BECN1, ATG, LC3, and favors autophagy. Red arrow: deactivation; Black arrow: activation
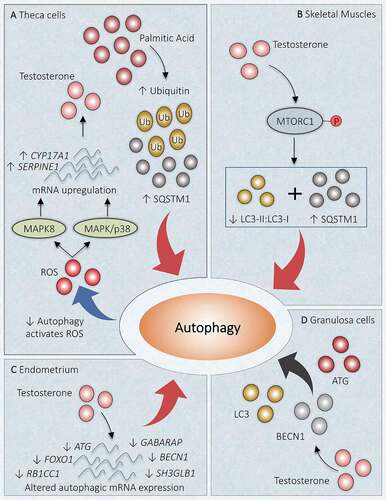
In a recent study in DHEA-induced PCOS mice, the role of MTORC1 was investigated in directing autophagy in skeletal muscles. This study has reported MTORC1 activation after INS administration which ultimately reduces the autophagy, evidenced by declined LC3-II:LC3-I and enhanced SQSTM1 expression. Similarly, testosterone exposure to in vitro culture of C2C12 myotubules induces the activation of MTORC1 (phosphorylation of MTORC1), which in turn reduces autophagy together with IR and mitochondrial dysfunction. However, impeding the MTORC1, we get the opposite results as mentioned before. Conclusively, the detrimental effect of HA specifically boosted testosterone level, is instigated by obstructing autophagy, showing that testosterone and autophagy are negatively associated with skeletal muscles () [Citation119].
Autophagy could exhibit a protective role in cyclical redesigning of the human endometrium [Citation120]. Another study has inspected mRNA level in endometrium and found decreased expression of genes incorporated in autophagy: FOXO1, ATG3, ATG5, ATG13, ATG14, RB1CC1, SH3GLB1, GABARAP, BECN1, indicate its linkage with HA in PCOS women (). Metformin is effective in the reversal of disrupted expression of some autophagic genes such as FOXO1, ATG3 in endometria [Citation121].
In PCOS women, increased mRNA abundance of ATG7, ATG5, BECN1, and an elevated ratio of autophagy marker protein LC3-II:LC3-I together with a declined abundance of SQSTM1 exhibits in GCs. Direct relation has been observed between mRNA level of BECN1 in GCs and testosterone level in vitro in a dose-dependent fashion in cultured GCs of PCOS women [Citation24], stating the correlation between androgen and autophagy. Autophagy in the GCs is detected to be enhanced on exposure to testosterone propionate by increasing BECN1 and LC3 (). In contrast, treatment with the Gui Zhu Yi Kun formula remarkably reduces BECN1 level in response to testosterone. The result of Xing et al. [Citation122] study explored the feasible mechanism involved in Gui Zhu Yi Kun formula action and suggested the activation of tumor protein TP53-AMP-activated protein kinase (AMPK) signaling succeeded by enhanced MTOR expression and consequently leads to autophagy inhibition.
Anovulation associated with PCOS and autophagy
Infertility caused by anovulation is defined by no ovum release due to hampered follicle bursting [Citation123]. In conjunction with HA, anovulation is termed as hyperandrogenic anovulation, identifiable by cysts formation in ovaries accompanied by an elevated LH to FSH ratio and decreased SHBG [Citation124]. PCOS is a diversified disorder with HA associated with persistent anovulation. Due to the failure of selecting dominant follicles as a result of cysts formation, PCOS women experience anovulatory infertility and menstrual irregularities. In accordance with WHO, PCOS women cover a maximum fraction of women under WHO class 2 ovulatory dysfunction marked by chronic anovulation coupled with normal levels of estradiol and FSH [Citation125].
Crosschecking the follicular population in ovaries of anovulatory and ovulatory women with PCOS demonstrates a reduced fraction of resting follicles and an increased fraction of growing follicles compared to healthy ovaries [Citation126]. Notably, in PCOS-associated anovulation, there is emerging evidence that supports deviation in folliculogenesis at the earliest stage, which is entirely gonadotropin independent. Therefore, an increased population of follicles at the initial growth phase and small pre-AFs are noticed in anovulatory PCOS. Four possible reasons explain the increased number of the small pre-AF in ovaries of anovulatory women with PCOS – (i) fetal ovaries are granted with a high percentage of PGCs, (ii) in fetal ovary oogonia experience high mitotic division, (iii) during pregnancy; follicle formation begins at 12th week which involves the arrangement of somatic cells around oocyte, this process might get pronounced in PCOS ovaries, and (iv) from fetal ovary till menopause there is a high rate of germ cell loss, which might get reduced in PCOS [Citation127]. Because follicles cannot undergo ovulation in PCOS, the development of PCOS ultimately leads to anovulatory infertility. Although the exact mechanism underlying PCOS-associated anovulation is unknown, evidence suggests that the arrest of follicles development at the antral stage (5–10 mm wide) and failure to go in pre-ovulatory phage is the possible cause behind anovulation. Despite detention in follicular growth, there is an abnormality in the secretory function of GCs leading to hypersecretion of estradiol, which might be a cause of hyperinsulinemia. Excessive release of INS, testosterone, and LH may be engaged in causing anovulation through a common mechanism involving cAMP production within GCs at the pre-ovulatory stage (). Plausibly, anovulation has its origin at the very beginning of folliculogenesis [Citation128].
Figure 6. Autophagy adaptations during anovulation associated with PCOS. An altered population of follicles at the initial stages of follicular development including, an increase in the number of primary follicles and pre-antral follicles. Autophagy causes granulosa cell death in the pre-antral follicle and leads to follicular atresia. Heightened mitophagy in the cumulus cells in response to oxidative stress in follicular fluid. Defective mitophagy causes oocyte damage causes deformation of oocytes. Autophagy prevents the transition of antral follicles to the pre-ovulatory stage and opposes ovulation. An increase in the level of INS, LH, and testosterone prevents ovulation by augmenting the cAMP level
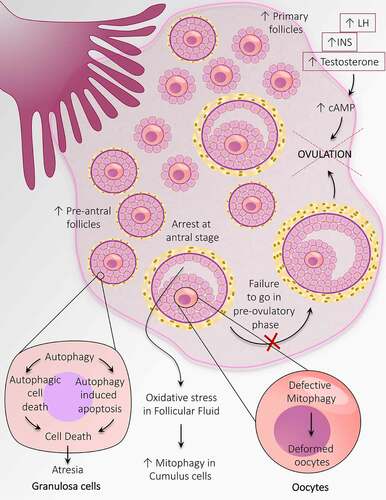
Early-stage follicles mainly secrete AMH specifically pre-AF and early-AF. Locally secreted AMH is majorly involved in disturbing follicular growth along with other local regulators such as IGF1 and sex hormones [Citation129]. Level of AMH is found to be elevated in PCOS women exhibiting anovulatory infertility, indicates a linkage between AMH level and malfunctioning in the ovary in anovulatory PCOS women [Citation125,Citation130]. Additionally, it has been noticed that disturbance in INS action is associated with an increase in body mass index. Obesity in PCOS women is more frequently associated with defaulted reproductive performance regarding ovulation and AE, leading to anovulatory infertility [Citation131]. However, the principal dissimilarity in PCOS women’s metabolic background with or without ovulation is the occurrence of hyperinsulinemia and IR in anovulatory sufferers [Citation132]. In conclusion, the fundamental cause of anovulation associated with PCOS is aberrant folliculogenesis at the initial stages.
Mitophagy, a type of selective autophagy which particularly destroys defective mitochondria to counteract stress conditions [Citation133]. It has been anticipated that maternal transfer of malformed mitochondria to fetus might be involved in the defective mitophagy in oocytes [Citation134,Citation135]. Spoiled mitophagy and decreased mitochondrial membrane potential contribute to deformed oocyte formation in the PCOS ovary. A discrepancy in oxidation-reduction potential associated with elevated mitophagy. It has been recognized that oxidative stress in FF is the cause of increased mitophagy in cumulus cells, which may be responsible for anovulation in PCOS [Citation136]. Ramly et al. [Citation137] have reported four PCOS-associated proteins engaged in mitophagy; E2F1, FOXO3, JUN, and MAPK9, and suggest PCOS association with hypertension.
The primary process by which follicles commit death by atresia differs according to the stage of follicles. It has been reported that atresia occurs in the pre-AFs, and the AFs are by GCs autophagy and apoptosis, respectively [Citation138]. As the population of follicles in the pre-antral stage is enhanced in PCOS-associated anovulation, it suggests that autophagy might play a part in anovulation associated with PCOS.
Disordered folliculogenesis, follicular cyst in PCOS, and autophagy
The primary difference between the normal ovary and polycystic ovary is disordered folliculogenesis at earlier stages of development, indicate that abnormality starts at the beginning stage of the follicular development only. In contrast to healthy ovaries, there is a 2-6-fold increase in primary, secondary, and small AFs in addition to AFs in polycystic ovaries [Citation139]. The follicle size ranging from 2–10 mm is observed more (=/>10 number) in polycystic ovaries [Citation127].
The imbalance in the female pituitary hormones (increased LH:FSH ratio) leading to disturbed folliculogenesis in ovaries ultimately causes cysts formation mainly in ovarian AFs. Cysts are primarily defined by follicle containing a large water-filled pouch and the altered density of follicular cells, which hinders the release of the ovum. Due to water deposition, the size of the cyst can expand as large as 10 mm wide, which ultimately increases the ovary size up to 10 cm wide. Hence, healthy egg transitions into a cyst named a functional cyst which hampers ovulation [Citation140]. Regardless of ovulation, the percentage of follicles in the growth phase indicates that there is an enhanced recruitment of follicles from the RP in polycystic ovaries.
Additionally, it is reported that inhibition of intra-ovarian HSD11B1 (hydroxysteroid 11-beta dehydrogenase 1) activity by androgens results in enhanced cystic follicle formation in anovulatory PCOS women [Citation141]. Taken together, the majority of studies have confirmed a surplus number of small AFs in PCOS along with hyperplasia of TCs (hyperthecosis), cortical thickening, and stromal hyperplasia and hypertrophy [Citation142]. As a result, disruption in the menstrual cycle results due to the blockade of ovulation, leading to the absence of menses called “amenorrhea.” Conclusively, due to disturbance in the female reproductive cycle, fertilization becomes impossible, leads to loss of fertility in women facing PCOS [Citation140].
In PCOS, the ovary acquired various changes in response to AE, promoting follicle recruitment, subsequently increasing the population of small follicles, morphological alteration, and follicular arrest. Folliculogenesis induced by androgen increases follicles population at an early stage promotes more AMH production per GCs [Citation61]. Indirectly arrest of follicles at maturation occurs due to enhanced follicles at an early stage, which is because of the inhibitory effect of AMH [Citation42]. In contrast to the mid-follicular phase in normal women, prematurely luteinized follicles are more likely to respond toward LH, which inappropriately secretes estradiol and androgen in PCOS women [Citation143]. Hindrance in follicle proliferation due to premature luteinization causes follicular arrest at the maturation phase, which further hampers the dominant follicle development [Citation144]. In the overproduction of androgens by TCs, dysfunction of GCs also found to give a contribution. Due to enhanced LH and INS levels, the GCs become more luteinized prematurely. Conclusively, intra-ovarian abnormality due to altered levels of hormones and other intra-ovarian factors contributes to disordered folliculogenesis in PCOS, indicating polycystic ovarian morphology [Citation129].
TUG1 (taurine up-regulated 1), a form of long non-coding RNA involved in cellular function like cell proliferation, apoptosis, and autophagy. A recent study reported enhanced expression of the TUG1 gene in GCs and contributes to the amplified AF population in PCOS ovaries [Citation145]. Although, the role of TUG1 is not explored much in PCOS. A study by Li and Chen et al. [Citation146] founded a notably high expression of TUG1 long non-coding RNA in PCOS GCs and was analogous to the AFs population. According to this study, TUG1 might hamper the MAPK/ERK pathway, hence, retard autophagy in GCs and support AFs survival (), stating the involvement of TUG1 in promoting the formation of AFs in the PCOS ovary.
Figure 7. Disturbed folliculogenesis in PCOS and its connection with autophagy. Different autophagic pathways are involved in altered folliculogenesis in the PCOS ovary. An increase in ROS promotes autophagy and the formation of cysts. An increase in TUG1 expression suppresses autophagy by obstructing the MAPK/ERK pathway and supports antral follicle survival. Activation of the MTOR pathway inhibits autophagy and promotes aberrant growth of follicles
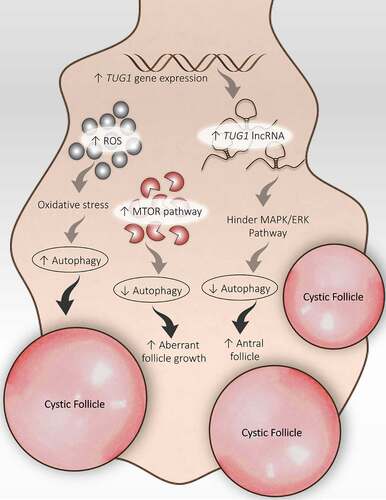
DHEA-induced PCOS model showed enhanced expression of MTORC1 and MTORC2 in the mouse ovary. Interruption in selecting dominant follicles during the ovarian cycle results in aberrant follicle growth; this disorganized ovarian physiology occurs in response to the MTOR signaling pathway in the PCOS ovary [Citation147]. However, in ovarian endometrial cysts (blood-filled ovarian cysts lined by endometriotic tissue), abnormal MTOR activity affects the autophagy process. In this particular situation, the autophagic induction of apoptosis is reduced by uninhibited MTOR activity [Citation148]. Altogether, it suggests that the MTOR-autophagy axis is involved in regulating ovarian folliculogenesis.
Oxidative stress caused by an elevated level of ROS can result from hypoxia/low partial pressure of oxygen in any organ. Hypoxia is responsible for inducing autophagy. A link between autophagy and ROS has been established already. Particularly in the ovary, a consequence of hypoxia has a very harmful impact on the reproductive functions of the ovary because of damaged ovarian follicles. Past studies demonstrate the involvement of hypoxia in the formation of cysts in the ovaries as well as modification of the estrous cycle in female rats resulting in decreased fertility in rats ovary, suggesting a detrimental effect on follicular development in the ovary [Citation149,Citation150].
Metabolic impact on the PCOS and autophagy role
A particular metabolic abnormality does not specify metabolic syndrome, but it is a fusion of diverse risk factors. The most noticed risk factors are obesity, hypertension, dyslipidemia, and hyperglycemia. PCOS is not only an endocrine disorder but also a metabolic disorder in which various metabolic abnormalities play a crucial role in PCOS progression. The potential pathogenic factor contributing to PCOS is IR, which serves as a connecting link between PCOS and metabolic syndrome (). Therefore, treatment targeted to control IR proved beneficial in PCOS [Citation151].
Figure 8. Autophagy role in metabolic abnormalities acquired during PCOS in various metabolic organs. Autophagy plays a crucial part in governing the metabolic response in affected tissues in PCOS. (A) Adipose tissue: Obesity-driven INS resistance because of adipocyte dysfunction and defaulted INS signaling pathway is suppressed via the induction of autophagy. (B) Ovary: Hyperinsulinemia and hyperandrogenism both share a direct relationship, where INS promotes the expression of steroidogenic enzymes ARK1C3 and CYP17A1. Regulation of autophagy inhibits INS resistance and hyperandrogenemia. (C) Skeletal muscles: Autophagy is attenuated in PCOS via activation of MTOR accounts for INS resistance. (D) Heart: Cardiac autophagy gets halted by decreased autophagy protein level and decreased AMPK phosphorylation responsible for causing hypertrophy in cardiac cells, leading to cardiovascular (CVS) complications. (E) Liver: Decline in turnover of autophagosomes is observed in response to decreased vitamin D level, contributes to liver disease which gets suppressed on autophagy stimulation
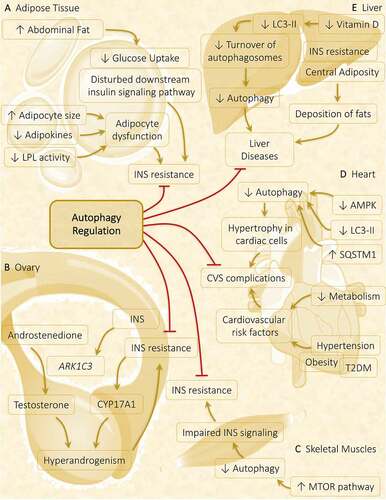
It is believed that with an increase in body mass index, the risk of acquiring metabolic syndromes rises by 14-fold in PCOS females. Previous researches suggest increased deposition of abdominal fat in 60–95% of PCOS women, which can undermine INS signaling and exacerbates IR [Citation152]. In all the INS-targeted tissues, impairment in the INS signaling pathway is noticed, whereas in the ovary, INS-mediated steroidogenesis remains functional. As a consequence of IR, there is an activation of CYP17A1 in TCs leading to functional ovarian hyperandrogenism. Additionally, INS directly upregulates the genetic expression of ARK1C3 via INS-facilitated promoter functions, which increases the production of testosterone from androstenedione [Citation153]. Thus, considering all the evidence together contributing to the development of PCOS, two features are found to be constant in the majority of PCOS-positive women are HA and hyperinsulinemia, where the development of both features are dependent on each other [Citation154].
Nevertheless, abnormal adiposity is not the only factor that contributes to IR; there are other factors also including alteration in adipokine level, disturbed downstream INS pathway, and default in glucose, lipid, and steroid metabolism [Citation155]. The size of the adipocytes is an essential criterion to predict the incidence of T2DM in women as it is correlated with impaired INS signaling. Observing the mean adipocyte size in PCOS women reveals hypertrophy in adipocytes. Despite its size, decreased adipokines level and decreased LPL (lipoprotein lipase) activity in adipocytes lead to adipocytes dysfunction. All these observations might play a crucial role in causing IR in PCOS women [Citation156].
Another metabolic factor prevalent in PCOS women is the cardiovascular risk factor [Citation157]. More precisely, in PCOS, cardiovascular disease risk is enhanced with deviation in the body’s metabolic status, such as defective metabolism of glucose and lipid along with low-grade inflammation, increased coagulation factors, and increased body weight [Citation158]. PCOS women have a strong possibility of being detected with cardiometabolic diseases, including T2DM, obesity, and hypertension, compared to healthy women without PCOS [Citation159]. The result of anthropometric studies and observation of metabolic defects in PCOS exactly corresponds to metabolic syndrome [Citation151].
Recent pieces of evidence have supported crosstalk between liver disease, specifically nonalcoholic fatty liver disease (NAFLD), and PCOS, which proposes a unique hepato-ovarian link. NAFLD is a chronic liver disease defined by the deposition of fats and is related to IR and central adiposity. Therefore, a possible link exists between NAFLD and PCOS as they share a common culprit for developing metabolic abnormalities. PCOS-associated fatty liver disease shows a higher waist-hip ratio, elevated fasting glucose level, lower high-density lipoprotein level, higher testosterone, and higher Homeostatic Model Assessment of IR (a measure of IR). In summation, the consequence of PCOS do not remain in the reproductive organ; instead, it gets spread to tissues playing a part in developing metabolic abnormalities [Citation160]
Burghen et al. [Citation161] were the first to focus attention on the connection between PCOS and IR. For the development process of follicles, the ovary requires energy derived from the INS pathway. On account of previous studies, impairment in the downward/post-receptor INS signaling causes IR in the PCOS ovary [Citation162]. The role of autophagy is well established in the normal functioning of the INS signaling pathway in various metabolic disorders such as obesity [Citation163,Citation164]. In INS-targeted tissues, autophagy shows divergent responses in diabetes – autophagy stimulates the INS signaling in skeletal muscles, adipose, and liver; conversely, persistent autophagy destroys β-cell in the pancreas and decreases INS secretion. Hence, precise autophagy is required for regulating INS action [Citation165]. Similarly, regulating the autophagy in adipose () and ovary () might inhibit IR in PCOS. In skeletal muscles of DHEA-treated mice, suppression of autophagy is spotted in response to INS exposure via activation of MTOR1 following decreased phosphorylation of AKT. Data suggests hyperactivation of MTORC1 on excessive androgen exposure, independent of AKT activation, inhibits autophagy, thereby promotes glucose intolerance and mitochondrial dysfunction leading to IR (). Therefore, this MTORC1-autophagy pathway might produce IR in the skeletal muscles in PCOS [Citation119].
Acupuncture is believed to be effective in PCOS suggested by many clinical examinations [Citation166–169]. Data indicates that electroacupuncture is found effective in relieving symptoms associated with PCOS such as IR, mitochondrial dysfunction, ER stress, and hyperandrogenism by promoting autophagy in skeletal muscles. A possible mechanism involved in activating autophagy is via suppressing the hyperactivated MTOR-EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1) signaling pathway [Citation170]. A cardiac study unveils that mitochondrial damage can be repaired by fixing mitophagy [Citation171]. Similarly, controlling mitophagy in PCOS rats will reduce mitochondrial dysfunction, in which electroacupuncture is found to be effective [Citation170].
Overgrowth is fostered in cardiac cells due to depletion in autophagy is observed in hypertrophied cardiac tissue [Citation172]. Diminished cardiac cell autophagy is linked with decreased AMPK activity in the myocardium of PCOS-induced mice. Therefore, autophagy plays a crucial role in cardiac tissue; unbalanced autophagy is concomitant with cardiac hypertrophy. The involvement of macrophage migration inhibitory factor in governing MTOR activity is associated with inducing autophagy, which safeguards the myocytes during hypertrophy [Citation173]. Interestingly, estimating autophagy in myocardiocytes of PCOS mice showed – (i) decreased expression of autophagy protein LC3-II with increased autophagy substrate SQSTM1, (ii) reduced expression of activated AMPK, suggests depleted autophagy (). Observed facts reflect cardiac impairment in PCOS mice correlated with autophagic interference [Citation18,Citation174].
PCOS is correlated with the damage associated with autophagy in the hepatic cells of nonalcoholic steatohepatitis patients [Citation175]. In the PCOS rat model, a link has been established between autophagosome formation and unorganized INS signaling coupled with nitrative stress in liver cells. Vitamin D deficiency is widespread among infertile women; frequently correlated with PCOS [Citation176,Citation177]. According to some reports, hyperandrogenic animals with reduced vitamin D levels manifest a lower turnover of autophagosomes because of the dropped level of activated LC3-II () [Citation178]. Malfunctioning of autophagy in the liver is due to vitamin D deficiency [Citation178] Taken together, all the evidence encourages further research in targeting autophagy in PCOS pathogenesis.
Interplay between different conditions associated with PCOS progression and autophagy
To overcome unfavorable conditions, SIRT1 (sirtuin 1) works complementarily with AMPK to achieve the desired cellular function [Citation179]. Elevated AMPK level and the existence of autophagy marker evince the correlation between SIRT1-AMPK signaling and autophagy. When SIRT1-AMPK-Autophagy relation was explored in PCOS by Di Emidio et al. [Citation180] result stipulates an increased level of activated phosphorylated AMPK with increased SIRT1 in DHEA-treated ovaries besides the presence of autophagy marker. Hence, the result displays the SIRT1-AMPK axis in managing autophagy in PCOS ovaries. Deacetylation of crucial autophagy mediator protein LC3 was found to be regulated by SIRT1, which implies its role in controlling autophagy [Citation181]. SIRT1 mediated activation of LC3 by deacetylation took place inside the nucleus during nutrient unavailability. Deacetylation performed by SIRT1 at K49 and K51 position permit LC3 to communicate with nuclear cofactor called TP53INP2/DOR (tumor protein p53 inducible nuclear protein 2), which sequentially returns it to the cytoplasm where it can chain with autophagy promoting factors like ATG7 and finally initiate the formation of autophagic membrane following conjugation with phosphatidylethanolamine (). Therefore, in PCOS the acetylation-deacetylation cycle of LC3 plays a significant role in regulating autophagy to subjugate starvation [Citation182].
Figure 9. Mechanisms governing autophagy during different conditions related to PCOS. In PCOS, autophagy is depicted in response to different conditions such as (A) Nutrient deprivation: In ovaries, phosphorylation of AMPK induces deacetylation of LC3 via SIRT1 activation. Activated LC3 binds with TP53INP2, which promotes its translocation from the nucleus to the cytoplasm, where it can bind with ATG7 and initiates autophagosome formation. (B) Inflammation: Autophagy activation is accompanied by inflammation in PCOS via an increase in HMGB1, which stimulates autophagy in granulosa cells and causes its death. Whereas, an enhanced level of IGF1 is involved in governing autophagy via the PI3K-AKT-FOXO1-MTOR pathway. (C) Endometrial hyperplasia: Defaulted autophagy triggers hyperplasia in the endometrium in PCOS, which is reduced by induction of autophagy and autophagy induced apoptosis
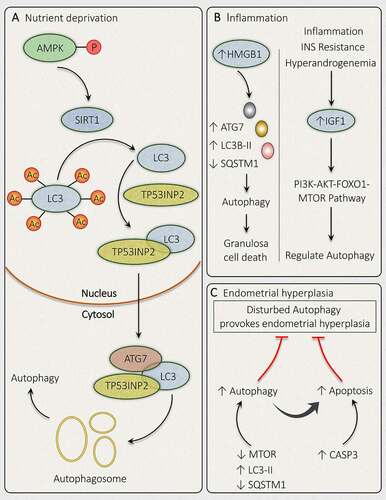
Evidence from previous studies proved PCOS to be a pro-inflammatory condition. In PCOS ovarian tissue, IR and HA are found to be correlated with a low grade of inflammation. Increased intranuclear expression of NFKB (nuclear factor kappa B) and reduced NFKBIA (NFKB inhibitor alpha) in mononuclear cells in PCOS women supports the association of inflammation with PCOS [Citation183,Citation184]. HMGB1 (high mobility group box 1) acts as an inflammation mediator; its level is measured to be raised in the blood of T2DM patients. HMGB1 exerts its effect via NFKB signaling, stating its role in IR [Citation185]. An elevated level of HMGB1 is observed in the blood circulation and FF in PCOS women with IR, suggesting the contribution of HMGB1 in developing IR in PCOS with associated inflammation [Citation186,Citation187]. Recently, the role of HMGB1 in regulating the autophagy inside the ovaries is being noticed, where it induces the autophagic reactions in GCs, resulting in a declined GCs population (). Exacerbated autophagy in response to HMGB1 is due to its action on autophagic proteins including an enhanced LC3B-II:LC3B-I ratio, ATG7 level, and reduced SQSTM1 level. HMGB1 affects INS signaling by influencing SLC2A4/GLUT4 (solute carrier family 2 member 4) translocation, NFKB signaling, AKT phosphorylation, IRS1 as well as glucose uptake in GCs. All these actions can be revoked by occluding HMGB1-mediated autophagy [Citation20].
IGF1, a hormone that can induce inflammatory cytokines production, is found to be elevated in PCOS and appear to have an impact on autophagy. Many aspects can contribute to increased IGF1, including inflammation, IR, HA, all are the features of PCOS [Citation24]. A recent study on zebrafish ovary reveals that locally produced intra-ovarian Igf1 is involved in the growth and development of PFs [Citation188]. Therefore, IGF1 might play a potential role in the PCOS ovary. Hyperglycemic diet is hypothesized to induce IGF1 and INS secretion in acne conditions. Furthermore, a reduced amount of transcription factor FOXO1 in the nucleus, activation of PI3K-AKT pathway, increased expression of MTOR is noticed in acne patients. However, it has been reported that INS, IGF1, and androgen play an essential role in controlling the above PI3K-AKT-FOXO1-MTOR signaling pathway. Similarly, PCOS is also associated with PI3K-AKT-FOXO1-MTOR signaling [Citation189,Citation190], therefore suggests its relation with an autophagic impairment.
Disorganized autophagy is reported to be accountable for endometrial hyperplasia (EH) associated with PCOS [Citation121,Citation191]. A study conducted by Wang et al. [Citation192] checked the effect of metformin and sorafenib in regulating programmed cell death in PCOS induced EH. In vitro study in endometrial cells report that metformin reduces MTORC1 and SQSTM1 expression and sorafenib reduces MTORC2 and SQSTM1 expression, whereas both significantly enhance markers of autophagy (LC3-II) and apoptosis (CASP3) (). Hence, data supports autophagy involvement in EH-associated with PCOS.
Furthermore, to overcome the stressed microenvironment in the ovarian cells, autophagy plays a potential role by regulating the expression of autophagic genes, supported by real-time polymerase chain reaction. Regression analysis reveals a possible network responsible for activating autophagy in PCOS, which includes genes: FOXO1, MAPK1, MAPK9, IGF1, TP53, NFKB1, ERBB2, EGFR. In response to stress conditions, the involvement of these genes are observed in particular cellular mechanisms and are associated with signaling pathways, namely INS, MTOR, ERBB, and involvement of endoplasmic reticulum in the processing of proteins. Hence, this study supports that the alteration in autophagic genes is related to PCOS pathogenesis [Citation193]. Conclusively, all the evidence supported the importance of autophagy in altered physiology in PCOS conditions.
Concluding remarks and future perspectives
Evidence from the previous researches signifies the unique role of autophagy in governing follicle’s fate. Death of follicular cells is constant activity, where autophagy, autophagic cell death, and autophagy induced apoptosis are all noticed in the ovary throughout their lifespan. Recently, the impact of autophagy in ovarian follicular development and follicular atresia has caught a lot of attention, evincing its role in reproductive functions [Citation194]. Autophagy has become noticeable in regulating functions of major female sex hormones (progesterone and estrogen), which play a crucial role in maintaining many physiological processes necessary for reproduction [Citation195]. Plenty of proofs from preclinical researches reveal that exorbitant/inadequate autophagy might be involved in boosting cell death [Citation196]. Therefore, proper tuning of autophagy is required in maintaining cellular homeostasis. Exploring the autophagy involvement in disease progression and/or retrogression is an interesting domain for understanding the pathology of diseases.
Several studies have established the participation of autophagy in PCOS development, where the inhibition and/or activation of autophagy appears to ameliorate the PCOS progression depending upon the type of cells affected. As discussed above in PCOS, in some cells, autophagy is found to be pronounced (GCs), while in others it is suppressed (TCs). In PCOS, apoptosis is evident in follicular turn of events for a long time, but its relation with autophagy has recently gained more attention. As described above, impaired autophagy machinery is involved in declining GCs population, disturbed folliculogenesis, and associated metabolic abnormalities. Various researches have proposed possible mechanisms involved in altered autophagy in PCOS. However, there is still a need to establish: is there any other mechanism overseeing autophagy associated cell death and/or cell survival in ovarian tissue? As of now, a profound understanding of autophagy and its link-up with PCOS development remains unaccounted for.
Perturbed circadian rhythmicity provokes IR and which augments the chances of acquiring metabolic syndromes like, T2DM [Citation197], NAFLD [Citation198], and obesity [Citation199]. Similarly, circadian arrhythmicity is linked with IR and HA in PCOS [Citation200]. Recent investigations define the pivotal role of chronobiology in promoting PCOS development [Citation201]. As per the literature available to date, hyperinsulinemia and HA are the two major pathological contributors of PCOS [Citation202] and emerging evidence shows their influence on autophagy during PCOS development. Also, both of these factors contribute to the abnormal expression of circadian clock genes leads to the imbalanced circadian rhythm in PCOS [Citation203]. In PCOS, hyperandrogenism is recognized in modifying the expression of clock genes in hepatic and ovarian cells in rats, where it is involved in metabolic and reproductive functions, respectively [Citation204].
Lately, the interplay between the circadian clock and autophagy has become apparent [Citation205]. Autophagy can control circadian biology and can be controlled by circadian rhythm. Sometimes, circadian proteins can get deteriorated by autophagy which influences circadian biology [Citation206]. Circadian rhythm supervises various cellular processes to cope with the metabolic demands, including control over the transcription of ATG genes [Citation207,Citation208]. Additionally, mRNA expression of key autophagy protein BECN1 and the conversion of LC3-I to LC3-II are also under rhythmic control [Citation209,Citation210]. Some proteins are known to control autophagy as well as circadian biology, for instance, SIRT1, as discussed above is involved in managing autophagy in PCOS via deacetylation of LC3 and can also stimulate autophagy by impeding MTOR activation [Citation211]. Additionally, SIRT1 controls the expression of circadian proteins like ARNTL/BMAL1, PER, and CRY [Citation212]. Therefore, targeting these proteins gives better insight into circadian-autophagy-rhythm in PCOS progression. Therefore, the possible mechanism involved in circadian control over ATG genes or vice versa involved in PCOS pathogenesis convey a novel pathogenic mechanism underlying PCOS pathogenesis. Till now, no study has referenced the link between circadian biology and autophagy in PCOS. Whether the circadian rhythm signals the cell to increase/decrease the rate of autophagy in ovarian tissue in PCOS is remains to be determined.
According to our visionary, elucidating the connection between autophagy machinery and circadian biology in PCOS pathogenesis would be a great idea to focus on in future studies. Based on the suggestion for future studies, we have made a pictorial representation () for a better understanding of the circadian-autophagy-rhythm in PCOS, how circadian biology is linked with autophagy in PCOS. Targeting the circadian rhythm, the main dominator over the majority of the pathophysiological causes in PCOS may provide us a network between HA, IR, obesity, endocrine disturbances, and ovarian physiology concerning autophagy. Furthermore, future researches should look over cellular processes and molecular pathways through which circadian rhythm directs autophagy in PCOS.
Figure 10. Depicting the role of Circadian-autophagy-rhythm in PCOS. Under normal physiology, the circadian biology of the body regulates autophagy by governing the rhythmic expression of the autophagic genes. Conversely, autophagy also governs the circadian rhythm. Therefore, a balance between autophagy and circadian biology is necessary for normal physiology. In PCOS: Disturbance in circadian rhythm affects the normal physiology of the body and resulted in INS resistance which causes hyperandrogenemia, metabolic abnormalities, and damages autophagic responses leading to PCOS pathogenesis. Where hyperandrogenemia and IR affect circadian biology by hindering the expression of clock genes. Altogether, states a direct link between circadian arrhythmicity and damaged autophagy in PCOS progression
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the author(s).
Acknowledgments
SK is thankful to the Indian Council of Medical Research (ICMR) for the research fellowships, VU is thankful to the University Grant Commission (UGC) for the research fellowship. JRG is thankful to DBT and CSIR for financial support. My special thanks to all the lab members of JRG lab for helping me. CDRI communication number: 10254.
References
- Goodman NF, Cobin RH, Futterweit W, et al. American association of clinical endocrinologists, American college of endocrinology, and androgen excess and pcos society disease state clinical review: guide to the best practices in the evaluation and treatment of polycystic ovary syndrome - Part 1. Endocr Pract. 2015;21(11):1291–1300.
- Escobar-Morreale HF. Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol. 2018;14(5):270–284.
- Stein IF, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29(2):181–191.
- Goodarzi MO, Dumesic DA, Chazenbalk G, et al. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7(4):219–231.
- Lizneva D, Suturina L, Walker W, et al. Criteria, prevalence, and phenotypes of polycystic ovary syndrome. Fertil Steril. 2016;106(1):6–15.
- Picton HM, Balen AH. Transgenerational PCOS transmission. Nat Med. 2019;25(12):1818–1820.
- Ganie MA, Vasudevan V, Wani IA, et al. Epidemiology, pathogenesis, genetics & management of polycystic ovary syndrome in India. Indian J Med Res. 2019;150(4):333–344.
- Teede HJ, Misso ML, Costello MF, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum Reprod. 2018;33(9):1602–1618.
- Amato MC, Galluzzo A, Finocchiaro S, et al. The evaluation of metabolic parameters and insulin sensitivity for a more robust diagnosis of the polycystic ovary syndrome. Clin Endocrinol. 2008;69(1):52–60.
- Wolf WM, Wattick RA, Kinkade ON, et al. Geographical prevalence of polycystic ovary syndrome as determined by region and race/ethnicity. Int J Environ Res Public Health. 2018;15(11):1–13.
- Azziz R, Carmina E, Dewailly D, et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Vol. 91.Fertil Steril. 2009;91(2):456–488.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741.
- Doherty J, Baehrecke EH. Life, death and autophagy. Nat Cell Biol. 2018;20(10):1110–1117.
- Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26(4):605–616.
- Kim J, Lim YM, Lee MS. The role of autophagy in systemic metabolism and human-type diabetes. Mol Cells. 2018;41(1):11–17.
- Wang F, Jia J, Rodrigues B. Autophagy, metabolic disease, and pathogenesis of heart dysfunction. Can J Cardiol. 2017;33(7):850–859.
- Ren J, Sowers JR, Zhang Y. Metabolic stress, autophagy, and cardiovascular aging: from pathophysiology to Therapeutics. Trends Endocrinol Metab. 2018;29(10):699–711.
- Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14(6):356–376.
- Yang JS, Lu CC, Kuo SC, et al. Autophagy and its link to type II diabetes mellitus. BioMed. 2017;7(2):1–12.
- Zhang C, Hu J, Wang W, et al. HMGB1-induced aberrant autophagy contributes to insulin resistance in granulosa cells in PCOS. FASEB J. 2020;34(7):9563–9574.
- Zhou J, Peng X, Mei S. Autophagy in ovarian follicular development and Atresia. Int J Biol Sci. 2019;15(4):726–737.
- Choi JY, Jo MW, Lee EY, et al. The role of autophagy in follicular development and atresia in rat granulosa cells. Fertil Steril. 2010;93(8):2532–2537.
- Bailly Y, editor. Autophagy-A double-edged sword: Cell survival or death? In: Chapter 19, role of autophagy in the ovary cell death in mammals. Escobar ML, Echeverría OM, Vázquez-Nin GH. eds. London (UK): IntechOpen; 2013. 423–441.
- Li X, Qi J, Zhu Q, et al. The role of androgen in autophagy of granulosa cells from PCOS. Gynecol Endocrinol. 2019;35(8):669–672.
- Crichton EG, Krutzsch PH, editors. Reproductive biology of bats. In: Chapter 5, anatomy and physiology of the female reproductive tract, 1st, Elsevier Academic Press: [place unknown]. 2000. 157–219.
- De Felici M, Scaldaferri ML, Lobascio M, et al. Experimental approaches to the study of primordial germ cell lineage and proliferation. Hum Reprod Update. 2004;10(3):197–206.
- Oktem O, Urman B. Understanding follicle growth in vivo. Hum Reprod. 2010;25(12):2944–2954.
- Abir R, Orvieto R, Dicker D, et al. Preliminary studies on apoptosis in human fetal ovaries. Fertil Steril. 2002;78(2):259–264.
- Grive KJ, Freiman RN. The developmental origins of the mammalian ovarian reserve. Development. 2015;142(15):2554–2563.
- Wallace WHB, Kelsey TW. Human ovarian reserve from conception to the menopause. PLoS One. 2010;5(1):e8772.
- Lintern-Moore S, Moore GPM. The initiation of oocyte growth in the mouse ovary. Ann Biol Anim Biochim Biophys. 1979;19(5):1399–1407.
- Oktem O, Oktay K. The ovary: anatomy and function throughout human life. Ann N Y Acad Sci. 2008;1127(1):1–9.
- Cox E, Takov V. Embryology, ovarian follicle development. StatPearls [Internet]. 2020.
- Lass A, Silye R, Abrams DC, et al. Follicular density in ovarian biopsy of infertile women: a novel method to assess ovarian reserve. Hum Reprod. 1997;12(5):1028–1031.
- Abraham GE, Odell WD, Swerdloff RS, et al. Simultaneous radioimmunoassay of plasma FSH, LH, progesterone, 17-hydroxyprogesterone, and estradiol-17β during the menstrual cycle. J Clin Endocrinol Metab. 1972;34(2):312–318.
- Goodman AL, Descalzi CD, Johnson DK, et al. Composite pattern of circulating lh, fsh, estradiol, and progesterone during the menstrual cycle in cynomolgus monkeys. Proc Soc Exp Bio Med. 1977;155(4):479–481.
- McGee EA, Hsueh AJW. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000;21(2):200–214.
- Baerwald AR, Adams GP, Pierson RA. Characterization of ovarian follicular wave dynamics in women. Biol Reprod. 2003;69(3):1023–1031.
- De Castro FC, Cruz MHC, Leal CLV. Role of growth differentiation factor 9 and bone morphogenetic protein 15 in ovarian function and their importance in mammalian female fertility—a review. Asian-Australas J Anim Sci. 2016;29(8):1065.
- Durlinger ALL, Kramer P, Karels B, et al. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology. 1999;140(12):5789–5796.
- Rosenfield RL, Ehrmann DA. The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr Rev. 2016;37(5):467–520.
- Dewailly D, Andersen CY, Balen A, et al. The physiology and clinical utility of anti-Müllerian hormone in women. Hum Reprod Update. 2014;20(3):370–385.
- Peters AE, Mihalas BP, Bromfield EG, et al. Autophagy in female fertility: a role in oxidative stress and aging. Antioxid Redox Signal. 2020;32(8):550–568.
- Adastra KL, Chi MM, Riley JK, et al. A differential autophagic response to hyperglycemia in the developing murine embryo. Reproduction. 2011;141(5):607–615.
- Sonigo C, Beau I, Grynberg M, et al. AMH prevents primordial ovarian follicle loss and fertility alteration in cyclophosphamide-treated mice. FASEB J. 2019;33(1):1278–1287.
- Sun YC, Wang YY, Sun XF, et al. The role of autophagy during murine primordial follicle assembly. Aging (Albany NY). 2018;10(2):197–211.
- Escobar ML, Echeverría OM, Sánchez-Sánchez L, et al. Analysis of different cell death processes of prepubertal rat oocytes in vitro. Apoptosis. 2010;15(4):511–526.
- Cao B, Camden AJ, Parnell LA, et al. Autophagy regulation of physiological and pathological processes in the female reproductive tract. Am J Reprod Immunol. 2017;77(5):1–7.
- Song ZH, Yu HY, Wang P, et al. Germ cell-specific Atg7 knockout results in primary ovarian insufficiency in female mice. Cell Death Dis. 2015;6(1):1–11.
- Gawriluk TR, Hale AN, Flaws JA, et al. Autophagy is a cell survival program for female germ cells in the murine ovary. Reproduction. 2011;141(6):759–765.
- Rankin T, Familari M, Lee E, et al. Mice homozygous for an insertional mutation in the Zp3 gene lack a zona pellucida and are infertile. Development. 1996;122(9):2903–2910.
- Roy SK. Regulation of ovarian follicular development: a review of microscopic studies. Micros Res Tech. 1994;27(2):83–96.
- Bassett DL. The changes in the vascular pattern of the ovary of the albino rat during the estrous cycle. Am J Anat. 1943;73(2):251–291.
- Eppig JJ. Oocyte control of ovarian follicular development and function in mammals. Reproduction. 2001;122(6):829–838.
- Knight PG, Glister C. TGF-β superfamily members and ovarian follicle development. Reproduction. 2006;132(2):191–206.
- Fortune JE, Eppig JJ. Effects of gonadotropins on steroid secretion by infantile and juvenile mouse ovaries in vitro. Endocrinology. 1979;105(3):760–768.
- Zeleznik AJ. The physiology of follicle selection. Reprod Biol Endocrinol. 2004;2(1):1–7.
- Xu Z, Allen Garverick H, Smith GW, et al. Expression of follicle-stimulating hormone and luteinizing hormone receptor messenger ribonucleic acids in bovine follicles during the first follicular wave. Biol Reprod. 1995;53(4):951–957.
- Fortune JE. Ovarian follicular growth and development in mammals. Biol Reprod. 1994;50(2):225–232.
- Chang RJ, editor. Polycystic ovary syndrome. In: Chapter 2, dynamics of follicle development in the human ovary. New York (NY): Springer; 1996. 21–36.
- Jonard S, Dewailly D. The follicular excess in polycystic ovaries, due to intra-ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update. 2004;10(2):107–117.
- Robker RL, Richards JS. Hormonal control of the cell cycle in ovarian cells: proliferation versus differentiation. Biol Reprod. 1998;59(3):476–482.
- Jeppesen JV, Kristensen SG, Nielsen ME, et al. LH-receptor gene expression in human granulosa and cumulus cells from antral and preovulatory follicles. J Clin Endocrinol Metab. 2012;97(8):E1524–31.
- Tao T, Xu H. Autophagy: biology and diseases: Clinical science. In: Chapter 33, autophagy and obesity-related reproductive dysfunction, 1st, Springer: Singapore (SG). 2019. 463–466.
- Choi J, Jo M, Lee E, et al. AKT is involved in granulosa cell autophagy regulation via mTOR signaling during rat follicular development and atresia. Reproduction. 2014;147(1):73–80.
- Choi JY, Jo MW, Lee EY, et al. The role of autophagy in corpus luteum regression in the rat. Biol Reprod. 2011;85(3):465–472.
- Asselin E, Wang Y, Tsang BK. X-linked inhibitor of apoptosis protein activates the phosphatidylinositol 3-Kinase/Akt pathway in rat granulosa cells during follicular development*. Endocrinology. 2001;142(6):2451–2457.
- Zheng Y, Ma L, Liu N, et al. Autophagy and apoptosis of porcine ovarian granulosa cells during follicular development. Animals. 2019;9(12):1–11.
- Lai Q, Xiang W, Li Q, et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Front Med. 2018;12(5):518–524.
- Duerrschmidt N, Zabirnyk O, Nowicki M, et al. Lectin-like oxidized low-density lipoprotein receptor-1-mediated autophagy in human granulosa cells as an alternative of programmed cell death. Endocrinology. 2006;147(8):3851–3860.
- Gougeon A. Croissance folliculaire dans l’ovaire humain: de l’entrée en croissance du follicule primordial jusqu’à la maturation préovulatoire [Human ovarian follicular development: from activation of resting follicles to preovulatory maturation]. Ann Endocrinol. 2010;71(3):132–143. French.
- Richards JS, Ireland JJ, Rao MC, et al. Ovarian follicular development in the rat: hormone receptor regulation by estradiol, follicle stimulating hormone and luteinizing hormone1. Endocrinology. 1976;99(6):1562–1570.
- Gougeon A, Lefevre B. Evolution of the diameters of the largest healthy and atretic follicles during the human menstrual cycle. J Reprod Fertil. 1983;69(2):497–502.
- Rothchild I. Recent progress in hormone research. In: Greep RO, editor. Chapter 4, The regulation of the mammalian corpus luteum, 1st, Elsevier Academic Press: [place unknown]. 1981. 183–298.
- Stocco C, Telleria C, Gibori G. The molecular control of corpus luteum formation, function, and regression. Endocr Rev. 2007;28(1):117–149.
- Kang W, Ishida E, Yamatoya K, et al. Autophagy-disrupted LC3 abundance leads to death of supporting cells of human oocytes. Biochem Biophys Reports. 2018;15:107–114.
- Yamamoto A, Mizushima N, Tsukamoto S. Fertilization-induced autophagy in mouse embryos is independent of mTORC1. Biol Reprod. 2014;91(1):1–7.
- Randall Armant D. Autophagy’s expanding role in development: implantation is next. Endocrinology. 2011;152(5):1739–1741.
- Gawriluk TR, Ko CM, Hong X, et al. Beclin-1 deficiency in the murine ovary results in the reduction of progesterone production to promote preterm labor. Proc Natl Acad Sci U S A. 2014;111(40):E4194–203.
- Grzesiak M, Michalik A, Rak A, et al. The expression of autophagy-related proteins within the corpus luteum lifespan in pigs. Domest Anim Endocrinol. 2018;64:9–16.
- Aboelenain M, Kawahara M, Balboula AZ, et al. Status of autophagy, lysosome activity and apoptosis during corpus luteum regression in cattle. J Reprod Dev. 2015;61(3):229–236.
- Tanida I, Ueno T, Kominami E. Autophagosome and Phagosome. In: Chapter 4, LC3 and autophagy, Deretic V, editor. 1st, Springer Humana Press: United States (US). 2008. 77–88.
- Gaytán M, Morales C, Sánchez-Criado JE, et al. Immunolocalization of beclin 1, a bcl-2-binding, autophagy-related protein, in the human ovary: possible relation to life span of corpus luteum. Cell Tissue Res. 2008;331(2):509–517.
- Teeli AS, Leszczyński P, Krishnaswamy N, et al. Possible mechanisms for maintenance and regression of corpus luteum through the ubiquitin-proteasome and autophagy system regulated by transcriptional factors. Front Endocrinol. 2019;10:1–11.
- Choi JY, Jo MW, Lee EY, et al. ERK1/2 is involved in luteal cell autophagy regulation during corpus luteum regression via an mTOR-independent pathway. Mol Hum Reprod. 2014;20(10):972–980.
- Paavola LG. The corpus luteum of the guinea pig. III. Cytochemical studies on the Golgi complex and GERL during normal postpartum regression of luteal cells, emphasizing the origin of lysosomes and autophagic vacuoles. J Cell Biol. 1978;79(1):59–73.
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151.
- Kiriakidou M, Mcallister JM, Sugawara T, et al. Expression of steroidogenic acute regulatory protein (StAR) in the human ovary. Clin Endocrinol Metab. 1996;81(11):4122–4128.
- Karsch FJ. Seasonal reproduction: a sage of reversible fertility. Physiologist. 1980;23(6):29–38.
- Jonassen JA, Richards JS. Granulosa cell desensitization: effects of gonadotropins on antral and preantral follicles. Endocrinology. 1980;106(6):1786–1794.
- Fortune JE, Quirk SM. Regulation of steroidogenesis in bovine preovulatory follicles. J Anim Sci. 1988;66(suppl_2):1–8.
- Badinga L, Driancourt MA, Savio JD, et al. Endocrine and ovarian responses associated with the first-wave dominant follicle in cattle. Biol Reprod. 1992;47(5):871–883.
- Texada MJ, Malita A, Rewitz K, et al. Autophagy regulates steroid production by mediating cholesterol trafficking in endocrine cells. Autophagy. 2019;15(8):1478–1480.
- Zhang B, Shozu M, Okada M, et al. Insulin-Like growth factor I enhances the expression of aromatase P450 by inhibiting autophagy. Endocrinology. 2010;151(10):4949–4958.
- Sirotkin A, Alexa R, Kádasi A, et al. Resveratrol directly affects ovarian cell sirtuin, proliferation, apoptosis, hormone release and response to follicle-stimulating hormone (FSH) and insulin-like growth factor I (IGF-I). Reprod Fertil Dev. 2019;31(8):1378–1385.
- Ding Y, Zhu Q, He Y, et al. Induction of autophagy by Beclin-1 in granulosa cells contributes to follicular progesterone elevation in ovarian endometriosis. Transl Res. 2020;227:15–29.
- Hirshfield AN. Development of follicles in the mammalian ovary. Int Rev Cytol. 1991;124:43–101.
- Tilly JL, Kowalski KI, Johnson AL, et al. Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology. 1991;129(5):2799–2801.
- Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocr Rev. 1996;17(2):121–155.
- Involvement of pro-apoptotic and pro-autophagic proteins in granulosa cell death. Cell Biol. 2013;1(1):9.
- Cao Y, Shen M, Jiang Y, et al. Melatonin reduces oxidative damage in mouse granulosa cells via restraining JNK-dependent autophagy. Reproduction. 2018;155(3):307–319.
- Shen M, Jiang Y, Guan Z, et al. FSH protects mouse granulosa cells from oxidative damage by repressing mitophagy. Sci Rep. 2016;6(1):1–13.
- Shen M, Jiang Y, Guan Z, et al. Protective mechanism of FSH against oxidative damage in mouse ovarian granulosa cells by repressing autophagy. Autophagy. 2017;13(8):1364–1385.
- Vilser C, Hueller H, Nowicki M, et al. The variable expression of lectin-like oxidized low-density lipoprotein receptor (LOX-1) and signs of autophagy and apoptosis in freshly harvested human granulosa cells depend on gonadotropin dose, age, and body weight. Fertil Steril. 2010;93(8):2706–2715.
- Choi J, Jo M, Lee E, et al. Induction of apoptotic cell death via accumulation of autophagosomes in rat granulosa cells. Fertil Steril. 2011;95(4):1482–1486.
- Barbieri RL, Smith S, Ryan KJ. The role of hyperinsulinemia in the pathogenesis of ovarian hyperandrogenism. Fertil Steril. 1988;50(2):197–212.
- Yao K, Bian C, Zhao X. Association of polycystic ovary syndrome with metabolic syndrome and gestational diabetes: aggravated complication of pregnancy. Exp Ther Med. 2017;14(2):1271–1276.
- Rosenfield RL, Barnes RB, Jose’F C, et al. Dysregulation of cytochrome P450c17α as the cause of polycystic ovarian syndrome. Fertil Steril. 1990;53(5):785–791.
- Ehrmann DA, Rosenfield RL, Barnes RB, et al. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327(3):157–162.
- Nelson VL, Qin K, Rosenfield RL, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86(12):5925–5933.
- Nelson VL, Legro RS, Strauss JF, et al. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13(6):946–957.
- Hotamisligil GS, Peraldi P, Budavari A, et al. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α-and obesity-induced insulin resistance. Science. 1996;271(5249):665–670.
- Komatsu M, Waguri S, Koike M, et al. Homeostatic Levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131(6):1149–1163.
- Kobayashi M, Yoshino O, Nakashima A, et al. Inhibition of autophagy in theca cells induces CYP17A1 and PAI-1 expression via ROS/p38 and JNK signalling during the development of polycystic ovary syndrome. Mol Cell Endocrinol. 2020;508(November2019):110792.
- Vanholder T, Lmr Leroy J, Van Soom A, et al. Effect of non-esterified fatty acids on bovine theca cell steroidogenesis and proliferation in vitro. Anim Reprod Sci. 2006;92(1–2):51–63.
- Niu Z, Lin N, Gu R, et al. Associations between insulin resistance, free fatty acids, and oocyte quality in polycystic ovary syndrome during in vitro fertilization. J Clin Endocrinol Metab. 2014;99(11):E2269–76.
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545.
- Li H, Li D, Ma Z, et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy. 2018;14(10):1726–1741.
- Song X, Shen Q, Fan L, et al. Dehydroepiandrosterone-induced activation of mTORC1 and inhibition of autophagy contribute to skeletal muscle insulin resistance in a mouse model of polycystic ovary syndrome. Oncotarget. 2018;9(15):11905–11921.
- Choi J, Jo M, Lee E, et al. The role of autophagy in human endometrium. Biol Reprod. 2012;86(3):1–10.
- Sumarac-Dumanovic M, Apostolovic M, Janjetovic K, et al. Downregulation of autophagy gene expression in endometria from women with polycystic ovary syndrome. Mol Cell Endocrinol. 2017;440:116–124.
- Xing Y, Liu YX, Liu X, et al. Effects of Gui Zhu Yi Kun formula on the P53/AMPK pathway of autophagy in granulosa cells of rats with polycystic ovary syndrome. Exp Ther Med. 2017;13(6):3567–3573.
- Group TECW. Anovulatory infertility*. Hum Reprod. 1995;10(6):1549–1553.
- Ben-Shlomo I, Homburg R, Shalev E. Hyperandrogenic anovulation (the polycystic ovary syndrome) - Back to the ovary? Hum Reprod Update. 1998;4(3):296–300.
- Laven JSE, Mulders AGMGJ, Visser JA, et al. Anti-müllerian hormone serum concentrations in normoovulatory and anovulatory women of reproductive age. J Clin Endocrinol Metab. 2004;89(1):318–323.
- Franks S, Hardy K. Aberrant follicle development and anovulation in polycystic ovary syndrome. Ann Endocrinol (Paris). 2010;71(3):228–230.
- Webber LJ, Stubbs S, Stark J, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362(9389):1017–1021.
- Franks S, Mason H, White D, et al. Etiology of anovulation in polycystic ovary syndrome. Steroids. 1998;63(5–6):306–307.
- Franks S, Stark J, Hardy K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum Reprod Update. 2008;14(4):367–378.
- Mulders AG, Laven JSE, Eijkemans MJC, et al. Changes in anti-Müllerian hormone serum concentrations over time suggest delayed ovarian ageing in normogonadotrophic anovulatory infertility. Hum Reprod. 2004;19(9):2036–2042.
- Balen AH, Platteau P, Andersen AN, et al. The influence of body weight on response to ovulation induction with gonadotrophins in 335 women with World Health Organization group II anovulatory infertility. BJOG An Int J Obstet Gynaecol. 2006;113(10):1195–1202.
- Dunaif A, Graf M, Mandeli J, et al. Characterization of groups of hyperaiidrogenic women with acanthosis nigricans, impaired glucose tolerance, and/or hyperinsulinemia*. J Clin Endocrinol Metab. 1987;65(3):499–507.
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–564.
- Jia L, Li J, He B, et al. Abnormally activated one-carbon metabolic pathway is associated with mtDNA hypermethylation and mitochondrial malfunction in the oocytes of polycystic gilt ovaries. Sci Rep. 2016;6(1):1–11.
- Boudoures AL, Saben J, Drury A, et al. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev Biol. 2017;426(1):126–138.
- Zhang J, Bao Y, Zhou X, et al. Polycystic ovary syndrome and mitochondrial dysfunction. Reprod Biol Endocrinol. 2019;17(1):1–15.
- Ramly B, Afiqah-Aleng N, Mohamed-Hussein ZA. Protein–protein interaction network analysis reveals several diseases highly associated with polycystic ovarian syndrome. Int J Mol Sci. 2019;20(12):2959.
- Meng L, Jan SZ, Hamer G, et al. Preantral follicular atresia occurs mainly through autophagy, while antral follicles degenerate mostly through apoptosis. Biol Reprod. 2018;99(4):853–863.
- Norman RJ, Dewailly D, Legro RS, et al. Polycystic ovary syndrome. Lancet. 2007;370(9588):685–697.
- Patel S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. J Steroid Biochem Mol Biol. 2018;182:27–36.
- Michael AE, Glenn C, Wood PJ, et al. Ovarian 11β-hydroxysteroid dehydrogenase (11βHSD) activity is suppressed in women with anovulatory polycystic ovary syndrome (PCOS): apparent role for ovarian androgens. J Clin Endocrinol Metab. 2013;98(8):3375–3383.
- Hughesdon PE. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called” hyperthecosis”. Obstet Gynecol Surv. 1982;37(2):59–77.
- Hirshfeld-Cytron J, Barnes RB, Ehrmann DA, et al. Characterization of functionally typical and atypical types of polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94(5):1587–1594.
- Willis DS, Watson H, Mason HD, et al. Premature response to luteinizing hormone of granulosa cells from anovulatory women with polycystic ovary syndrome: relevance to mechanism of anovulation. J Clin Endocrinol Metab. 1998;83(11):3984–3991.
- Li Y, Zhang J, Liu YD, et al. Long non-coding RNA TUG1 and its molecular mechanisms in polycystic ovary syndrome. RNA Biol. 2020;17(12):1798–1810.
- Li Y, Chen S. Upregulation of the long non-coding RNA TUG1 inhibits granulosa cell apoptosis and autophagy in polycystic ovary syndrome by regulating ERK/MAPK pathway. Fertil Steril. 2019;112(3):e245–6.
- Yaba A, Demir N. The mechanism of mTOR (mammalian target of rapamycin) in a mouse model of polycystic ovary syndrome (PCOS). J Ovarian Res. 2012;5(1):1–12.
- Choi JY, Jo MW, Lee EY, et al. Differential induction of autophagy by mtor is associated with abnormal apoptosis in ovarian endometriotic cysts. Mol Hum Reprod. 2014;20(4):309–317.
- Martin IH, Costa LE. Reproductive function in female rats submitted to chronic hypobaric hypoxia. Arch Int Physiol Biochim Biophys. 1992;100(5):327–330.
- Yadav AK, Yadav PK, Chaudhary GR, et al. Autophagy in hypoxic ovary. Cell Mol Life Sci. 2019;76(17):3311–3322.
- Ehrmann DA, Liljenquist DR, Kasza K, et al. Prevalence and predictors of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(1):48–53.
- Moghetti P, Tosi F, Bonin C, et al. Divergences in insulin resistance between the different phenotypes of the polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98(4):E628–37.
- Du X, Rosenfield RL, Qin K. KLF15 is a transcriptional regulator of the human 17β-hydroxysteroid dehydrogenase type 5 gene. A potential link between regulation of testosterone production and fat stores in women. J Clin Endocrinol Metab. 2009;94(7):2594–2601.
- Amato MC, Vesco R, Vigneri E, et al. Hyperinsulinism and polycystic ovary syndrome (PCOS): role of insulin clearance. J Endocrinol Invest. 2015;38(12):1319–1326.
- Dumesic DA, Oberfield SE, Stener-Victorin E, et al. Scientific statement on the diagnostic criteria, epidemiology, pathophysiology, and molecular genetics of polycystic ovary syndrome. Endocr Rev. 2015;36(5):487–525.
- Mannerås-Holm L, Leonhardt H, Kullberg J, et al. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab. 2011;96(2):E304–11.
- Wild S, Pierpoint T, McKeigue P, et al. Cardiovascular disease in women with polycystic ovary syndrome at long‐term follow‐up: a retrospective cohort study. Clin Endocrinol. 2000;52(5):595–600.
- Dokras A. Cardiovascular disease risk in women with PCOS. Steroids. 2013;78(8):773–776.
- Wekker V, Van DL, Koning A, et al. Long-term cardiometabolic disease risk in women with PCOS: a systematic review and meta-analysis. Hum Reprod Update. 2020;26(6):942–960.
- Karoli R, Fatima J, Chandra A, et al. Prevalence of hepatic steatosis in women with polycystic ovary syndrome. J Hum Reprod Sci. 2013;6(1):9–14.
- Burghen GA, Givens JR, Kitabchi AE. Correlation of hyperandrogenism with hyperinsulinism in poly cystic ovarian disease. J Clin Endocrinol Metab. 1980;50(1):113–116.
- Rice S, Christoforidis N, Gadd C, et al. Impaired insulin-dependent glucose metabolism in granulosa-lutein cells from anovulatory women with polycystic ovaries. Hum Reprod. 2005;20(2):373–381.
- Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–478.
- Kang YH, Cho MH, Kim JY, et al. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget. 2016;7(24):35577–35591.
- Yamamoto S, Kuramoto K, Wang N, et al. Autophagy differentially regulates insulin production and insulin sensitivity. Cell Rep. 2018;23(11):3286–3299.
- Wang Q, Deng H, Cheng K, et al. Manual acupuncture for the infertile female with polycystic ovary syndrome (PCOS): study protocol for a randomized sham-controlled trial. Trials. 2019;20(1):1–9.
- Zhang H, Li D, Li R, et al. Treatment of polycystic ovary syndrome with acupuncture. Acupunct Med. 2018;36(4):269–270.
- Lim CED, Wong WSF. Current evidence of acupuncture on polycystic ovarian syndrome. Gynecol Endocrinol. 2010;26(6):473–478.
- Zhang H-L, Huo Z-J, Wang H-N, et al. [Acupuncture ameliorates negative emotion in PCOS patients: a randomized controlled trial]. Zhongguo Zhen Jiu. 2020;40(4):385–390.
- Peng Y, Guo L, Gu A, et al. Electroacupuncture alleviates polycystic ovary syndrome-like symptoms through improving insulin resistance, mitochondrial dysfunction, and endoplasmic reticulum stress via enhancing autophagy in rats. Mol Med. 2020;26(1):73.
- Shirakabe A, Zhai P, Ikeda Y, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133(13):1249–1263.
- Nishida K, Kyoi S, Yamaguchi O, et al. The role of autophagy in the heart. Cell Death Differ. 2009;16(1):31–38.
- Xu X, Hua Y, Nair S, et al. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension. 2014;63(3):490–499.
- Gao L, Cao JT, Liang Y, et al. Calcitriol attenuates cardiac remodeling and dysfunction in a murine model of polycystic ovary syndrome. Endocrine. 2016;52(2):363–373.
- Zhang Y, Meng F, Sun X, et al. Hyperandrogenism and insulin resistance contribute to hepatic steatosis and inflammation in female rat liver. Oncotarget. 2018;9(26):18180–18197.
- Davis EM, Peck JD, Hansen KR, et al. Associations between vitamin D levels and polycystic ovary syndrome (PCOS) phenotypes. Minerva Endocrinol. 2019;44(2):176–184.
- Mogili KD, Karuppusami R, Thomas S, et al. Prevalence of vitamin D deficiency in infertile women with polycystic ovarian syndrome and its association with metabolic syndrome – a prospective observational study. Eur J Obstet Gynecol Reprod Biol. 2018;229:15–19.
- Lajtai K, Nagy CT, Tarszabó R, et al. Effects of vitamin D deficiency on proliferation and autophagy of ovarian and liver tissues in a rat model of polycystic ovary syndrome. Biomolecules. 2019;9(9):471.
- Fulco M, Sartorelli V. Comparing and contrasting the roles of AMPK and SIRT1 in metabolic tissues. Cell Cycle. 2008;7(23):3669–3679.
- Di Emidio G, Placidi M, Rea F, et al. Methylglyoxal-dependent glycative stress and deregulation of SIRT1 functional network in the ovary of PCOS mice. Cells. 2020;9(1):209.
- Kitada M, Ogura Y, Koya D. The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging (Albany NY). 2016;8(10):2290–2307.
- Huang R, Xu Y, Wan W, et al. Deacetylation of nuclear LC3 drives autophagy initiation under starvation. Mol Cell. 2015;57(3):456–466.
- Gonzalez F. Insulin resistance and ovarian dysfunction. Steroids. 2012;77(4):300–305.
- González F, Rote NS, Minium J, et al. Increased activation of nuclear factor κB triggers inflammation and insulin resistance in polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91(4):1508–1512.
- Wang Y, Zhong J, Zhang X, et al. The Role of HMGB1 in the Pathogenesis of Type 2 Diabetes. J Diabetes Res. 2016;2016;1-11.
- Cirillo F, Catellani C, Lazzeroni P, et al. HMGB1 is increased in adolescents with polycystic ovary syndrome (PCOS) and decreases after treatment with myo-inositol (MYO) in combination with alpha-lipoic acid (ALA). Gynecol Endocrinol. 2020;36(7):588–593.
- Cirillo F, Catellani C, Sartori C, et al. CFTR and FOXO1 gene expression are reduced and high mobility group box 1 (HMGB1) is increased in the ovaries and serum of women with polycystic ovarian syndrome. Gynecol Endocrinol. 2019;35(10):842–846.
- Katti PA, Narvekar SS, Goundadkar BB, et al. IGF1 stimulates differentiation of primary follicles and their growth in ovarian explants of zebrafish (Danio rerio) cultured in vitro. J Biosci. 2017;42(4):647–656.
- Ju Q, Tao T, Hu T, et al. Sex hormones and acne. Clin Dermatol. 2017;35(2):130–137.
- Mirdamadi Y, Thielitz A, Wiede A, et al. Insulin and insulin-like growth factor-1 can modulate the phosphoinositide-3-kinase/Akt/FoxO1 pathway in SZ95 sebocytes in vitro. Mol Cell Endocrinol. 2015;415:32–44.
- Zhang Y, Hu M, Meng F, et al. Metformin ameliorates uterine defects in a rat model of polycystic ovary syndrome. EBioMedicine. 2017;18:157–170.
- Wang QQ, Guo XC, Li L, et al. Treatment with metformin and sorafenib alleviates endometrial hyperplasia in polycystic ovary syndrome by promoting apoptosis via synergically regulating autophagy. J Cell Physiol. 2020;235(2):1339–1348.
- Li D, You Y, Bi -F-F, et al. Autophagy is activated in the ovarian tissue of polycystic ovary syndrome. Reproduction. 2018;155(1):85–92.
- Nakashima A, Aoki A, Kusabiraki T, et al. Role of autophagy in oocytogenesis, embryogenesis, implantation, and pathophysiology of pre-eclampsia. J Obstet Gynaecol Res. 2017;43(4):633–643.
- Park J, Shin H, Song H, et al. Autophagic regulation in steroid hormone-responsive systems. Steroids. 2016;115:177–181.
- Petherick KJ, Williams AC, Lane JD, et al. Autolysosomal β‐catenin degradation regulates Wnt‐autophagy‐p62 crosstalk. EMBO J. 2013;32(13):1903–1916.
- Mason IC, Qian J, Adler GK, et al. Impact of circadian disruption on glucose metabolism: implications for type 2 diabetes. Diabetologia. 2020;63(6):462–472.
- Shetty A, Hsu JW, Manka PP, et al. Role of the circadian clock in the metabolic syndrome and nonalcoholic fatty liver disease. Dig Dis Sci. 2018;63(12):3187–3206.
- Shi SQ, Ansari TS, McGuinness OP, et al. Circadian disruption leads to insulin resistance and obesity. Curr Biol. 2013;23(5):372–381.
- Simon SL, McWhirter L, Diniz Behn C, et al. Morning circadian misalignment is associated with insulin resistance in girls with obesity and polycystic ovarian syndrome. J Clin Endocrinol Metab. 2019;104(8):3525–3534.
- Zhang J, Liu J, Zhu K, et al. Effects of BMAL1–SIRT1-positive cycle on estrogen synthesis in human ovarian granulosa cells: an implicative role of BMAL1 in PCOS. Endocrine. 2016;53(2):574–584.
- Wang J, Wu D, Guo H, et al. Hyperandrogenemia and insulin resistance: the chief culprit of polycystic ovary syndrome. Life Sci. 2019;236:116940.
- Li S, Zhai J, Chu W, et al. Altered circadian clock as a novel therapeutic target for constant darkness-induced insulin resistance and hyperandrogenism of polycystic ovary syndrome. Transl Res. 2020;219:13–29.
- Sellix MT, Murphy ZC, Menaker M. Excess androgen during puberty disrupts circadian organization in female rats. Endocrinology. 2013;154(4):1636–1647.
- Zhang TW, Li ZF, Dong J, et al. The circadian rhythm in intervertebral disc degeneration: an autophagy connection. Exp Mol Med. 2020;52(1):31–40.
- Wang X, Xu Z, Cai Y, et al. Rheostatic balance of circadian rhythm and autophagy in metabolism and disease. Front Cell Div Biol. 2020;8:1–12.
- Ma D, Li S, Molusky MM, et al. Circadian autophagy rhythm: a link between clock and metabolism? Trends Endocrinol Metab. 2012;23(7):319–325.
- Hale AN, Ledbetter DJ, Gawriluk TR, et al. Autophagy: regulation and role in development. Autophagy. 2013;9(7):951–972.
- Ma D, Lin JD. Circadian regulation of autophagy rhythm through transcription factor C/EBPβ. Autophagy. 2012;8(1):124–125.
- Ueda HR, Chen W, Adachi A, et al. A transcription factor response element for gene expression during circadian night. Nature. 2002;418(6897):534–539.
- Wu Y, Li W, Hu Y, et al. Suppression of sirtuin 1 alleviates airway inflammation through mTOR‑mediated autophagy. Mol Med Rep. 2020;22(3):2219–2226.
- Chung S, Yao H, Caito S, et al. Regulation of SIRT1 in cellular functions: role of polyphenols. Arch Biochem Biophys. 2010;501(1):79–90.