
ABSTRACT
Lysosomes are the primary degradative compartment within cells and there have been significant advances over the past decade toward understanding how lysosome homeostasis is maintained. Lysosome repopulation ensures sustained autophagy function, a fundamental process that protects against disease. During macroautophagy/autophagy, cellular debris is sequestered into phagophores that mature into autophagosomes, which then fuse with lysosomes to generate autolysosomes in which contents are degraded. Autophagy cannot proceed without the sufficient generation of lysosomes, and this can be achieved via their de novo biogenesis. Alternatively, during autophagic lysosome reformation (ALR), lysosomes are generated via the recycling of autolysosome membranes. During this process, autolysosomes undergo significant membrane remodeling and scission to generate membrane fragments, that mature into functional lysosomes. By utilizing membranes already formed during autophagy, this facilitates an efficient pathway for re-deriving lysosomes, particularly under conditions of prolonged autophagic flux. ALR dysfunction is emerging as an important disease mechanism including for neurodegenerative disorders such as hereditary spastic paraplegia and Parkinson disease, neuropathies including Charcot-Marie-Tooth disease, lysosome storage disorders, muscular dystrophy, metabolic syndrome, and inflammatory and liver disorders. Here, we provide a comprehensive review of ALR, including an overview of its dynamic spatiotemporal regulation by MTOR and phosphoinositides, and the role ALR dysfunction plays in many diseases.
Introduction
Autophagy is a fundamental catabolic pathway integral to health. This homeostatic process mobilizes amino acids during starvation, ensures organelle quality control, removes intracellular debris and is an essential cellular stress response [Citation1]. Autophagy inhibition occurs in a range of severe diseases, including those caused by mutations in key autophagy regulatory genes (recently reviewed in [Citation2–4]). During macroautophagy (herein called autophagy), cargo is encapsulated within autophagosomes (APs), and is degraded once autophagosomes fuse with multiple lysosomes to form autolysosomes (ALs) [Citation1]. Lysosomes are heavily utilized during this process, and the inability to sustain lysosome homeostasis markedly reduces autophagy function [Citation5,Citation6]. Two pioneering discoveries have revealed how lysosomes are repopulated to sustain autophagy function. The first is the de novo lysosome biogenesis pathway controlled by the MiTF (microphthalmia-associated transcription/melanocyte inducing transcription factor) family of transcription factors, including TFEB (transcription factor EB) and TFE3 (transcription factor E3) [Citation7–9]. The second pathway, called autophagic lysosome reformation (ALR), is energetically favorable as it recycles autolysosome membranes already formed during autophagy to re-derive lysosomes () [Citation10]. During ALR, the autolysosome membrane forms a bud, which is then extruded along microtubules to generate membrane tubules, called “reformation tubules” [Citation10–13]. The scission of reformation tubules generates membrane fragments (“proto-lysosomes”) that mature into functional lysosomes [Citation10].
Figure 1. Overview of the autophagic lysosome reformation pathway. (A) Autophagy commences with phagophore and autophagosome formation, which encapsulate cellular debris including damaged organelles and protein aggregates. Autophagosomes fuse with lysosomes forming autolysosomes where contents are degraded by lysosomal hydrolases. When starvation-induced autophagy is prolonged, the efflux of amino acids from autolysosomes initiates autophagic lysosome reformation (ALR). During ALR, the autolysosome membrane undergoes budding and extrusion to generate membranous tubules called “reformation tubules”. The scission of reformation tubules generates membrane fragments called proto-lysosomes, which mature into functional lysosomes. (B) Confocal laser scanning microscopy image of a reformation tubule formed at autolysosomes during ALR in an intact cell. As we described previously [Citation6], myoblasts were exposed to prolonged starvation-induced autophagy by culturing for 8 h in Earle’s balanced salt solution (EBSS). Cells were fixed and co-stained for LAMP1 to identify autolysosomes/lysosomes and DAPI to define nuclei. Each individual punctum present around the nucleus represents autolysosomes, and a single reformation tubule extends from an autolysosome, which is also shown at high magnification. Scale bar: 10 μm.
![Figure 1. Overview of the autophagic lysosome reformation pathway. (A) Autophagy commences with phagophore and autophagosome formation, which encapsulate cellular debris including damaged organelles and protein aggregates. Autophagosomes fuse with lysosomes forming autolysosomes where contents are degraded by lysosomal hydrolases. When starvation-induced autophagy is prolonged, the efflux of amino acids from autolysosomes initiates autophagic lysosome reformation (ALR). During ALR, the autolysosome membrane undergoes budding and extrusion to generate membranous tubules called “reformation tubules”. The scission of reformation tubules generates membrane fragments called proto-lysosomes, which mature into functional lysosomes. (B) Confocal laser scanning microscopy image of a reformation tubule formed at autolysosomes during ALR in an intact cell. As we described previously [Citation6], myoblasts were exposed to prolonged starvation-induced autophagy by culturing for 8 h in Earle’s balanced salt solution (EBSS). Cells were fixed and co-stained for LAMP1 to identify autolysosomes/lysosomes and DAPI to define nuclei. Each individual punctum present around the nucleus represents autolysosomes, and a single reformation tubule extends from an autolysosome, which is also shown at high magnification. Scale bar: 10 μm.](/cms/asset/58ff46f5-6270-4d63-be27-5cfb3408d8f2/kaup_a_2128019_f0001_oc.jpg)
MTOR, a molecular switch that coordinates lysosome repopulation during autophagy
The nutrient-sensor MTOR (mechanistic target of rapamycin kinase) coordinates both autophagy induction and the activation of lysosome repopulation [Citation8,Citation10,Citation14–16]. When cellular amino acid levels are high, MTOR is recruited to lysosomes by the RRAG-Ragulator complex facilitating its activation by RHEB (Ras homolog, MTORC1 binding), which in turn inhibits autophagy [Citation17–20]. Conversely, nutrient deprivation is rapidly sensed by MTOR, leading to MTOR suppression and the stimulation of autophagy via induction of ULK1 (unc-51 like autophagy activating kinase 1)-dependent autophagosome formation [Citation21]. Intriguingly, MTOR also functions as a molecular switch, that determines whether lysosome repopulation during autophagy occurs via TFEB-dependent de novo lysosome biogenesis or by membrane recycling via ALR. This is because MTOR has opposing effects on these two distinct lysosome repopulation pathways during autophagy ().
Figure 2. Opposing roles of MTOR as a master regulator of both de novo lysosome biogenesis and autophagic lysosome reformation. Amino acid depletion during starvation results in MTOR inactivation, which induces autophagosome formation (1). MTOR inactivation also activates TFEB (and TFE3; not shown), which is dephosphorylated by PPP3/calcineurin and dissociates from YWHA/14-3-3 (2). TFEB (or TFE3) then translocate to the nucleus where they induce the transcription of genes required for de novo lysosome formation (3). Lysosomes fuse with autophagosomes to generate autolysosomes in which the contents are degraded and recycled (4). If autophagy activation is prolonged, the pervasive degradation of macromolecules within autolysosomes results in the release of amino acids into the cytosol via efflux transporters and also requires the function of the sugar transporter SPNS1/spin. The efflux of amino acids induces localized MTOR reactivation and initiates autophagic lysosome reformation (ALR) (5). Following initiation of ALR, reformation tubules form at autolysosomes and undergo scission to generate proto-lysosomes (6), that mature into functional lysosomes and can re-enter the autophagy pathway (7).
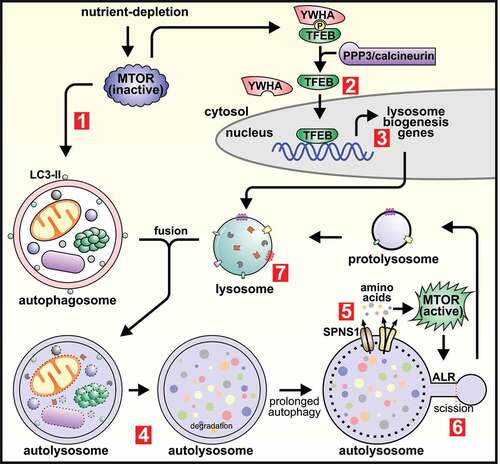
MTOR inhibition is initially required for de novo lysosome biogenesis, to enable activation of TFEB and TFE3, master transcriptional regulators of the lysosomal system (CLEAR gene network) [Citation7,Citation8,Citation14,Citation15,Citation22]) (). Under nutrient-rich conditions, TFEB is phosphorylated by MTOR, promoting its association with members of the YWHA/14-3-3 (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein) family of proteins and restricting its localization to the cytosol [Citation14–16,Citation23]. MTOR inhibition upon autophagy induction promotes TFEB activation, whereby TFEB dephosphorylation by PPP3/calcineurin (protein phosphatase 3) permits its dissociation from the YWHA complex and subsequent translocation to the nucleus [Citation24,Citation25]. In the nucleus TFEB promotes the transcription of genes required to generate new lysosomes [Citation7]. TFE3 also regulates MTOR-dependent lysosome biogenesis in a similar manner [Citation8].
If starvation-induced autophagy is prolonged, MTOR becomes reactivated which induces ALR () [Citation10]. The protracted degradation of macromolecules within autolysosomes/lysosomes via lysosomal hydrolases generates amino acids that are released into the cytosol by transmembrane efflux reporters. This amino acid signal induces localized MTOR reactivation on autolysosome membranes which initiates ALR [Citation10]. In Drosophila, MTOR reactivation and ALR initiation also requires the function of the sugar transporter spin (spinster), a role also conserved for the mammalian homolog SPNS1 (sphingolipid transporter 1 (putative)) [Citation26]. When MTOR reactivation is prevented, either by the inhibitor rapamycin, or via blockade of the degradative function of autophagy using lysosomal hydrolase inhibitors, then ALR is not initiated [Citation10]. As such, experimental conditions used to study ALR require the reactivation of MTOR during autophagy, and prolonged starvation is the preferred autophagy-stimulus for initiating ALR in cell culture studies [Citation10]. However, mechanistic understanding of MTOR reactivation and hence ALR initiation during autophagy is still emerging. For example, starvation also activates the general amino acid control (GAAC) pathway, which increases amino acid transporters including SLC7A5 (solute carrier family 7 member 5) at the plasma membrane [Citation27,Citation28]. This enhances cellular amino acid uptake and provides an additional route for MTOR reactivation, but whether this pathway has a role in ALR is yet to be established. Alternatively, lysosomal calcium efflux is emerging as an important regulator of autophagy [Citation29] and is implicated in ALR regulation. Prolonged starvation activates the lysosomal Ca2+ channel MCOLN1/TRPML1 (mucolipin TRP cation channel 1), which causes the reactivation of MTOR in a CALM (calmodulin)-dependent manner [Citation30]. A separate study demonstrated that MCOLN1 is required for ALR, suggesting that Ca2+ efflux may also be an important MTOR reactivation signal to initiate ALR during autophagy [Citation31].
The reliance of ALR initiation on the amino acid-dependent reactivation of MTOR, allows the cell to fine-tune the extent to which ALR is activated in response to the efflux of amino acids from autolysosomes, and hence the amplitude of autophagic flux. It is possible that this mechanism enables cells to prioritize membrane recycling, to efficiently produce new lysosomes when autophagy function is prolonged or increased. However, there are still significant knowledge gaps in understanding the physiological contexts in which de novo lysosome biogenesis versus ALR are required, particularly in vivo. A recent study also identified a new process called autophagosomal components recycling (ACR), whereby protein components from the outer autolysosome membrane are also recycled during autophagy [Citation32]. ACR is mechanistically distinct, being regulated via a SNX4 (sorting nexin 4)-SNX5-SNX17 multi-protein “recycler complex” and occurs prior to ALR during autophagy. Loss of ACR function also inhibits autophagy, reinforcing the new concepts that both membrane (ALR) and protein (ACR) recycling at autolysosomes are necessary to sustain autophagy function.
Phosphoinositide-dependent regulation of autophagic lysosome reformation
Autolysosome membrane remodeling during ALR is controlled by the transient and reversible generation of a suite of membrane-bound phosphoinositides in a precise spatiotemporal manner. This includes phosphatidylinositol (PtdIns) 3-phosphate (PtdIns3P) [Citation33], phosphatidylinositol 4-phosphate (PtdIns4P) [Citation34,Citation35] and phosphatidylinositol 4,5-bisphosphate (PtdIns[Citation4,Citation5]P2) [Citation6,Citation11–13,Citation36]. Each phosphoinositide species is generated via reversible phosphorylation of the inositol head-group of PtdIns at one of three positions (numbered D3, D4 or D5), which are added or removed by PtdIns-kinase or -phosphatase enzymes respectively. These enzymes also exhibit specificity for the position(s) of the phosphate on the ring (reviewed in [Citation37,Citation38]). Once generated on autolysosome membranes, phosphoinositides recruit specific membrane-deforming effector proteins [Citation39] that drive ultrastructural membrane changes during ALR [Citation6,Citation11–13,Citation36]. Dysregulation of these phosphoinositide-dependent processes inhibits ALR, leading to lysosome depletion and autophagy inhibition [Citation6,Citation34]. How the generation of these phosphoinositides are linked to the MTOR-dependent activation of ALR remains a significant unanswered question. Furthermore, the functions for each phosphoinositide species during ALR have been dissected through loss of function studies on PtdIns-kinases or -phosphatases that generate or degrade the phosphoinositides respectively. However, as this approach often leads to interconversion between different PtdIns species [Citation36], the specific roles of each enzyme is complex, somewhat overlapping and remains to be fully elucidated. The complexity of this system is further compounded by evidence that multiple PtdIns-kinases and -phosphatases phosphorylate or dephosphorylate the same phosphate on the inositol head group of PtdIns. Hence the specificity of function remains to be fully determined for the PtdIns-kinases, -phosphatases and phosphoinositides themselves during ALR. The roles of each phosphoinositide, the enzymes responsible for their generation and their effector proteins involved in ALR regulation are summarized in and discussed in further detail below.
Table 1. Known and predicted phosphoinositide-regulatory enzyme and effector protein functions in ALR.
PtdIns4P and PtdIns(4,5)P2 are master regulators of membrane dynamics during ALR
During ALR, PtdIns4P is generated from PtdIns on autolysosomes by two distinct PtdIns-4-kinases, PI4K2A (phosphatidylinositol 4-kinase type 2 alpha) or PI4KB/PI4K3B (phosphatidylinositol 4-kinase beta) [Citation34,Citation35] (). In turn, PtdIns4P serves as a substrate for the generation of PtdIns(4,5)P2 by two PtdIns4P-5kinases, PIP5K1A (phosphatidylinositol-4-phosphate 5-kinase type 1 alpha) or PIP5K1B (phosphatidylinositol-4-phosphate 5-kinase type 1 beta) [Citation11]. PtdIns(4,5)P2 is hydrolyzed at the D5-position phosphate by INPP5K/SKIP (inositol polyphosphate 5-phosphatase K) forming PtdIns4P [Citation6].
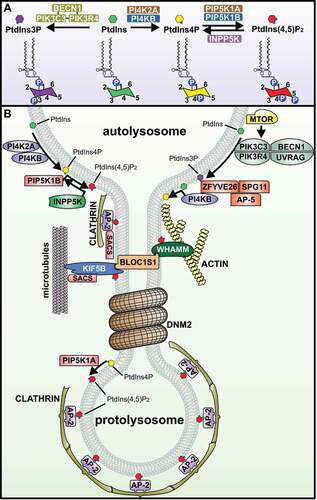
Figure 3. Membrane dynamics during autophagic lysosome reformation are under tight spatiotemporal control by phosphoinositides. (A) The ultrastructural membrane changes that take place at autolysosomes during ALR are driven by the localized generation of membrane-associated phosphoinositides, and are regulated by phosphatidylinositol-kinase and -phosphatase enzymes. PtdIns3P is generated from PtdIns by the class III phosphoinositide 3-kinase complex that includes PIK3C3/VPS34 (catalytic subunit), PIK3R4/VPS15 (regulatory subunit) and BECN1. In a successive pathway, the generation of PtdIns4P from PtdIns is controlled by the phosphatidylinositol 4-kinases PI4K2A or PI4KB/PI4K3B. In turn, PtdIns4P serves as a substrate for the synthesis of PtdIns(4,5)P2 by the PtdIns4P-5kinases, PIP5K1A or PIP5K1B. Interconversion between PtdIns(4,5)P2 and PtdIns4P on autolysosome membranes is important for ALR, whereby PtdIns(4,5)P2 is hydrolyzed by the inositol polyphosphate 5-phosphatase INPP5K to form PtdIns4P. (B) The generation of PtdIns(4,5)P2 at discrete foci on the autolysosome membrane initiates a succession of ultrastructural changes through the recruitment of specific effector proteins. This commences with the AP-2-clathrin complex which stimulates membrane budding, followed by KIF5B and WHAMM to extrude membrane buds into reformation tubules. KIF5B a motor protein, “pulls” membranes along microtubules, and WHAMM forces membranes into a tubular structure by stimulating localized actin polymerization. BLOC1S1 interacts with both KIF5B and WHAMM to co-ordinate this membrane extrusion stage. SACS is also important for reformation tubule formation possibly through its regulation of microtubule dynamics and/or binding to the AP-2-clathrin complex and KIF5B. DNM2 drives the scission of reformation tubules to form proto-lysosomes. MTOR activation of UVRAG (UV radiation resistance associated)-PIK3C3/VPS34 results in generation of PtdIns3P from PtdIns, which has also been implicated in the scission of reformation tubules to generate lysosomes. The hereditary spastic paraplegia protein complex AP-5-SPG11-ZFYVE26, is recruited to autolysosomes via ZFYVE26 binding to PtdIns3P and is required for ALR. ZFYVE26 also binds PI4KB/PI4K3B and has a role in regulating PtdIns4P at autolysosomes.
PtdIns4P is more than just a precursor for PtdIns(4,5)P2 generation during ALR
PtdIns4P is present predominantly at the Golgi under nutrient-rich conditions [Citation43], and during ALR PI4K2A or PI4KB/PI4K3B generate PtdIns4P from PtdIns, where PtdIns4P is localized along the entire autolysosome membrane including reformation tubules () [Citation11,Citation34,Citation35]. PtdIns4P generated by PI4K2A or PI4KB/PI4K3B not only serves as a precursor for generating PtdIns(4,5)P2, but also appears to have an active and complex role during ALR [Citation34,Citation35]. PI4KB/PI4K3B-derived PtdIns4P mediates the retention of lumenal proteins within the main autolysosome body and thereby their exclusion from reformation tubules [Citation35]. As such, the scission of reformation tubules generates proto-lysosomes that contain few lumenal components [Citation10]. Proto-lysosmes then undergo a little characterized maturation process, including acidification and acquisition of lysosomal hydrolases, to form functional lysosomes [Citation10].
A reduction in PtdIns4P at autolysosomes/lysosomes also increases the formation of extended reformation tubules [Citation34,Citation35]. A current model predicts that reduced PtdIns4P, due to loss of PI4KB/PI4K3B, prevents the unnecessary efflux of lysosomal vesicles from autolysosomes/lysosomes, hence leading to longer membrane tubules [Citation35]. However, a separate study suggests that this occurs because PtdIns4P, generated by PI4K2A, is required for the scission of reformation tubules to produce lysosomes during ALR [Citation34]. Therefore, in the absence of PtdIns4P reformation tubules do not undergo scission, causing their hyper-extension. Clarification of the complex roles PtdIns4P plays during ALR will be illuminated with the identification of PtdIns4P-binding effector proteins involved in this process. Furthermore, the scission of reformation tubules during ALR is a complex event, that requires further investigation given that PtdIns3P [Citation33] and PtdIns(4,5)P2 [Citation6,Citation11] are also implicated in this step (discussed in further detail in a following section).
PtdIns(4,5)P2 is required for membrane budding and tubulation during ALR
Generation of PtdIns(4,5)P2 is critical for multiple stages of ALR, including the budding of autolysosome membranes, the extrusion of the bud to form reformation tubules, and may also be required for the scission of reformation tubules to produce lysosomes (). In a sequential pathway, the PtdIns4P-5-kinase PIP5K1B first generates PtdIns(4,5)P2 at discrete foci on the autolysosome membrane, which initiates recruitment of the adaptor protein-2 (AP-2)-clathrin complex [Citation11]. Formation of this clathrin lattice stimulates membrane budding, and also serves as a membrane-platform for the further enrichment of AP-2-PtdIns(4,5)P2 microdomains, that promote KIF5B (kinesin family member 5B) protein recruitment and clustering [Citation11,Citation12]. KIF5B is a PtdIns(4,5)P2-binding kinesin-motor protein that “pulls” autolysosome membrane buds along the length of microtubule filaments to form reformation tubules [Citation12]. This membrane extrusion stage also requires localized actin polymerization by the PtdIns(4,5)P2-binding protein WHAMM (WASP homolog associated with actin, golgi membranes and microtubules), that forces regions of the autolysosome membrane into a tubular structure [Citation13]. This membrane extrusion step is further coordinated by BLOC1S1/BLOS1/BORCS1/GCN5L1 (biogenesis of lysosomal organelles complex 1 subunit 1), part of the biogenesis of lysosome-related organelles complex-1 (BLOC-1), which recruits KIF5B to autolysosomes while concomitantly also binding WHAMM [Citation44]. Finally, during the terminal stage of ALR, PIP5K1A-dependent generation of PtdIns(4,5)P2 on reformation tubules is thought to be required for their scission to generate proto-lysosomes, as loss of PIP5K1A results in hyper-extended reformation tubules [Citation11].
The complex roles of PtdIns4P, PtdIns(4,5)P2 and PtdIns3P in reformation tubule scission during ALR
The final stage of ALR, whereby autolysosome reformation tubules undergo scission to generate membrane fragments that become lysosomes, is not well understood. Proto-lysosomes may be generated through clathrin-dependent membrane budding, as clathrin is recruited by PtdIns(4,5)P2 to regions along the length of reformation tubules () [Citation6,Citation11]. The membrane-severing large GTPase protein DNM2 (dynamin 2) is also recruited to reformation tubules and is critical for their scission [Citation40]. Dynamins oligomerize into helical filaments on tubular membranes, and following their GTP hydrolysis, provide the mechanical torque necessary to sever membranes [Citation45]. Suppression of DNM2 function results in hyper-extended reformation tubules at autolysosomes, and a failure to generate new lysosomes, which is consistent with a defect in membrane scission [Citation40]. The pleckstrin homology (PH) domain of DNM2 binds PtdIns(4,5)P2 [Citation46], but whether this phosphoinositide specifically drives reformation tubule scission remains to be shown, given that PtdIns3P and PtdIns4P are also implicated in this scission step [Citation33,Citation34] and can bind dynamins [Citation46,Citation47].
Depletion of PtdIns4P or PtdIns(4,5)P2, via knockdown of PI4K2A or PIP5K1A respectively, both result in the hyper-extension of autolysosome reformation tubules, phenocopying the effects of DNM2 inhibition [Citation11,Citation34,Citation40]. Conversely increased PtdIns4P, due to PI4K2A overexpression, leads to shorter reformation tubules with increased association of clathrin and DNM2, suggesting that membrane budding and/or scission is enhanced [Citation34]. Collectively, these studies raise questions on whether PtdIns4P or PtdIns(4,5)P2 drive the scission of reformation tubules. Some clarification has come from a recent study showing that the inositol polyphosphate 5-phosphatase INPP5K is required for ALR completion [Citation6]. Loss of INPP5K results in an inability to convert PtdIns(4,5)P2 to PtdIns4P on autolysosomes that leads to increased association of clathrin on reformation tubules, which possibly affects accessibility of the scission machinery. As a result, autolysosome reformation tubules are long-lived and hyper-extended, together with a failure to maintain lysosome homeostasis during autophagy. This defect can be reversed by normalization of PtdIns4P and PtdIns(4,5)P2 levels in INPP5K-depleted cells, indicating that bidirectional interconversion between PtdIns4P and PtdIns(4,5)P2 on autolysosomes is necessary for the successful completion of ALR [Citation36].
PtdIns3P, generated by the class III PtdIns-3-kinase complex including PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), PIK3R4/VPS15 (phosphoinositide 3-kinase regulatory subunit 4) and BECN1 (beclin 1), is also implicated in the scission of reformation tubules to generate lysosomes during ALR () [Citation33]. Intriguingly, PIK3C3/VPS34 activity during ALR is controlled by MTOR, indicating an expanded role for MTOR in both the initiation [Citation10] and completion of ALR [Citation33]. However, PtdIns3P-binding effector proteins during ALR are yet to be fully identified. It has been suggested that PtdIns3P-dependent recruitment of DNM2 is important for reformation tubule scission during ALR, but is yet to be proven experimentally [Citation33]. Another likely effector protein is ZFYVE26/SPG15/FYVE-CENT/spastizin (zinc finger FYVE-type containing 26), which binds PtdIns3P via its FYVE domain and is required for ALR [Citation41,Citation42,Citation48,Citation49]. ZFYVE26 is mutated in herediatary spastic paraplegia [Citation50] and its role in ALR is discussed in detail in the next section.
The involvement of autophagic lysosome reformation defects in disease
ALR dysfunction is associated with an increasing number of diseases, and these represent a new sub-category of lysosome-based disorders, whereby the inability to maintain lysosome homeostasis leads to marked autophagy inhibition [Citation5,Citation6]. This was first identified in lysosome-storage disorders [Citation10], but has rapidly expanded into many other diseases as mechanistic understanding of the ALR process has evolved (summarized in ). These diseases typically affect tissues with a high autophagy dependence, including neurons, skeletal muscle and liver, indicating that ALR is integral to the efficient generation of lysosomes and sustained autophagy function in these tissues [Citation5,Citation6,Citation40,Citation42,Citation54,Citation79]. ALR inhibition has a potent suppressive effect on autophagy, resulting in several characteristic features including the depletion of lysosomes, that are not sufficiently repopulated [Citation5,Citation6]. Autophagosomes also accumulate as their maturation is impeded, due to the insufficient availability of lysosomes to fuse with and form autolysosomes. The marked accumulation of enlarged autolysosomes (immune-reactive for both MAP1LC3/LC3 [microtubule associated protein 1 light chain 3] and LAMP1 [lysosomal associated membrane protein 1]) is also highly characteristic of ALR inhibition, possibly occurring as the growing autolysosome membranes are not recycled to form lysosomes [Citation6,Citation10,Citation12,Citation42]. The role of ALR dysfunction in specific disorders is discussed below.
Table 2. Disorders showing inhibition of autophagic lysosome reformation.
Lysosome storage disorders
Lysosome storage disorders (LSDs) are a genetically heterogeneous group of multisystem disorders, often caused by mutations in genes encoding lysosomal hydrolases, which are essential components of lysosomal degradative functions [Citation81]. LSDs can be highly debilitating, with progressive neurological decline and death. A shared histopathological feature of LSDs is the accumulation of stored substances within lysosomes due to their impaired degradative functions [Citation81], together with autophagy suppression [Citation82].
Fibroblasts from patients with the LSDs, Scheie syndrome (mucopolysaccharidosis type I), Fabry disease and aspartylglucosaminuria show suppression of ALR [Citation10]. Scheie syndrome is caused by a deficiency of alpha-L-iduronidase (IDUA), resulting in the accumulation of glycosaminoglycans (GAGs) within lysosomes [Citation51]. It is a multisystem disorder with skeletal deformities, cardiac complications, and cognitive decline. Fabry disease is caused by mutations in the lysosomal hydrolase GLA (galactosidase alpha), and is associated with episodic/chronic pain, as well as kidney, cardiac, and cerebrovascular complications that can be fatal [Citation52]. Aspartylglucosaminuria results from deficient activity of the lysosomal enzyme, aspartylglucosaminidase (AGA) [Citation53]. This childhood-onset disorder manifests as learning and behavioral issues, and with a rapid progression in adult life including skeletal abnormalities, poor oral health, epileptic seizures, and psychiatric disorders. Due to the deleterious effects on lysosomal hydrolases in LSDs, it is not surprising that fibroblasts from patients with these disorders show ALR inhibition due to impaired MTOR reactivation [Citation10]. The failure of lysosomal hydrolases to degrade autolysosome contents during autophagy prevents the generation and efflux of amino acids, thereby suppressing the process that is required for MTOR reactivation and initiation of ALR [Citation10,Citation26].
There are more than 50 different LSDs [Citation81], and it is likely that others may be associated with inhibition of ALR. For example, spin (spinster) is a sugar transporter that is required for MTOR reactivation during autophagy and hence ALR initiation [Citation26]. Disruption of spin in Drosophila [Citation83] or its homolog in zebrafish (spns1/nrs, spinster homolog 1 {Drosophila}) [Citation84] results in progressive LSD-like neurodegeneration. Expression of the human homolog SPNS1 is reduced in fibroblasts from patients with the LSD Niemann-Pick type C disease-1(NPC), together with abnormalities consistent with an ALR defect, including autophagy inhibition and the accumulation of enlarged autolysosomes/lysosomes [Citation62]. These abnormalities can be rescued by increasing the expression of SPNS1 or supplementing cells with L-leucine [Citation62], a potent amino acid-inducer of MTOR activation [Citation85]. However, some LSDs show evidence of MTOR hyperactivation, which suppresses both autophagy and TFEB-dependent lysosome biogenesis [Citation86,Citation87]. The effects of this sustained MTOR activation on ALR are unknown but this is an interesting question for future study. Given that sustained MTOR activation suppresses autophagy it is likely this prevents the formation of autolysosomes, the site at which ALR occurs.
Parkinson disease
Parkinson disease (PD) is a severe and prevalent neurodegenerative disease that affects approximately 1% of the population aged over 60 [Citation88], and has a strong association with autophagy inhibition [Citation89]. Genetic risk factors are identified in approximately 5–10% of cases of PD [Citation88], and associated genes include GBA1 (glucosylceramidase beta 1) and GAK (cyclin G associated kinase) [Citation63–65], which are required for ALR [Citation54,Citation66]. GBA1 deficiency leads to reduced lysosome function [Citation90] and autophagy inhibition [Citation91,Citation92] that correlates with neurodegeneration [Citation93]. GBA1 deficiency also results in an inability to maintain lysosome homeostasis [Citation54,Citation94], that has been attributed to reduced activation of MITF/TFEB-dependent de novo lysosome biogenesis [Citation94], but GBA1 is also required for ALR [Citation54]. Patient-derived fibroblasts with GBA1 mutations show reduced ALR, resulting from a defect in lysosomal acidification and degradative function that prevents MTOR reactivation during autophagy [Citation54]. The accumulation of α-synuclein also coincides with ALR suppression [Citation54]. This is a key finding since α-synuclein should normally be cleared by autophagy, but its accumulation within Lewy bodies in neurons is a key pathological hallmark in PD [Citation95]. Recent studies suggest other genetic loci associated with increased risk of PD also show an association with ALR dysfunction including GAK [Citation66].
The GBA1-dependent ALR pathway has implications not only for PD, but also for the LSD Gaucher disease, where GBA1 mutations are also causative [Citation55,Citation56]. In Gaucher disease, the buildup of an un-degraded glycosphingolipid (glucocerebroside), largely in macrophage lysosomes, leads to inflammation, hepatosplenomegaly, impaired hematopoiesis, bone abnormalities and in some cases severe neurological complications [Citation56]. Sap C (saposin C) is derived from cleavage of the precursor protein PSAP (prosaposin), which in turn is an activator of the GBA1 enzyme and is also required ALR [Citation57,Citation58]. PSAP mutations cause a rare form of Gaucher disease [Citation56,Citation59,Citation60]. Treatment of GBA1-deficient MEFs with imiglucerase (Cerezyme), an enzyme-replacement therapy for Gaucher disease patients, is sufficient to restore MTOR reactivation and lysosome homeostasis [Citation54]. Similarly, treatment of Sap C-deficient patient-derived fibroblasts with (2-hydroxypropyl)-β-cyclodextrin (HP-β-CD), a drug in clinical trials for LSD, increases lysosomal enzyme function and restores ALR [Citation61]. Therefore, drug-targeting of ALR may have future therapeutic potential not only for LSDs but also PD.
Hereditary spastic paraplegia and spastic ataxia
Hereditary spastic paraplegias (HSPs) are a group of genetically heterogeneous, neurodegenerative disorders characterized by lower limb spasticity and weakness, due to degeneration of the upper motor neuron axons of the corticospinal tract [Citation96]. Mutations in more than 80 different genetic loci cause HSP, many of which are called “SPG” genes (for spastic gait) and are numbered according to the order of their discovery. A causal link between ALR inhibition and HSP has emerged [Citation5,Citation34,Citation42,Citation67–69], including for mutations in SPG11/spatacsin and ZFYVE26/SPG15 which are the most common causes of autosomal recessive HSP [Citation50,Citation70,Citation96], and also AP5Z1/SPG48 (adaptor related protein complex 5 subunit zeta 1) [Citation71]. AP5Z1 is a subunit of the AP-5 (adaptor protein 5) complex. SPG11 mutations also cause a rare form of the peripheral neuropathy Charcot-Marie-Tooth (CMT) [Citation97], and the severe neurodegenerative disorder juvenile amyotrophic lateral sclerosis (ALS) [Citation98]. SPG11, ZFYVE26 and AP5Z1 gene mutations cause HSP subtypes that are clinically similar, including early-onset Parkinsonism, thinning of the corpus callosum, cognitive impairment, white matter lesions, cerebellar ataxia, retinal abnormalities, and lens opacity [Citation99]. This clinical overlap likely arises as the ZFYVE26, SPG11 and AP-5 proteins form a complex [Citation49,Citation71] that functions within shared pathological pathways, including ALR [Citation5,Citation34,Citation42,Citation67–69].
Features characteristic of ALR inhibition are present in fibroblasts from patients with HSP caused by SPG11 or ZFYVE26 mutations [Citation100,Citation101]. This includes the accumulation of autophagosomes and enlarged LAMP1-positive organelles (autolysosomes), and in neurons from Spg11, Zfyve26 or Ap5z1 knockout mouse models where these features precede evidence of neurodegeneration [Citation5,Citation34,Citation68]. Cells with SPG11, ZFYVE26 or AP5Z1 depletion are unable to recover lysosome numbers during prolonged starvation-induced autophagy due to ALR inhibition [Citation5,Citation34,Citation42,Citation68], which prevents the appropriate clearance of autophagic material and contributes to the pathophysiology of HSP [Citation5,Citation34,Citation68,Citation69]. This includes the deposition of gangliosides; glycosphingolipids, which in the nervous system are the main carriers for sialic acid, but are salvaged and recycled following their degradation within lysosomes [Citation69]. Accumulation of gangliosides has multiple deleterious cellular effects including the alteration of membrane properties, abnormal Ca2+ homeostasis and apoptosis [Citation102]. Lipidomics of brain tissue from the ALR-deficient Spg11 knockout mouse model of HSP revealed ganglioside accumulation occurs at disease onset, concomitant with ALR inhibition, and is also evident in brain tissue and organoids from HSP patients with the SPG11 subtype [Citation69]. Decreasing ganglioside synthesis prevents neurodegeneration in spg11 zebrafish models, supporting a causal association between ALR dysfunction, ganglioside accumulation and neurodegeneration in HSP [Citation69].
Mechanistically the AP-5-SPG11-ZFYVE26 complex likely regulates ALR through association with both MTOR and phosphoinositide signaling however, the specific details are still emerging. Recruitment of the AP-5-SPG11-ZFYVE26 complex to lysosomes/autolysosomes is facilitated by the nutrient-responsive RRAG GTPase complex, which also regulates MTOR activation [Citation19,Citation41]. This suggests that the same nutrient-sensing pathway that controls MTOR reactivation to initiate ALR, could also be responsible for directing the AP-5-SPG11-ZFYVE26 complex to autolysosomes/lysosomes. Autolysosome/lysosome recruitment of the AP-5-SPG11-ZFYVE26 complex also requires binding of the FYVE domain of ZFYVE26 to PtdIns3P [Citation41,Citation42,Citation49], and this recruitment is essential for the regulation of ALR [Citation42,Citation67,Citation68]. However, whether this complex directly affects PtdIns3P-dependent functions during ALR, including the proposed scission of reformation tubules to generate lysosomes remains unknown [Citation33]. This is a possibility given that in zebrafish oocytes, the ZFYVE26 homolog Zfyve26/Souffle/Suf promotes vesicle scission from lysosome-related structures called cortical granules [Citation103]. However, a separate study suggests an alternate mechanism that SPG11 and ZFYVE26 are required for the initiation of reformation tubule formation at autolysosomes during ALR [Citation42].
ALR regulation by AP-5-SPG11-ZFYVE26 also involves PtdIns4P, given that ZFYVE26 binds PI4KB/PI4K3B [Citation42], a PtdIns-4-kinase required for ALR [Citation35]. A second PtdIns-4-kinase involved in ALR, PI4K2A, also accumulates at autolysosomes and in undegraded material in the brains of Spg11 and Zfyve26 knockout mice [Citation34]. PI4K2A accumulation at autolysosomes occurs prior to the onset of neurodegeneration, and also correlates with increased PtdIns4P, clathrin and DNM2 at this site. This suggests that disruption of these HSP-associated genes interferes with the tight regulation of PtdIns4P signaling during ALR. Interestingly, mice lacking the catalytic domain of PI4K2A develop a neurodegenerative HSP-like phenotype [Citation104], suggesting that PI4K2A inactivation, and hence reduced generation of PtdIns4P, may contribute to HSP pathogenesis. However, although PI4K2A mutations are yet to be identified in HSP patients [Citation105], PI4KA/PI4K3A (phosphatidylinositol 4-kinase alpha) mutations occur in individuals with a neurodevelopmental syndrome that includes spastic paraplegia [Citation106]. Whether the PtdIns 4-kinase PI4KA/PI4K3A is required for ALR remains to be shown.
These studies raise the interesting question of whether ALR defects also occur in any of the other 80-subtypes of HSP, or other inherited neurodegenerative disorders, but this is yet to be comprehensively examined. Cellular studies have reported adaptor protein 4 (AP-4) is required for ALR [Citation11], and mutations in the AP4E1/SPG51 (adaptor related protein complex 4 subunit epsilon 1) gene are causative of HSP [Citation107]. DNM2 is also required for ALR [Citation40], and when mutated causes HSP [Citation108]. However, both AP-4 and DNM2 have roles outside of ALR regulation, suggesting that the underlying pathogenic mechanisms for these subtypes of HSP is complex.
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a fatal, early-onset neurodegenerative disorder that is associated with the loss of cerebellar Purkinje neurons, leading to spasticity and ataxia [Citation72–74,Citation109,Citation110], and is also recently linked to ALR dysfunction [Citation75]. ARSACS is the second most common form of recessively inherited ataxia [Citation111], and is caused by mutations in the SACS (sacsin molecular chaperone) gene [Citation72,Citation73]. Loss of SACS prevents the formation of reformation tubules at autolysosomes during ALR, as SACS may be required for autolysosome membrane budding through interaction with the AP-2/clathrin complex [Citation11,Citation75]. SACS also binds the motor-protein KIF5B [Citation75], which is known to extrude autolysosome membrane buds along microtubules to form reformation tubules during ALR [Citation12]. Interestingly, SACS itself regulates microtubule dynamics, a role that not only has implications for ALR, but also for the regulation of lysosome positioning [Citation75]. Therefore, the regulation of ALR by SACS likely occurs via its effects on microtubule dynamics and/or interaction with known autolysosome membrane remodeling effector proteins. However, knowledge on the effects of SACS dysfunction on autophagy are currently limited. LC3 levels increase in the brains of Sacs knockout mice [Citation75], with studies in SACS knockout cells identifying a block in autophagic flux [Citation112], but whether this is caused by suppression of ALR remains to be conclusively shown.
Muscular dystrophy
Skeletal muscle is reliant upon the cytoprotective function of autophagy [Citation113] and its level of basal autophagy is high [Citation114]. Autophagy inhibition leads to muscle disease [Citation113] and mutations in critical autophagy regulatory genes cause inherited skeletal muscle disorders, called muscular dystrophies or myopathies (recently reviewed [Citation115,Citation116]). In skeletal muscle, enhanced autophagy activation is an important physiological response to both fasting [Citation113] and exercise [Citation117]. Increased autophagic degradation of muscle proteins during fasting mobilizes amino acids for use elsewhere in the body when their nutritional intake is low [Citation118]. Enhanced autophagy activation during post-exercise recovery is also required for mitochondrial quality control [Citation117]. The responsiveness of these adaptive autophagy processes likely requires efficient lysosome repopulation, to enable increased and continued autophagy function.
ALR dysfunction has severe consequences in skeletal muscle and is causative of muscular dystrophy [Citation6]. Mutations in the inositol polyphosphate 5-phosphatase INPP5K cause a multisystem disorder that shows clinical overlap with Marinesco-Sjögren syndrome [Citation76,Citation77]. Affected individuals exhibit congenital muscular dystrophy [Citation76,Citation77], cataracts and variable penetrance of other effects including developmental delay, intellectual disability, microcephaly and short stature [Citation76–78,Citation119,Citation120]. Muscle weakness is present from birth, with loss of ambulation commencing in childhood, and progressively worsens in adulthood [Citation76,Citation77]. INPP5K mutations occur either within the 5-phosphatase domain reducing its catalytic function toward PtdIns(4,5)P2, or within the membrane-targeting SKIP C-terminal homology (SKICH) domain [Citation76,Citation77].
Initial studies suggested no involvement for autophagy dysfunction in INPP5K congenital muscular dystrophy [Citation76], however, the SKICH domain first identified in INPP5K [Citation121], is common to other well-known autophagy-regulatory proteins including CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), TAX1BP1 (Tax1 binding protein 1) and CALCOCO1 (calcium binding and coiled-coil domain 1) [Citation121–127]. Histopathological features in muscle from patients with INPP5K mutations also suggested the involvement of autophagy dysregulation, including the accumulation of rimmed vacuoles and SQSTM1/p62 [Citation76]. Mice with conditional, skeletal muscle-specific loss of Inpp5k show an early-onset and severe muscle disease, with marked autophagy inhibition and lysosome depletion that is due to suppression of ALR [Citation6]. In muscle cells, INPP5K is recruited to autolysosomes during ALR, where it dephosphorylates PtdIns(4,5)P2 to form PtdIns4P (). Loss of INPP5K results in the accumulation of PtdIns(4,5)P2 on autolysosomes with a corresponding decrease in PtdIns4P, that alters the dynamic association of clathrin with reformation tubules and reduces the production of lysosomes. Critically, ALR does not occur in muscle cells that express INPP5K disease mutants with reduced catalytic function toward PtdIns(4,5)P2. This is the first account of a muscular dystrophy associated with inhibition of ALR, and provides important rationale for the future screening of other muscular dystrophy/myopathy genes for effects on ALR function.
Metabolic syndrome and liver disorders
Metabolic syndrome encompasses high blood pressure, high triglyceride and low HDL cholesterol, excessive abdominal obesity and high blood sugar, and are risk factors for developing type 2 diabetes, heart attack or stroke [Citation128]. ALR dysfunction is associated with several aspects of metabolic syndrome including insulin resistance [Citation129] and liver steatosis [Citation40,Citation79]. Iron overload is causally linked to diabetes and this was first recognized in individuals with the iron hyper-absorption disorder hemochromatosis, or in those with thalassemia or excessive dietary iron-intake [Citation130]. If circulating iron levels exceed the binding capacity of transferrin, then the overload of unbound iron contributes to insulin resistance and diabetes. Skeletal muscle is a key metabolic tissue and a primary consumer of glucose. In mice, prolonged iron overload via intravenous delivery decreases glucose uptake and causes insulin resistance in skeletal muscle, and is associated with a block in RHEB-mediated MTOR reactivation on autolysosomes [Citation129]. This leads to the suppression of ALR, causing lysosome depletion and autophagy inhibition. Insulin sensitivity can be restored in muscle cells by the reactivation of MTOR or by iron withdrawal, which both reinstate ALR and replenish lysosomes.
ALR inhibition may also contribute to liver steatosis [Citation40], a condition considered the hepatic component of metabolic syndrome, and is characterized by the abnormal buildup of fat within hepatic tissue that has deleterious consequences for liver function [Citation80]. Risk factors for liver steatosis include obesity, type 2 diabetes, excessive alcohol consumption and drug-induced liver-toxicity. Autophagy protects against non-alcoholic/NAFLD and alcoholic/AFLD fatty liver disease, by removing fat storage organelles known as lipid droplets in hepatocytes (“lipophagy”) [Citation131,Citation132]. Under physiological conditions, these lipid droplets function as an energy reservoir within the liver, that can be accessed when nutrient availability is low. However, when autophagy is inhibited, lipid droplets accumulate excessively in the liver causing steatosis. DNM2 promotes lipophagy in hepatocytes through its requirement for ALR [Citation40]. Suppression of DNM2 prevents the scission of reformation tubules at autolysosomes during ALR, thereby reducing the generation of lysosomes. As a consequence, insufficient lysosomes are available to sustain autophagy function and lipid droplets are not cleared via autophagy, leading to their accumulation. Chronic alcohol intake, a known risk factor for liver steatosis [Citation80], reduces lipophagy in rats due to suppression of DNM2 function during ALR [Citation79].
Nanomaterials offer significant and diverse potential in the future development of biomedicines and medical imaging technologies [Citation133,Citation134]. While this has proven highly beneficial in some instances, complications can arise with nanoparticle toxicity causing liver damage, that may be linked to autophagy inhibition [Citation135]. Upconverting nanoparticles (UCN) are of particular interest for biomedical imaging due to key properties including their highly efficient cellular uptake, photo conversion and high optical penetrating power [Citation136]. These applications are also being expanded toward developing photoactivatable therapies that target localized tumors in cancer. However, recently it was shown that UCN and materials used to develop nanoparticles (e.g. SiO2), inhibit ALR in hepatocytes by interfering with the autolysosome recruitment of PIP5K1B, an enzyme required to convert PtdIns4P to PtdIns(4,5)P2 [Citation137].
Immune disorders
Bidirectional crosstalk exists between the autophagy and immune pathways. Autophagy ensures normal immune function, and immune signaling also regulates autophagy [Citation138]. A breakdown of normal immune homeostasis leads to inflammatory diseases. TNFAIP8L2/TIPE2 (TNF alpha induced protein 8 like 2), a negative regulator of innate and adaptive immunity, prevents hyper-responsiveness of the immune system [Citation139]. TNFAIP8L2 is preferentially expressed in lymphoid tissues and its deletion in mice causes severe multi-organ inflammation, splenomegaly and premature death [Citation139]. TNFAIP8L2 is also abnormally expressed in many human autoimmune disorders including asthma, systemic lupus erythematosus, myasthenia gravis, rheumatoid arthritis, chronic rhinosinusitis with nasal polyps/CRSwNP, type 2 diabetes and cancer [Citation140]. In a lipopolysaccharide/LPS-induced endotoxemia mouse model, TNFAIP8L2-deficiency exacerbated the inflammatory response, an effect associated with suppression of MTOR reactivation on autolysosomes and inhibition of ALR during autophagy [Citation141]. TNFAIP8L2/TIPE2 also binds PtdIns(4,5)P2 [Citation142,Citation143] and functions as a phosphoinositide-transfer protein that extracts PtdIns(4,5)P2 from the lipid bilayer, transferring it to the aqueous intracellular environment [Citation143]. Whether TNFAIP8L2 regulates PI(4,5)P2-dependent events during ALR, has not been examined directly. Collectively this establishes an interesting new link between ALR and auto-immune disorders that warrants further examination.
Autophagic lysosome reformation and longevity
By 2050, approximately 1 in 6 people will be aged over 60 (World Population Prospects 2019, the United Nations), creating a critical need to understand the molecular basis of health decline with aging. There are clear links between sustained autophagy function and longevity [Citation144–146]. Diminishing autophagy function contributes to age-associated conditions including sarcopenia (loss of skeletal muscle mass and strength) [Citation147,Citation148], neurological decline including Alzheimer disease [Citation149], osteoporosis [Citation150], and metabolic conditions (insulin resistance, type 2 diabetes and obesity) [Citation151]. Autophagy dysfunction impinges on many cellular processes that contribute to aging including loss of proteostasis, maladaptive responses to cellular stress, disrupted cellular metabolism, persistent cellular senescence, stem cell dysfunction, telomere attrition, the accumulation of intracellular debris and oxidative tissue damage [Citation152]. Proper lysosomal function is also critical for longevity [Citation153]. The progressive accumulation of un-degraded material called lipofuscin occurs in tissues when lysosome function is insufficient, and is a prominent feature of aging [Citation154].
A recent study identified a new link between lipid metabolism, ALR regulation and longevity [Citation155]. The fatty acid elongase ELO-3 is required for ALR, with downstream consequences for MTOR and SKN-1 (SKiNhead 1) activation, that affects longevity in C. elegans. ELO-3 is required for biosynthesis of the sphingolipid glucosylceramide, a component of intracellular membranes that enables adaptor protein-1 (AP-1)-clathrin membrane association. Reduced glucosylceramide levels inhibit ALR by blocking the recruitment of clathrin to autolysosomes, thereby preventing the membrane budding required to form reformation tubules. An intriguing downstream consequence of this ALR inhibition is an increase in MTOR activation, proposed to occur as cells attempt to restore lysosome homeostasis. It is known that MTOR hyper-activation reduces lifespan in many animal models [Citation156] and critically, lifespan is improved in ALR-deficient C. elegans by treatment with MTOR inhibitors [Citation155]. Decreased SKN-1 activation also occurs as consequence of increased MTOR activation in ALR-deficient C. elegans, further leading to deleterious consequences in oxidative stress. The mammalian homolog of SKN-1 is NFE2L2/Nrf2 (NFE2 like bZIP transcription factor 2), a master transcriptional regulator of anti-oxidant defense, with a major cytoprotective role against aging [Citation157].
As the ALR field advances, it will be important to know how these effects observed in C. elegans translate to humans and age-related health conditions. Indeed, the stimulation of autophagy is considered a key anti-aging target, including by proponents of intermittent fasting and caloric restriction mimetics [Citation158], but the role ALR plays in this beneficial process remains to be shown in humans.
Future perspectives and conclusions
The past decade has seen substantial advances toward understanding the molecular processes that regulate ALR. Research on ALR is now moving into a new era aimed at understanding how defects in this cellular process contribute to disease, but this field is still in its infancy. For example, it is not certain if other phosphoinositide regulatory enzymes or lipids such as PtdIns(3,5)P2 also play a role in ALR, and how this impacts disease. Components of the PIKFYVE complex that generate PtdIns(3,5)P2, including FIG4 (FIG4 phosphoinositide 5-phosphatase) and VAC14 (VAC14 component of PIKFYVE complex) [Citation159,Citation160], are mutated in neurological disorders where detrimental effects on lysosome function are frequently observed [Citation161–166]. This includes Yunis-Varón Syndrome [Citation167,Citation168], amyotrophic lateral sclerosis (ALS) [Citation169], Charcot Marie-tooth 4J (CMT4J) [Citation170], familial epilepsy [Citation171], Parkinsonism [Citation172], and a pediatric neurological disorder called striatonigral degeneration [Citation161,Citation173,Citation174]. PIKFYVE mutations cause hereditary ocular disorders including François-Neetens mouchetée fleck corneal dystrophy [Citation175] and congenital cataracts [Citation176]. There are recent reports that PtdIns(3,5)P2 may regulate ALR via the effector MCOLN1/TRPML1 [Citation31], or possibly via effects on MTOR reactivation [Citation177]. GDAP1 (ganglioside induced differentiation associated protein 1), which is mutated in Charcot-Marie-Tooth neuropathy [Citation178], may also be required for PIKFYVE-dependent regulation of ALR [Citation179]. However, PIKFYVE also promotes TFEB-dependent lysosomal gene expression [Citation180]. During autophagy PIKFYVE is required for lysosome reformation from endolysosomes [Citation181,Citation182] and its generation of phosphatidylinositol-5-phosphate (PtdIns5P) promotes autophagosome formation [Citation183,Citation184]. Therefore, dissection of the role ALR dysfunction plays in PIKFYVE complex-associated disorders is challenging.
There has been a rapid expansion of the number of disorders that are linked to ALR dysfunction, including severe neurodegenerative conditions and muscular dystrophy. Collectively, these studies to date provide a strong rationale for screening currently known or newly discovered ALR regulatory genes for their roles in disease. For many ALR-associated disorders there also remains an unmet clinical need for treatments, highlighting a new focus on developing therapeutic strategies for overcoming ALR suppression. In a promising new advance, a recent study utilizing patient-derived cells and a Drosophila model of the SPG15 HSP subtype to screen a library of autophagy-modulating compounds, identified several candidates including small molecule enhancer of rapamycin 28 (SMER28), which was sufficient to restore ALR, reinstate lysosome homeostasis and improve locomotor function [Citation185]. Finally, given that ALR inhibition leads to lysosome depletion, particularly when autophagic flux is high, in future studies it will also be important to consider how ALR defects impact other critical lysosome-dependent processes.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018 Mar;20(3):233–242. PubMed PMID: 29476151.
- Klionsky DJ, Petroni G, Amaravadi RK, et al. Autophagy in major human diseases. EMBO J. 2021 Oct 1;40(19):e108863. PubMed PMID: 34459017.
- Lei Y, Klionsky DJ. The emerging roles of autophagy in human diseases. Biomedicines. 2021 Nov 9;9(11):1651. PubMed PMID: 34829881.
- Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020 Oct 15; 383(16):1564–1576. PubMed PMID: 33053285.
- Varga RE, Khundadze M, Damme M, et al. In vivo evidence for lysosome depletion and impaired autophagic clearance in hereditary spastic paraplegia type SPG11. PLoS Genet. 2015 Aug 15;11(8):e1005454. PubMed PMID: 26284655.
- McGrath MJ, Eramo MJ, Gurung R, et al. Defective lysosome reformation during autophagy causes skeletal muscle disease. J Clin Invest. 2021 Jan 4;131(1):e135124. PubMed PMID: 33119550.
- Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009 Jul 24;325(5939):473–477. PubMed PMID: 19556463.
- Martina JA, Diab HI, Lishu L, et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci Signal. 2014 Jan 21;7(309):ra9. PubMed PMID: 24448649.
- Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011 Jun 17;332(6036):1429–1433. PubMed PMID: 21617040.
- Yu L, McPhee CK, Zheng L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010 Jun 17;465(7300):942–946. PubMed PMID: 20526321.
- Rong Y, Liu M, Ma L, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nat Cell Biol. 2012 Sep;14(9):924–934. PubMed PMID: 22885770.
- Du W, Su QP, Chen Y, et al. Kinesin 1 drives autolysosome tubulation. Dev Cell. 2016 May 23;37(4):326–336. PubMed PMID: 27219061.
- Dai A, Yu L, Wang HW. WHAMM initiates autolysosome tubulation by promoting actin polymerization on autolysosomes. Nat Commun. 2019 Aug 16;10(1):3699. PubMed PMID: 31420534.
- Settembre C, Zoncu R, Medina DL, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012 Mar 7;31(5):1095–1108. PubMed PMID: 22343943.
- Roczniak-Ferguson A, Petit CS, Froehlich F, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012 Jun 12;5(228):ra42. PubMed PMID: 22692423.
- Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012 Jun;8(6):903–914. PubMed PMID: 22576015.
- Condon KJ, Sabatini DM. Nutrient regulation of mTORC1 at a glance. J Cell Sci. 2019 Nov 13;132(21):jcs222570. PubMed PMID: 31722960.
- Sancak Y, Peterson TR, Shaul YD, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008 Jun 13;320(5882):1496–1501. PubMed PMID: 18497260.
- Sancak Y, Bar-Peled L, Zoncu R, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010 Apr 16;141(2):290–303. PubMed PMID: 20381137.
- Kim E, Goraksha-Hicks P, Li L, et al. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008 Aug;10(8):935–945. PubMed PMID: 18604198.
- Dossou AS, Basu A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers (Basel). 2019 Sep 24;11(10):1422. PubMed PMID: 31554253.
- Zhu SY, Yao RQ, Li YX, et al. The role and regulatory mechanism of transcription factor EB in health and diseases. Front Cell Dev Biol. 2021 Aug 13;9:667750. PubMed PMID: 34490237.
- Xu Y, Ren J, He X, et al. YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization. Autophagy. 2019 Jun;15(6):1017–1030. PubMed PMID: 30653408.
- Medina DL, Di Paola S, Peluso I, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015 Mar;17(3):288–299. PubMed PMID: 25720963.
- Puertollano R, Ferguson SM, Brugarolas J, et al. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018 Jun 1;37(11):e98804. PubMed PMID: 29764979.
- Rong Y, McPhee CK, Deng S, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A. 2011 May 10;108(19):7826–7831. PubMed PMID: 21518918.
- Chen R, Zou Y, Mao D, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014 Jul 21;206(2):173–182. PubMed PMID: 25049270.
- Nicklin P, Bergman P, Zhang B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009 Feb 6;136(3):521–534. PubMed PMID: 19203585.
- Medina DL. Lysosomal calcium and autophagy. Int Rev Cell Mol Biol. 2021 Apr 20;362:141–170. PubMed PMID: 34253294.
- Sun X, Yang Y, Zhong XZ, et al. A negative feedback regulation of MTORC1 activity by the lysosomal Ca(2+) channel MCOLN1 (mucolipin 1) using a CALM (calmodulin)-dependent mechanism. Autophagy. 2018 Feb 20;14(1):38–52. PubMed PMID: 29460684.
- Li X, Rydzewski N, Hider A, et al. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat Cell Biol. 2016 Apr;18(4):404–417. PubMed PMID: 26950892.
- Zhou C, Wu Z, Du W, et al. Recycling of autophagosomal components from autolysosomes by the recycler complex. Nat Cell Biol. 2022 Apr;24(4):497–512. PubMed PMID: 35332264. 10.1038/s41556-022-00861-8
- Munson MJ, Allen GF, Toth R, et al. mTOR activates the VPS 34– UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J. 2015 Sep 2;34(17):2272–2290. PubMed PMID: 26139536.
- Khundadze M, Ribaudo F, Hussain A, et al. Mouse models for hereditary spastic paraplegia uncover a role of PI4K2A in autophagic lysosome reformation. Autophagy. 2021 Nov;17(11):3690–3706. PubMed PMID: 33618608.
- Sridhar S, Patel B, Aphkhazava D, et al. The lipid kinase PI4KIIIbeta preserves lysosomal identity. EMBO J. 2013 Feb 6;32(3):324–339. PubMed PMID: 23258225.
- Eramo MJ, Gurung R, Mitchell CA, et al. Bidirectional interconversion between PtdIns4P and PtdIns(4,5)P2 is required for autophagic lysosome reformation and protection from skeletal muscle disease. Autophagy. 2021 May;17(5):1287–1289. PubMed PMID: 33879025.
- Dyson JM, Fedele CG, Davies EM, et al. Phosphoinositide phosphatases: just as important as the kinases. Subcell Biochem. 2012;58:215–279. PubMed PMID: 22403078.
- Posor Y, Jang W, Haucke V. Phosphoinositides as membrane organizers. Nat Rev Mol Cell Biol. 2022 May 19:1–20. PubMed PMID: 35589852.
- Pemberton JG, Balla T. Polyphosphoinositide-Binding domains: insights from peripheral membrane and lipid-transfer proteins. Adv Exp Med Biol. 2019;1111:77–137. PubMed PMID: 30483964.
- Schulze RJ, Weller SG, Schroeder B, et al. Lipid droplet breakdown requires dynamin 2 for vesiculation of autolysosomal tubules in hepatocytes. J Cell Biol. 2013 Oct 28;203(2):315–326. PubMed PMID: 24145164.
- Hirst J, Hesketh GG, Gingras AC, et al. Rag GTPases and phosphatidylinositol 3-phosphate mediate recruitment of the AP-5/SPG11/SPG15 complex. J Cell Biol. 2021 Feb 1;220(2):e202002075. PubMed PMID: 33464297.
- Chang J, Lee S, Blackstone C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J Clin Invest. 2014 Dec;124(12):5249–5262. PubMed PMID: 25365221.
- Boura E, Nencka R. Phosphatidylinositol 4-kinases: function, structure, and inhibition. Exp Cell Res. 2015 Oct 1;337(2):136–145. PubMed PMID: 26183104.
- Wu K, Seylani A, Wu J, et al. BLOC1S1/GCN5L1/BORCS1 is a critical mediator for the initiation of autolysosomal tubulation. Autophagy. 2021 Nov;17(11):3707–3724. PubMed PMID: 33629936.
- Jimah JR, Hinshaw JE. Structural insights into the mechanism of dynamin superfamily proteins. Trends Cell Biol. 2019 Mar;29(3):257–273. PubMed PMID: 30527453.
- Klein DE, Lee A, Frank DW, et al. The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J Biol Chem. 1998 Oct 16;273(42):27725–27733. PubMed PMID: 9765310.
- Salim K, Bottomley MJ, Querfurth E, et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 1996 Nov 15;15(22):6241–6250. PubMed PMID: 8947047.
- Sagona AP, Nezis IP, Pedersen NM, et al. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol. 2010 Apr;12(4):362–371. PubMed PMID: 20208530.
- Hirst J, Borner GH, Edgar J, et al. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 2013 Aug;24(16):2558–2569. PubMed PMID: 23825025.
- Hanein S, Martin E, Boukhris A, et al. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including kjellin syndrome. Am J Hum Genet. 2008 Apr;82(4):992–1002. PubMed PMID: 18394578.
- Hampe CS, Wesley J, Lund TC, et al. Mucopolysaccharidosis type I: current treatments, limitations, and prospects for improvement. Biomolecules. 2021;11(2):189. PubMed PMID: 33572941.
- Dinu IR, Ş G Firu. Fabry disease - current data and therapeutic approaches. Rom J Morphol Embryol. 2021 Jan-Mar;62(1):5–11. PubMed PMID: 34609404.
- Arvio M, Mononen I. Aspartylglycosaminuria: a review. Orphanet J Rare Dis. 2016 Dec 1;11(1):162. PubMed PMID: 27906067.
- Magalhaes J, Gegg ME, Migdalska-Richards A, et al. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to parkinson disease. Hum Mol Genet. 2016 Aug 15;25(16):3432–3445. PubMed PMID: 27378698.
- Ginns EI, Brady RO, Pirruccello S, et al. Mutations of glucocerebrosidase: discrimination of neurologic and non-neurologic phenotypes of gaucher disease. Proc Natl Acad Sci U S A. 1982 Sep;79(18):5607–5610. PubMed PMID: 6957882.
- Roh J, Subramanian S, Weinreb NJ, et al. Gaucher disease - more than just a rare lipid storage disease. J Mol Med (Berl). 2022 Apr;100(4):499–518. PubMed PMID: 35066608.
- Tatti M, Motta M, Di Bartolomeo S, et al. Cathepsin-Mediated regulation of autophagy in saposin C deficiency. Autophagy. 2013 Feb 1;9(2):241–243. PubMed PMID: 23108186.
- Tatti M, Motta M, Di Bartolomeo S, et al. Reduced cathepsins B and D cause impaired autophagic degradation that can be almost completely restored by overexpression of these two proteases in sap C-deficient fibroblasts. Hum Mol Genet. 2012 Dec 1;21(23):5159–5173. PubMed PMID: 22949512.
- Harzer K, Paton BC, Poulos A, et al. Sphingolipid activator protein deficiency in a 16-week-old atypical gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur J Pediatr. 1989 Oct;149(1):31–39. PubMed PMID: 2514102.
- Schnabel D, Schröder M, Sandhoff K Mutation in the sphingolipid activator protein 2 in a patient with a variant of gaucher disease. FEBS Lett. 1991 Jun 17;284(1):57–59. PubMed PMID: 2060627.
- Tatti M, Motta M, Scarpa S, et al. BCM-95 and (2-hydroxypropyl)-β-cyclodextrin reverse autophagy dysfunction and deplete stored lipids in sap C-deficient fibroblasts. Hum Mol Genet. 2015 Aug 1;24(15):4198–4211. PubMed PMID: 25926625.
- Yanagisawa H, Ishii T, Endo K, et al. L-Leucine and SPNS1 coordinately ameliorate dysfunction of autophagy in mouse and human niemann-pick type C disease. Sci Rep. 2017 Nov 21;7(1):15944. PubMed PMID: 29162837.
- Avenali M, Blandini F, Cerri S. Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front Aging Neurosci. 2020 Apr;12:97. PubMed PMID: 32372943.
- Latourelle JC, Pankratz N, Dumitriu A, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009 Sep 22;10(1):98. PubMed PMID: 19772629.
- Do CB, Tung JY, Dorfman E, et al. Web-Based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011 Jun 23;7(6):e1002141. PubMed PMID: 21738487.
- Miyazaki M, Hiramoto M, Takano N, et al. Targeted disruption of GAK stagnates autophagic flux by disturbing lysosomal dynamics. Int J Mol Med. 2021 Oct;48(4):195. PubMed PMID: 34468012.
- Vantaggiato C, Panzeri E, Castelli M, et al. ZFYVE26/SPASTIZIN and SPG11/SPATACSIN mutations in hereditary spastic paraplegia types AR-SPG15 and AR-SPG11 have different effects on autophagy and endocytosis. Autophagy. 2019 Jan;15(1):34–57. PubMed PMID: 30081747.
- Khundadze M, Ribaudo F, Hussain A, et al. A mouse model for SPG48 reveals a block of autophagic flux upon disruption of adaptor protein complex five. Neurobiol Dis. 2019 Jul;127:419–431. PubMed PMID: 30930081.
- Boutry M, Branchu J, Lustremant C, et al. Inhibition of lysosome membrane recycling causes accumulation of gangliosides that contribute to neurodegeneration. Cell Rep. 2018 Jun 26;23(13):3813–3826. PubMed PMID: 29949766.
- Stevanin G, Santorelli FM, Azzedine H, et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007 Mar;39(3):366–372. PubMed PMID: 17322883.
- Słabicki M, Theis M, Krastev DB, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010 Jun 29;8(6):e1000408. PubMed PMID: 20613862.
- Bouchard JP, Barbeau A, Bouchard R, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. 1978 Feb;5(1):61–69. PubMed PMID: 647499.
- Engert JC, Bérubé P, Mercier J, et al. ARSACS, a spastic ataxia common in northeastern québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000 Feb;24(2):120–125. PubMed PMID: 10655055.
- Bagaria J, Bagyinszky E, An SSA Genetics of autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) and role of sacsin in neurodegeneration. Int J Mol Sci. 2022 Jan 4;23(1):552. PubMed PMID: 35008978.
- Francis V, Alshafie W, Kumar R, et al. The ARSACS disease protein sacsin controls lysosomal positioning and reformation by regulating microtubule dynamics. J Biol Chem. 2022 Sep; 298(9):102320. PubMed PMID: 35933016.
- Wiessner M, Roos A, Munn CJ, et al. Mutations in INPP5K, encoding a phosphoinositide 5-phosphatase, cause congenital muscular dystrophy with cataracts and mild cognitive impairment. Am J Hum Genet. 2017 Mar 2;100(3):523–536. PubMed PMID: 28190456.
- Osborn DPS, Pond HL, Mazaheri N, et al. Mutations in INPP5K cause a form of congenital muscular dystrophy overlapping Marinesco-Sjogren syndrome and dystroglycanopathy. Am J Hum Genet. 2017 Mar 2;100(3):537–545. PubMed PMID: 28190459.
- D’Amico A, Fattori F, Nicita F, et al. A recurrent pathogenic variant of INPP5K underlies autosomal recessive congenital muscular dystrophy with cataracts and intellectual disability: evidence for a founder effect in Southern Italy. Front Genet. 2020 Sep 18;11:565868. PubMed PMID: 33193651.
- Rasineni K, Donohue TM Jr., Thomes PG, et al. Ethanol-Induced steatosis involves impairment of lipophagy, associated with reduced dynamin2 activity. Hepatol Commun. 2017 Aug;1(6):501–512. PubMed PMID: 29152606.
- Lazarus JV, Mark HE, Anstee QM, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol 2021. Jan;19(1):60–78. PubMed PMID: 34707258.
- Parenti G, Medina DL, Ballabio A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol Med. 2021 Feb 5;13(2):e12836. PubMed PMID: 33459519.
- Myerowitz R, Puertollano R, Raben N. Impaired autophagy: the collateral damage of lysosomal storage disorders. EBioMedicine. 2021 Jan;63:103166. PubMed PMID: 33341443.
- Nakano Y, Fujitani K, Kurihara J, et al. Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in drosophila melanogaster. Mol Cell Biol. 2001 Jun;21(11):3775–3788. PubMed PMID: 11340170.
- Kishi S, Bayliss PE, Uchiyama J, et al. The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet. 2008 Aug 15;4(8):e1000152. PubMed PMID: 18704191.
- Chen J, Ou Y, Luo R, et al. SAR1B senses leucine levels to regulate mTORC1 signalling. Nature. 2021 Aug;596(7871):281–284. PubMed PMID: 34290409.
- Brown RA, Voit A, Srikanth MP, et al. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis Model Mech. 2019 Oct 16;12(10):dmm038596. PubMed PMID: 31519738.
- Bartolomeo R, Cinque L, De Leonibus C, et al. mTORC1 hyperactivation arrests bone growth in lysosomal storage disorders by suppressing autophagy. J Clin Invest. 2017 Oct 2;127(10):3717–3729. PubMed PMID: 28872463.
- Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm (Vienna). 2017 Aug;124(8):901–905. PubMed PMID: 28150045.
- Zhang L, Dong Y, Xu X, et al. The role of autophagy in Parkinson’s disease. Neural Regen Res. 2012 Jan 15;7(2):141–145. PubMed PMID: 25767490.
- Brooker SM, Krainc D. Glucocerebrosidase dysfunction in neurodegenerative disease. Essays Biochem. 2021 Sep 16;65(7):873–883. PubMed PMID: 34528667.
- Schöndorf DC, Aureli M, McAllister FE, et al. iPSC-Derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun. 2014 Jun 6;5(1):4028. PubMed PMID: 24905578.
- Kuo SH, Tasset I, Cheng MM, et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv. 2022 Feb 11;8(6):eabm6393. PubMed PMID: 35138901.
- Kinghorn KJ, Grönke S, Castillo-Quan JI, et al. A drosophila model of neuronopathic Gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J Neurosci. 2016 Nov 16;36(46):11654–11670. PubMed PMID: 27852774.
- Awad O, Sarkar C, Panicker LM, et al. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum Mol Genet. 2015 Oct 15;24(20):5775–5788. PubMed PMID: 26220978.
- Teixeira M, Sheta R, Idi W, et al. Alpha-Synuclein and the endolysosomal system in Parkinson’s disease: guilty by association. Biomolecules. 2021 Sep 9;11(9):1333. PubMed PMID: 34572546.
- Murala S, Nagarajan E, Bollu PC. Hereditary spastic paraplegia. Neurol Sci. 2021 Mar;42(3):883–894. PubMed PMID: 33439395.
- Montecchiani C, Pedace L, Lo Giudice T, et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal charcot-marie-tooth disease. Brain. 2016 Jan;139(1):73–85. PubMed PMID: 26556829.
- Daoud H, Zhou S, Noreau A, et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging. 2012 Apr;33(4): 839.e5-9. PubMed PMID: 22154821.
- Pensato V, Castellotti B, Gellera C, et al. Overlapping phenotypes in complex spastic paraplegias SPG11, SPG15, SPG35 and SPG48. Brain. 2014 Jul;137(7):1907–1920. PubMed PMID: 24833714.
- Vantaggiato C, Crimella C, Airoldi G, et al. Defective autophagy in spastizin mutated patients with hereditary spastic paraparesis type 15. Brain. 2013 Oct;136(10):3119–3139. PubMed PMID: 24030950.
- Renvoisé B, Chang J, Singh R, et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann Clin Transl Neurol. 2014 Jun 1;1(6):379–389. PubMed PMID: 24999486.
- Kalinichenko LS, Gulbins E, Kornhuber J, et al. Sphingolipid control of cognitive functions in health and disease. Prog Lipid Res. 2022 Apr;86:101162. PubMed PMID: 35318099.
- Kanagaraj P, Gautier-Stein A, Riedel D, et al. Souffle/spastizin controls secretory vesicle maturation during zebrafish oogenesis. PLoS Genet. 2014 Jun 26;10(6):e1004449. PubMed PMID: 24967841.
- Simons JP, Al-Shawi R, Minogue S, et al. Loss of phosphatidylinositol 4-kinase 2alpha activity causes late onset degeneration of spinal cord axons. Proc Natl Acad Sci U S A. 2009 Jul 14;106(28):11535–11539. PubMed PMID: 19581584.
- Cleeter M, Houlden H, Simons P, et al. Screening for mutations in the phosphatidylinositol 4-kinase 2-alpha gene in autosomal recessive hereditary spastic paraplegia. Amyotroph Lateral Scler. 2011 Mar;12(2):148–149. PubMed PMID: 21190509.
- Verdura E, Rodríguez-Palmero A, Vélez-Santamaria V, et al. Biallelic PI4KA variants cause a novel neurodevelopmental syndrome with hypomyelinating leukodystrophy. Brain. 2021 Oct 22;144(9):2659–2669. PubMed PMID: 34415322.
- Kong XF, Bousfiha A, Rouissi A, et al. A novel homozygous p.R1105x mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS One. 2013 March 5;8(3):e58286. PubMed PMID: 23472171.
- Sambuughin N, Goldfarb LG, Sivtseva TM, et al. Adult-Onset autosomal dominant spastic paraplegia linked to a GTPase-effector domain mutation of dynamin 2. BMC Neurol. 2015 Oct 30;15:1223. PubMed PMID: 26517984.
- Larivière R, Gaudet R, Gentil BJ, et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum Mol Genet. 2015 Feb 1;24(3):727–739. PubMed PMID: 25260547.
- Girard M, Larivière R, Parfitt DA, et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci U S A. 2012 Jan 31;109(5):1661–1666. PubMed PMID: 22307627.
- Vermeer S, Meijer RP, Pijl BJ, et al. ARSACS in the dutch population: a frequent cause of early-onset cerebellar ataxia. Neurogenetics. 2008 Jul;9(3):207–214. PubMed PMID: 18465152.
- Morani F, Doccini S, Sirica R, et al. Functional transcriptome analysis in ARSACS KO cell model reveals a role of sacsin in autophagy. Sci Rep. 2019 Aug 15;9(1):11878. PubMed PMID: 31417125.
- Masiero E, Agatea L, Mammucari C, et al. Autophagy is required to maintain muscle mass. Cell Metab. 2009 Dec;10(6):507–515. PubMed PMID: 19945408.
- Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004 Mar;15(3):1101–1111. PubMed PMID: 14699058.
- Margeta M. Autophagy defects in skeletal myopathies. Annu Rev Pathol. 2020 Jan 24;15:261–285. PubMed PMID: 31594457.
- Franco-Romero A, Sandri M. Role of autophagy in muscle disease. Mol Aspects Med. 2021 Dec;82:101041. PubMed PMID: 34625292.
- Lo Verso F, Carnio S, Vainshtein A, et al. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy. 2014 Oct 30;10(11):1883–1894. PubMed PMID: 25483961.
- Brook MS, Wilkinson DJ, Atherton PJ. Nutrient modulation in the management of disease-induced muscle wasting: evidence from human studies. Curr Opin Clin Nutr Metab Care. 2017 Nov;20(6):433–439. PubMed PMID: 28832372.
- Yousaf S, Sheikh SA, Riazuddin S, et al. INPP5K variant causes autosomal recessive congenital cataract in a Pakistani family. Clin Genet. 2018 Mar;93(3):682–686. PubMed PMID: 28940338.
- Homma TK, Freire BL, Honjo Kawahira RS, et al. Genetic disorders in prenatal onset syndromic short stature identified by exome sequencing. J Pediatr. 2019 Dec;215:192–198. PubMed PMID: 31630891.
- Gurung R, Tan A, Ooms LM, et al. Identification of a novel domain in two mammalian inositol-polyphosphate 5-phosphatases that mediates membrane ruffle localization. The inositol 5-phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J Biol Chem. 2003 Mar 28;278(13):11376–11385. PubMed PMID: 12536145.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015 Aug 20;524(7565):309–314. PubMed PMID: 26266977.
- Fu T, Liu J, Wang Y, et al. Mechanistic insights into the interactions of NAP1 with the SKICH domains of NDP52 and TAX1BP1. Proc Natl Acad Sci U S A. 2018 Dec 11;115(50):E11651–11660. PubMed PMID: 30459273.
- Nthiga TM, Shrestha BK, Bruun JA, et al. Regulation of golgi turnover by CALCOCO1-mediated selective autophagy. J Cell Biol. 2021 Jun 7;220(6):e202006128. PubMed PMID: 33871553.
- Nthiga TM, Kumar Shrestha B, Sjøttem E, et al. CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. EMBO J. 2020 Aug 3;39(15):e103649. PubMed PMID 32525583.
- Stefely JA, Zhang Y, Freiberger EC, et al. Mass spectrometry proteomics reveals a function for mammalian CALCOCO1 in MTOR-regulated selective autophagy. Autophagy. 2020 Dec;16(12):2219–2237. PubMed PMID: 31971854.
- Furuya N, Kakuta S, Sumiyoshi K, et al. NDP52 interacts with mitochondrial RNA poly(a) polymerase to promote mitophagy. EMBO Rep. 2018 Dec;19(12):e46363. PubMed PMID: 30309841.
- Priest C, Tontonoz P. Inter-Organ cross-talk in metabolic syndrome. Nat Metab. 2019 Dec 9;1(12):1177–1188. PubMed PMID: 32694672.
- Jahng JWS, Alsaadi RM, Palanivel R, et al. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019 Oct 4;20(10):e47911. PubMed PMID: 31441223.
- Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013 Mar 5;17(3):329–341. PubMed PMID: 23473030.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009 Apr 30;458(7242):1131–1135. PubMed PMID: 19339967.
- Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013 May;58(5):993–999. PubMed PMID: 23339953.
- Selmani A, Kovačević D, Bohinc K. Nanoparticles: from synthesis to applications and beyond. Adv Colloid Interface Sci. 2022 May;303:102640. PubMed PMID: 35358806.
- Huang R, Zhou X, Chen G, et al. Advances of functional nanomaterials for magnetic resonance imaging and biomedical engineering applications. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2022 Jul;14:4e1800. PubMed PMID: 35445588.
- Zhou X, Jin W, Sun H, et al. Perturbation of autophagy: an intrinsic toxicity mechanism of nanoparticles. Sci Total Environ. 2022 Jun 1;823:153629. PubMed PMID: 35131247.
- Mendez-Gonzalez D, Lopez-Cabarcos E, Rubio-Retama J, et al. Sensors and bioassays powered by upconverting materials. Adv Colloid Interface Sci. 2017 Nov;249:66–87. PubMed PMID: 28641813.
- Zhang JQ, Zhou W, Zhu SS, et al. Persistency of enlarged autolysosomes underscores nanoparticle-induced autophagy in hepatocytes. Small. 2017 Feb;13(7):1602876. PubMed PMID: 27925395.
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013 Oct;13(10):722–737. PubMed PMID: 24064518.
- Sun H, Gong S, Carmody RJ, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008 May 2;133(3):415–426. PubMed PMID: 18455983.
- Gu Z, Cui X, Sun P, et al. Regulatory roles of tumor necrosis factor-α-induced protein 8 like-protein 2 in inflammation, immunity and cancers: a review. Cancer Manag Res. 2020 Dec 14;12:12735–12746. PubMed PMID: 33364825.
- Li W, Li Y, Guan Y, et al. TNFAIP8L2/TIPE2 impairs autolysosome reformation via modulating the RAC1-MTORC1 axis. Autophagy. 2021 Jun;17(6):1410–1425. PubMed PMID: 32460619.
- Fayngerts SA, Wu J, Oxley CL, et al. TIPE3 is the transfer protein of lipid second messengers that promote cancer. Cancer Cell. 2014 Oct 13;26(4):465–478. PubMed PMID: 25242044.
- Fayngerts SA, Wang Z, Zamani A, et al. Direction of leukocyte polarization and migration by the phosphoinositide-transfer protein TIPE2. Nat Immunol. 2017 Dec;18(12):1353–1360. PubMed PMID: 29058702.
- Kaushik S, Tasset I, Arias E, et al. Autophagy and the hallmarks of aging. Ageing Res Rev. 2021 Dec;72:101468. PubMed PMID: 34563704.
- Fernández ÁF, Sebti S, Wei Y, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018 Jun;558(7708):136–140. PubMed PMID: 29849149.
- Meléndez A, Tallóczy Z, Seaman M, et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003 Sep 5;301(5638):1387–1391. PubMed PMID: 12958363.
- Triolo M, Hood DA. Manifestations of age on autophagy, mitophagy and lysosomes in skeletal muscle. Cells. 2021 Apr 29;10(5):1054. PubMed PMID: 33946883.
- Jiao J, Demontis F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr Opin Pharmacol. 2017 Jun;34:1–6. PubMed PMID: 28407519.
- Zhang Z, Yang X, Song YQ, et al. Autophagy in Alzheimer’s disease pathogenesis: therapeutic potential and future perspectives. Ageing Res Rev. 2021 Dec;72:101464. PubMed PMID: 34551326.
- Li X, Xu J, Dai B, et al. Targeting autophagy in osteoporosis: from pathophysiology to potential therapy. Ageing Res Rev. 2020 Sep;62:101098. PubMed PMID: 32535273.
- Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol. 2021 Nov;17(11):647–661. PubMed PMID: 34508250.
- Barbosa MC, Grosso RA, Fader CM. Hallmarks of aging: an autophagic perspective. Front Endocrinol (Lausanne). 2019 Jan 9;9:790. PubMed PMID: 30687233.
- Carosi JM, Fourrier C, Bensalem J, et al. The mTOR-lysosome axis at the centre of ageing. FEBS Open Bio. 2022 Apr;12(4):739–757. PubMed PMID: 34878722.
- Korovila I, Hugo M, Castro JP, et al. Proteostasis, oxidative stress and aging. Redox Biol. 2017 Oct;13:550–567. PubMed PMID: 28763764.
- Wang F, Dai Y, Zhu X, et al. Saturated very long chain fatty acid configures glycosphingolipid for lysosome homeostasis in long-lived C. elegans. Nat Commun. 2021 Aug 20;12(1):5073. PubMed PMID: 34417467.
- Green CL, Lamming DW, Fontana L. Molecular mechanisms of dietary restriction promoting health and longevity. Nat Rev Mol Cell Biol. 2021 Jan 22;23(1):56–73. PubMed PMID: 34518687.
- Matsumaru D, Motohashi H. The KEAP1-NRF2 system in healthy aging and longevity. Antioxidants (Basel). 2021 Nov 30;10(12):1929. PubMed PMID: 34943032.
- Madeo F, Carmona-Gutierrez D, Hofer SJ, et al. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 2019 Mar 5;29(3):592–610. PubMed PMID: 30840912.
- Lees JA, Li P, Kumar N, et al. Insights into lysosomal PI(3,5)P2 homeostasis from a structural-biochemical analysis of the PIKfyve lipid kinase complex. Mol Cell. 2020 Nov 19;80(4):736–743.e4. PubMed PMID: 33098764.
- Zolov SN, Bridges D, Zhang Y, et al. In vivo, Pikfyve generates PI(3,5)P2, which serves as both a signaling lipid and the major precursor for PI5P. Proc Natl Acad Sci U S A. 2012 Oct 23;109(43):17472–17477. PubMed PMID: 23047693.
- Stutterd C, Diakumis P, Bahlo M, et al. Neuropathology of childhood-onset basal ganglia degeneration caused by mutation of VAC14. Ann Clin Transl Neurol. 2017 Nov 7;4(12):859–864. PubMed PMID: 29296614.
- Bao W, Wang X, Luo L, et al. The lysosomal storage disorder due to fig4a mutation causes robust liver vacuolation in zebrafish. Zebrafish. 2021 Jun;18(3):175–183. PubMed PMID: 33909505.
- Bharadwaj R, Cunningham KM, Zhang K, et al. FIG4 regulates lysosome membrane homeostasis independent of phosphatase function. Hum Mol Genet. 2016 Feb 15;25(4):681–692. PubMed PMID: 26662798.
- Katona I, Zhang X, Bai Y, et al. Distinct pathogenic processes between Fig4-deficient motor and sensory neurons. Eur J Neurosci. 2011 Apr;33(8):1401–1410. PubMed PMID: 21410794.
- Martyn C, Li J. Fig4 deficiency: a newly emerged lysosomal storage disorder? Prog Neurobiol. 2013 Feb-Mar;101-102:35–45. PubMed PMID: 23165282.
- Rivero-Ríos P, Weisman LS. Roles of PIKfyve in multiple cellular pathways. Curr Opin Cell Biol. 2022 June;76:102086. PubMed PMID: 35584589.
- Campeau PM, Lenk GM, Lu JT, et al. Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am J Hum Genet. 2013 May 2;92(5):781–791. PubMed PMID: 23623387.
- Lines MA, Ito Y, Kernohan KD, et al. Yunis-Varón syndrome caused by biallelic VAC14 mutations. Eur J Hum Genet. 2017 Sep;25(9):1049–1054. PubMed PMID: 28635952.
- Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009 Jan;84(1):85–88. PubMed PMID: 19118816.
- Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007 Jul 5;448(7149):68–72. PubMed PMID: 17572665.
- Baulac S, Lenk GM, Dufresnois B, et al. Role of the phosphoinositide phosphatase FIG4 gene in familial epilepsy with polymicrogyria. Neurology. 2014 Mar 25;82(12):1068–1075. PubMed PMID: 24598713.
- Zimmermann M, Schuster S, Boesch S, et al. FIG4 mutations leading to parkinsonism and a phenotypical continuum between CMT4J and Yunis Varón syndrome. Parkinsonism Relat Disord. 2020 May;74:6–11. PubMed PMID: 32268254.
- Kaur P, Bhavani GS, Raj A, et al. Homozygous variant, p.(Arg643Trp) in VAC14 causes striatonigral degeneration: report of a novel variant and review of VAC14-related disorders. J Hum Genet. 2019 Dec;64(12):1237–1242. PubMed PMID: 31591492.
- Lenk GM, Szymanska K, Debska-Vielhaber G, et al. Biallelic mutations of VAC14 in pediatric-onset neurological disease. Am J Hum Genet. 2016 Jul 7;99(1):188–194. PubMed PMID: 27292112.
- Li S, Tiab L, Jiao X, et al. Mutations in PIP5K3 are associated with François-Neetens mouchetée fleck corneal dystrophy. Am J Hum Genet. 2005 Jul;77(1):54–63. PubMed PMID: 15902656.
- Mei S, Wu Y, Wang Y, et al. Disruption of PIKFYVE causes congenital cataract in human and zebrafish. Elife. 2022 Jan 13;11:e71256. PubMed PMID: 35023829.
- Gan Q, Wang X, Zhang Q, et al. The amino acid transporter SLC-36.1 cooperates with PtdIns3P 5-kinase to control phagocytic lysosome reformation. J Cell Biol. 2019 Aug 5;218(8):2619–2637. PubMed PMID: 31235480.
- Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside-Induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat Genet. 2002 Jan;30(1):21–22. PubMed PMID: 11743579.
- Cantarero L, Juárez-Escoto E, Civera-Tregón A, et al. Mitochondria-lysosome membrane contacts are defective in GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet. 2021 Jan 21;29(22):3589–3605. PubMed PMID: 33372681.
- Choy CH, Saffi G, Gray MA, et al. Lysosome enlargement during inhibition of the lipid kinase PIKfyve proceeds through lysosome coalescence. J Cell Sci. 2018 May 21;131(10):jcs213587. PubMed PMID: 29661845.
- Bissig C, Hurbain I, Raposo G, et al. Pikfyve activity regulates reformation of terminal storage lysosomes from endolysosomes. Traffic. 2017 Nov;18(11):747–757. PubMed PMID: 28857423.
- Rodgers SJ, Jones EI, Arumugam S, et al. Endosome maturation links PI3Kα signaling to lysosome repopulation during basal autophagy. EMBO J. 2022 Oct 4;41(19):e110398. PubMed PMID: 35968799.
- Vicinanza M, Korolchuk VI, Ashkenazi A, et al. PI(5)P regulates autophagosome biogenesis. Mol Cell. 2015 Jan 22;57(2):219–234. PubMed PMID: 25578879.
- Karabiyik C, Vicinanza M, Son SM, et al. Glucose starvation induces autophagy via ULK1-mediated activation of PIKfyve in an AMPK-dependent manner. Dev Cell. 2021 Jul 12;56(13):1961–1975.e5. PubMed PMID: 34107300.
- Vantaggiato C, Orso G, Guarato G, et al. Rescue of lysosomal function as therapeutic strategy for SPG15 hereditary spastic paraplegia. Brain awac308. 2022 Aug 27. PubMed PMID: 36029068.