
Abstract
Context: Since the banning of dextropropoxyphene from the market, overdoses, and fatalities attributed to tramadol, a WHO step-2 opioid analgesic, have increased markedly. Tramadol overdose results not only in central nervous system (CNS) depression attributed to its opioid properties but also in seizures, possibly related to non-opioidergic pathways, thus questioning the efficiency of naloxone to reverse tramadol-induced CNS toxicity.
Objective: To investigate the most efficient antidote to reverse tramadol-induced seizures and respiratory depression in overdose.
Materials and methods: Sprague–Dawley rats overdosed with 75 mg/kg intraperitoneal (IP) tramadol were randomized into four groups to receive solvent (control group), diazepam (1.77 mg/kg IP), naloxone (2 mg/kg intravenous bolus followed by 4 mg/kg/h infusion), and diazepam/naloxone combination. Sedation depth, temperature, number of seizures, and intensity, whole-body plethysmography parameters and electroencephalography activity were measured.
Results: Naloxone reversed tramadol-induced respiratory depression (p < .05) but significantly increased seizures (p < .01) and prolonged their occurrence time. Diazepam abolished seizures but significantly deepened rat sedation (p < .05) without improving ventilation. Diazepam/naloxone combination completely abolished seizures, significantly improved rat ventilation by reducing inspiratory time (p < .05) but did not worsen sedation. None of these treatments significantly modified rat temperature.
Conclusions: Diazepam/naloxone combination is the most efficient antidote to reverse tramadol-induced CNS toxicity in the rat.
Introduction
To date, prescription opioids represent the major cause of drug-induced death in the US, mainly attributed to the WHO step-2 analgesics including tramadol [Citation1]. Following dextropropoxyphene withdrawal from the US (November 2010) and European (September 2011) market, tramadol prescriptions have risen exponentially, resulting in an increase in poisonings and fatalities [Citation2].
Tramadol overdose is responsible for consciousness impairment (∼30%), seizures and possible induced trauma (∼15%), agitation (∼10%), respiratory depression (∼8%), and serotonin syndrome (∼5%) [Citation3–6]. Tramadol-induced seizures represent an important issue contributing to ∼8% of the drug-induced seizures reported to poison control centers [Citation7]. Tramadol overdose typically produces brief, isolated, self-limiting generalized tonic-clonic seizures, most often occurring within 4–6 h of ingestion [Citation8]; however, electroencephalography (EEG) abnormalities are common and recurrent seizures or status epilepticus not exceptional [Citation8–10].
Management of tramadol-poisoned patients with naloxone remains controversial. Retrospective observational cohort studies reported decreased [Citation11], preserved [Citation12], or enhanced [Citation5,Citation13] risk of seizures with the use of naloxone to reverse neuro-respiratory toxicity. One randomized controlled study suggested that one single intravenous (IV) 0.8-mg dose of naloxone was responsible for seizurogenic effects in tramadol-overdosed patients [Citation14]. By contrast, another self-controlled study concluded to the efficacy and safety of 0.05 mg naloxone administered IV every 3–5 min after initial resuscitation to manage tramadol-intoxicated patients with postseizure complaints [Citation15]. In addition, tramadol-poisoned children are at risk of apnea and seizures highlighting the necessity to define an efficient and safe antidote to reverse central nervous system (CNS) toxicity, considering the European advice for the treatment of moderate pain in children suggesting limited codeine use and rather tramadol prescription [Citation16,Citation17].
Mechanisms of tramadol-induced CNS depression have been investigated showing that at least one of its metabolites, the O-desmethyl-tramadol (M1), has 200 times more affinity to μ-opioid-receptors than tramadol itself explaining increased toxicity in ultra-rapid cytochrome P450 2D6 metabolisers and variable geographical area-dependent rate of toxicity [Citation18]. In contrast, the exact mechanisms of tramadol-induced seizures are still poorly understood. Based on animal data, seizures are unlikely to be related to serotoninergic mechanisms [Citation19,Citation20] and alternative pathways including GABAergic [Citation21], histaminergic [Citation22], and opioidergic [Citation21] have been hypothesized, thus questioning the possible worsening role of naloxone in the treatment of tramadol overdose. Conformingly, in one observational study, naloxone administration was associated with increased probability of tracheal intubation [Citation11].
Interestingly, more limited seizing activity was observed in patients who co-ingested tramadol and benzodiazepines [Citation13], consistent with the abolishment of seizures we reported in our rat model of tramadol overdose following the combined administration of elevated doses of tramadol and diazepam [Citation20]. However, the benzodiazepine/tramadol combination consistently resulted in enhanced CNS depression, both in animals and humans [Citation20,Citation23].
Thus, due to all these existing controversial data, we designed a rat study to investigate the most efficient antidote to reverse tramadol-induced seizures and respiratory depression in overdose.
Materials and methods
Our experimental protocols were carried out within the ethical guidelines established by the National Institutes of Health and approved by Paris-Descartes University ethics committee for animal experimentation and by the French Ministry of Higher Education and Research (N°-APAFlS#5285-2017031714499381v2).
Animals and drugs
Male Sprague–Dawley rats (Janvier) weighing 250–350 g at the time of experimentation were used, housed for 7 days before experimentation in an environment maintained at 21 ± 0.5 °C with controlled humidity and light-dark cycle. Food and tap water were provided ad libitum. Tramadol hydrochloride (Grünenthal, Germany) and naloxone (Aguettant, France) were diluted in sterile water to obtain solutions of 44 and 0.4 mg/mL, respectively. Diazepam (Roche, France) was diluted in 4% Tween (Tween-20® diluted in 0.9% NaCl) to obtain a solution of 2 mg/mL.
Clinical measurements
Temperature was measured using intraperitoneal (IP) implanted temperature transmitters for the purpose of plethysmography study. Sedation level based on a four-stage scale from 0 (awake) to 3 (coma) was assessed [Citation24]: At stage 0 (awake), rats were completely awake and their gait and righting reflexes were intact. At stage 1 (lethargic), rats had reduced activity, showed light impairment of gait and intact righting reflex with diminished muscle tonus. At stage 2 (mildly comatose), rats were asleep or static and showed reduced righting reflex. At stage 3 (deeply comatose), rats were comatose and did not have any righting reflex. Seizure severity was graded according to the modified Racine Score [Citation25]: At stage 1, rats were immobile, with eyes closed, twitching of vibrissae, and facial clonus. At stage 2, rats had head nodding associated with more severe facial clonus. At stage 3, rats had clonus of one forelimb. At stage 4, rats had rearing, often accompanied by bilateral forelimb clonus. At stage 5, rats had rearing with loss of balance and falling accompanied by generalized clonic seizures.
Whole body plethysmography
Seven days before experimentation, temperature transmitters (TA-F10, DSI, Hertogenbosch, Netherlands) were implanted in the rat peritoneal cavity. Ventilatory parameters were recorded in a whole body plethysmograph by the barometric method described and validated in the rat [Citation26]. The first measurement was performed after a 30-min period of accommodation while the rat was quiet and not in deep or rapid eye movement sleep, as roughly estimated from their behavior, response to noise and pattern of breathing. Then, the rat was gently removed from the chamber for injection at T0 and replaced in the chamber for the remaining measurements. Ventilation was recorded at −30, −15, −5, 5, 10, 14, 20, 30, 40, 50, 60, 90, 120, 180, and 240 min, each record lasting about 60 s. The following parameters were measured: the tidal volume (VT), the inspiratory time (TI) and the expiratory time (TE). Additional parameters were calculated, including the respiratory frequency (f) and the minute volume (VM = VT × f).
Electroencephalography
Seven days before experimentation, epidural screw electrodes (TA10ETA-F20 implant, DSI, Hertogenbosch, Netherlands) for EEG recording were implanted as previously described [Citation20]. On the experimentation day, the rat was habituated to the recording box for one hour (to avoid any consequent stress due to this new environment) and allowed free movements. EEG signals were recorded continuously during the hour of habituation and 4 h after drug administration. EEG analysis was performed by a trained neurologist blinded to the treatments administered to the rats.
Study design
One week before the study, the rat jugular vein was catheterized as previously described [Citation20]. On the day of experimentation and before drug administration, the catheter was exteriorized, purged and its permeability checked. Tramadol dose was chosen to allow the onset of typical tramadol poisoning features in the rat as previously demonstrated [Citation20]. The tested antidotes were given at their usual pharmacologic doses. Rats received 75 mg/kg tramadol IP and were subsequently randomized into four groups (N = 6/group for each experiment) to be treated with either 4% Tween IP + saline IV (Control group); or diazepam (1.77 mg/kg IP, as previously reported [Citation27]) + saline IV (diazepam group); or 4% Tween IP + naloxone (2 mg/kg IV bolus followed by 4 mg/kg/h infusion, as previously described [Citation28]) (naloxone group); or diazepam/naloxone combination given according to the same protocols (diazepam/naloxone combination group). Antidotes were administered 15 min after tramadol injection at the time of the expected tramadol-induced peak CNS effects as previously reported [Citation20]. The clinical and plethysmography parameters described above were measured in the four groups of randomized rats. EEG was recorded in the diazepam/naloxone combination group.
Statistical analysis
The results are expressed as median (percentiles). For plethysmography, T0 was the mean of the three baseline measurements. To permit simultaneous analysis of the effect of time and treatments on sedation, temperature, and plethysmography parameters, we calculated for each animal and for each studied parameter, the area under the curve (AUC) from T0 to the completion of the measurement (240 min) using the trapezoid method [Citation29]. For each parameter, we compared the AUCs using Mann–Whitney tests for two by two comparisons between the four groups. Regarding the effects of treatments on seizures, comparisons were performed using two-way analysis of variance followed by multiple comparison tests using Bonferroni’s correction. All tests were performed using Prism version 6.0 (GraphPad Software, San Diego, CA). p Values < .05 were considered as significant.
Results
Tramadol overdose-induced sedation and hypothermia (), resulted in tonic–clonic seizures mainly of grade-3 on Racine’s scale up to 60 min after injection (), increased TI and reduced VM ().
Figure 1. Effects on sedation (A) and temperature (B) in Sprague–Dawley rats of intraperitoneal (IP) 75 mg/kg tramadol administration followed 15 min later by IP 4% tween + intravenous (IV) saline (Control group, black), IP 1.77 mg/kg diazepam + IV saline (diazepam group, horizontal lines), IP 4% tween + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (Naloxone group, vertical lines), or IP 1.77 mg/kg diazepam + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (diazepam/naloxone group, white). Results are expressed as median (percentiles; N = 6/group). *p < .05.
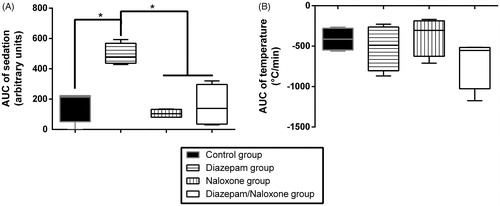
Figure 2. Effects on the time-course (A) and intensity (B) of seizures in Sprague–Dawley rats of intraperitoneal (IP) 75 mg/kg tramadol administration followed 15 min later by IP 4% tween + intravenous (IV) saline (Control group, black), IP 1.77 mg/kg diazepam + IV saline (diazepam group, horizontal lines), IP 4% tween + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (Naloxone group, vertical lines), or IP 1.77 mg/kg diazepam + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (diazepam/naloxone group, white). Each dot represents one seizure. Results are expressed as median (percentiles; N = 6/group). **p < .01 and ****p < .0001 between Naloxone and diazepam groups; °°p < .01 and °°°°p < .0001 between Naloxone and diazepam/naloxone groups; ##p < .01 between Naloxone and control groups.
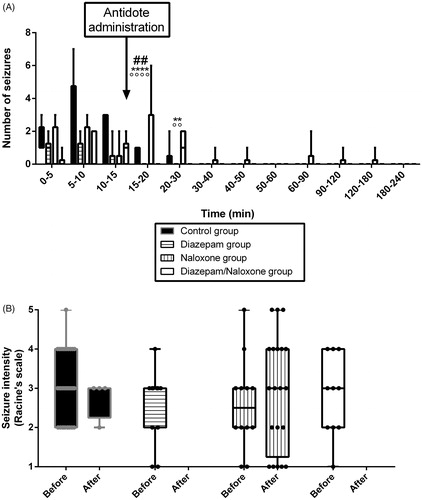
Figure 3. Effects on plethysmography parameters in Sprague–Dawley rats of intraperitoneal (IP) 75 mg/kg tramadol administration followed 15 min later by IP 4% tween + intravenous (IV) saline (control group, black), IP 1.77 mg/kg diazepam + IV saline (diazepam group, horizontal lines), IP 4% tween + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (naloxone group, vertical lines), or IP 1.77 mg/kg diazepam + IV 2 mg/kg bolus followed by 4 mg/kg/h naloxone (diazepam/naloxone group, white). TI represents the inspiratory time, TE the expiratory time, VT, the tidal volume and VM the minute volume. Areas under the effect-time curve (AUC) were determined. Results are expressed as median (percentiles). Comparisons between groups were performed two by two using Mann–Whitney tests. (N = 6/group). *p < .05.
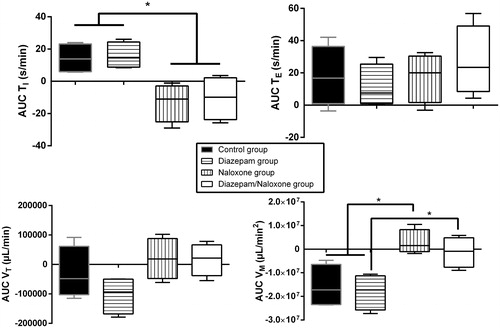
Naloxone administration significantly reversed TI increase and VM reduction (p < .05, ), but did not result in significant changes in sedation depth and hypothermia (). Seizures were significantly increased during the 5 min following naloxone bolus in comparison to the control rats (p < .001) and prolonged up to 3 h after tramadol injection ().
Diazepam administration significantly deepened sedation (p < .05, ) and completely abolished seizures (). Hypothermia () and plethysmography parameters () were not significantly modified in comparison to the control group.
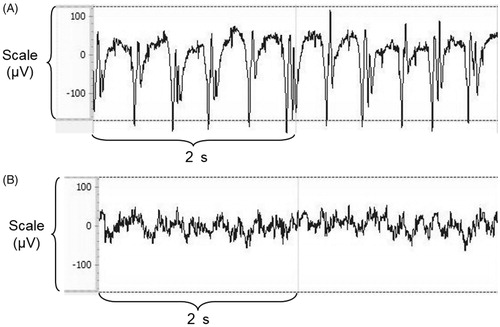
Diazepam/naloxone combination abolished seizures () and significantly increased TI (p < .05, ). There was a non-significant increase in VM (). No significant changes were observed in sedation depth and hypothermia (). Based on the EEG study, tramadol-treated rats experienced electro-clinical seizures including status epilepticus as soon as 5 min after the administration (). The electrical epileptic discharges lasting ∼15 s were characterized by spike-waves and polyspikes with progressively decreased frequencies and inter-critical phases of slow 1–4 Hz delta waves until the next crisis (median number of discharges = 6/rat). After diazepam/naloxone injection, EEG waveforms changed to normal patterns of waking (8–13 Hz alpha rhythm) and drowsiness (4–8 Hz theta rhythm). Tramadol-induced seizures and status epilepticus were completely abolished after treatment with diazepam/naloxone combination.
Figure 4. Rat electroencephalogram traces showing tramadol-induced electrical seizures (A) and its reversal after diazepam/naloxone administration (B).
Discussion
In this study, we demonstrated that naloxone is not an adequate antidote to reverse tramadol-induced neuro-respiratory toxicity in overdose in our rat model. We showed that the diazepam/naloxone combination is the most efficient treatment, able to abolish tramadol-induced seizures and improve ventilation by reversing tramadol-induced increase in TI.
Our rat model reliably reproduced life-threatening tramadol toxicity observed in humans [Citation3–5] including sedation, hypothermia, seizures, and respiratory depression. As previously published [Citation20], our tramadol-overdosed rats exhibited early-onset seizures occurring as soon as 5 min after tramadol injection and disappearing 45–60 min later, accompanied by spike-wave discharges supporting human observations [Citation8–10,Citation13]. Interestingly, in humans, seizures have been reported with therapeutic dosage of tramadol [Citation30] and discordant conclusions were published on the relationship between the risk of seizing and either the ingested dose or the plasma concentration of tramadol [Citation9–11,Citation13], suggesting the existence of individual vulnerability factors.
Naloxone, a competitive μ-opioid-receptor antagonist that reverses all signs of opioid intoxication, is the antidote for opioid overdose [Citation31]. In our rats, naloxone reversed the typical opioid-related increase in TI and its resulting decrease in VM (). Surprisingly, naloxone increased seizures and prolonged their occurrence time (). Experimental data regarding naloxone-related seizurogenic effects are controversial, depending on the animal model and experimental conditions. Most studies demonstrated rather protective effects against both opioid and non-opioid-related seizures. Naloxone markedly attenuated seizures induced by two opioids, meperidine, and normeperidine in mice [Citation32]. Similarly, the prior administration of naloxone (2 mg/kg) attenuated tramadol-exerted seizurogenic activity on pentylenetetrazole-treated mice and tramadol-exerted seizurogenic activity per se, suggesting an opioid-dependent GABA inhibitory pathway to explain tramadol-induced seizures [Citation21]. Interestingly, in a kainic acid-induced two-hit rat seizure model, 12-h infusion of naloxone resulted in dose-dependent protective effects attributed to significant reduction in cytokine production and glial activation [Citation33]. By contrast, few other studies reported no protective and even worsening effects of naloxone on seizure onset. In one rat study, naloxone pretreatment had no effect on seizures development in the course of pentylenetetrazol kindling [Citation34]. In another mouse study, tramadol-induced seizures were increased by naloxone, principally at high-tramadol doses; and this finding was attributed to non-equivalent competition at the opioid receptors between naloxone and the (−) versus (+) tramadol enantiomers [Citation35], known to exhibit different activities at non-opioid targets [Citation30].
In humans, seizure onset has been reported as adverse reactions following naloxone administration in opioid-poisoned patients and attributed to withdrawal symptoms in opioid-dependent individuals, confounding effects of other co-intoxicants or as a consequence of prolonged hypoxia [Citation36,Citation37]. In our rat study, we cannot rule out that naloxone-related increase in seizures was related to hypoxic brain injuries following tramadol-induced respiratory depression evidenced as soon as naloxone increased brain oxygen need [Citation38,Citation39]. At least, our study did not support the ability of naloxone to reverse tramadol-induced seizures, even if, as expected, it efficiently reversed respiratory depression. Interestingly in humans, the only available randomized study showed increased risk of seizures following low-dose naloxone administration to tramadol-poisoned patients [Citation14], consistent with our rat findings.
In tramadol-poisoned patients, seizures are successfully reversed by the usual anticonvulsant drugs including benzodiazepines [Citation4,Citation11,Citation13]. Benzodiazepine co-ingestion was also reported to be protective against tramadol-induced recurrent seizures [Citation13]. Our previous experimental data combining elevated doses of both diazepam and tramadol clearly showed the abolishment of tramadol-induced seizures by diazepam but also the onset of respiratory depression due to this deleterious drug–drug interaction [Citation20], supporting potential issues if diazepam alone is administered in tramadol-poisoned patients. With a pharmacologic dose of diazepam administered to the tramadol-overdosed rats, no respiratory depression was observed despite deepened sedation ( and ), in agreement with the usual safety of benzodiazepine administration for the emergence management of seizures in humans [Citation40]. In our rat model, this diazepam/naloxone combination efficiently abolished both clinical and electrical seizures and reversed tramadol-induced increase in TI. In addition, no enhanced sedation was observed, in contrast to diazepam alone.
Our experimental study has limitations. First, extension to humans of our rat findings should be cautious. However, investigating in vivo the treatment of CNS toxicity in tramadol overdose clearly represents a major strength. We used single doses of each antidote and did not investigate the shift in the dose-response curve with the combination of antidotes. However, naloxone was administered at rather a high-dose regimen able to completely reverse respiratory depression and diazepam at a pharmacologic dose unable by itself to induce significant alteration in the rat ventilation, as requested in the clinical setting with these antidotes administered to obviate tracheal intubation and mechanical ventilation. Consistently, using allometric scaling calculations, we used equivalent doses of 22 mg naloxone, 20 mg diazepam, and 847 mg tramadol in a 70-kg human, that would have been useful to check based on blood concentration assessment in our rats. One additional limitation consisted in the fact that we only recorded the EEG in the naloxone/diazepam group; however, we only aimed to check that no electrical seizing activity persisted with the abolishment of tramadol-induced clinical seizures.
Conclusions
Tramadol-induced CNS toxicity including respiratory depression and seizures can be efficiently reversed by the diazepam/naloxone combination in rats. This combination acts synergistically with diazepam abolishing seizures and naloxone improving ventilation. Our experimental data greatly encourage administering the diazepam/naloxone combination rather than naloxone alone as first-line antidote to reverse CNS toxicity in tramadol-poisoned patients.
Acknowledgments
The authors thank the animal housing facility team of the Pharmacy Faculty in Paris-Descartes University for their technical assistance. They also thank Mrs. Alison Good (Scotland, UK) for her helpful review of the manuscript.
Disclosure statement
The authors declare no competing interests.
Funding
Financial support was provided solely from institutional and/or departmental sources.
References
- Dart RC, Surratt HL, Cicero TJ, et al. Trends in opioid analgesic abuse and mortality in the United States. N Engl J Med. 2015;372:241–248.
- Hawton K, Bergen H, Simkin S, et al. Six-year follow-up of impact of co-proxamol withdrawal in England and Wales on prescribing and deaths: time-series study. PLoS Med. 2012;9:e1001213.
- Spiller HA, Gorman SE, Villalobos D, et al. Prospective multicenter evaluation of tramadol exposure. J Toxicol Clin Toxicol. 1997;35:361–364.
- Marquardt KA, Alsop JA, Albertson TE. Tramadol exposures reported to statewide poison control system. Ann Pharmacother. 2005;39:1039–1044.
- Shadnia S, Soltaninejad K, Heydari K, et al. Tramadol intoxication: a review of 114 cases. Hum Exp Toxicol. 2008;27:201–205.
- Farajidana H, Hassanian-Moghaddam H, Zamani N, et al. Tramadol-induced seizures and trauma. Eur Rev Med Pharmacol Sci. 2012;16:34–37.
- Thundiyil JG, Kearney TE, Olson KR. Evolving epidemiology of drug-induced seizures reported to a poison control center system. J Med Toxicol. 2007;3:15–19.
- Jovanović-Cupić V, Martinović Z, Nesić N. Seizures associated with intoxication and abuse of tramadol. Clin Toxicol.2006;44:143–146.
- Talaie H, Panahandeh R, Fayaznouri M, et al. Dose-independent occurrence of seizure with tramadol. J Med Toxicol. 2009;5:63–67.
- Taghaddosinejad F, Mehrpour O, Afshari R, et al. Factors related to seizure in tramadol poisoning and its blood concentration. J Med Toxicol. 2011;7:183–188.
- Eizadi-Mood N, Ozcan D, Sabzghabaee AM, et al. Does naloxone prevent seizure in tramadol intoxicated patients? Int J Prev Med. 2014;5:302–307.
- Tashakori A, Afshari R. Tramadol overdose as a cause of serotonin syndrome: a case series. Clin Toxicol.2010;48:337–431.
- Shadnia S, Brent J, Mousavi-Fatemi K, et al. Recurrent seizures in tramadol intoxication: implications for therapy based on 100 patients. Basic Clin Pharmacol Toxicol. 2012;111:133–136.
- Farzaneh A, Mostafazade B, Mehrpur O. Seizorugenic effects of low-dose naloxone in tramadol overdose. Iran J Pharmacol Ther. 2012;11:6–9.
- Saidi H, Ghadiri M, Abbasi S, et al. Efficacy and safety of naloxone in the management of postseizure complaints of tramadol intoxicated patients: a self-controlled study. Emerg Med J. 2010;27:928–930.
- Hassanian-Moghaddam H, Farnaghi F, Rahimi M. Tramadol overdose and apnea in hospitalized children, a review of 20 cases. Res Pharm Sci. 2015;10:544–552.
- Stassinos GL, Gonzales L, Klein-Schwartz W. Characterizing the toxicity and dose-effect profile of tramadol ingestions in children. Pediatr Emerg Care. 2017 [Feb 21]. DOI:10.1097/PEC.0000000000001084
- Hassanian-Moghaddam H, Farajidana H, Sarjami S, et al. Tramadol-induced apnea. Am J Emerg Med. 2013;31:26–31.
- Fujimoto Y, Funao T, Suehiro K, et al. Brain serotonin content regulates the manifestation of tramadol-induced seizures in rats: disparity between tramadol-induced seizure and serotonin syndrome. Anesthesiology. 2015;122:178–189.
- Lagard C, Chevillard L, Malissin I, et al. Mechanisms of tramadol-related neurotoxicity in the rat: Does diazepam/tramadol combination play a worsening role in overdose? Toxicol Appl Pharmacol. 2016;310:108–119.
- Rehni AK, Singh I, Kumar M. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent gamma-aminobutyric acid inhibitory pathway. Basic Clin Pharmacol Toxicol. 2008;103:262–266.
- Rehni AK, Singh TG, Singh N, et al. Tramadol-induced seizurogenic effect: a possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn-Schmied Arch Pharmacol. 2010;381:11–19.
- Clarot F, Goullé J, Vaz E, et al. Fatal overdoses of tramadol: is benzodiazepine a risk factor of lethality? Forensic Sci Int. 2003;134:57–61.
- Pirnay SO, Mégarbane B, Borron SW, et al. Effects of various combinations of benzodiazepines with buprenorphine on arterial blood gases in rats. Basic Clin Pharmacol Toxicol. 2008;103:228–239.
- Racine RJ. Modification of seizure activity by electrical stimulation. I. After-discharge thresholdElectroencephalogr Clin Neurophysiol. 1972;32:269–299.
- Bartlett DJ, Tenney S. Control of breathing in experimental anemia. Respir Physiol. 1970;10:384–395.
- Beaune S, Callebert J, Baud FJ, et al. Mechanisms of high-dose citalopram-induced death in a rat model. Toxicology. 2012;302:248–254.
- Haglind E. Effects of continuous naloxone infusion in intestinal ischemia shoch in the rat. Circ Shok. 1992;32:269–299.
- Tallarida R, Murray RB, Area under a curve: Simpson’s rule and trapezoidal rule. In: Tallarida R, Murray RB, editors. Manual of pharmacology calculations with computer programs. New York (NY): Springer-Verlag; 1981. p. 47–49.
- Grond S, Sablotzki A. Clinical pharmacology of tramadol. Clin Pharmacokinet. 2004;43:879–923.
- Boyer EW. Management of opioid analgesic overdose. N Engl J Med. 2012;367:146–155.
- Gilbert PE, Martin WR. Antagonism of the convulsant effects of heroin, d-propoxyphene, meperidine, normeperidine and thebaine by naloxone in mice. J Pharmacol Exp Ther. 1975;192:538–541.
- Yang L, Li F, Ge W, et al. Protective effects of naloxone in two-hit seizure model. Epilepsia. 2010;51:344–353.
- Omrani A, Ghadami MR, Fathi N, et al. Naloxone improves impairment of spatial performance induced by pentylenetetrazol kindling in rats. Neuroscience. 2007;145:824–821.
- Raffa RB, Stone DJ. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J Pharmacol Exp Ther. 2008;325:500–506.
- Buajordet I, Naess AC, Jacobsen D, et al. Adverse events after naloxone treatment of episodes of suspected acute opioid overdose. Eur J Emerg Med. 2004;11:19–23.
- Wermeling DP. Review of naloxone safety for opioid overdose: practical considerations for new technology and expanded public access. Ther Adv Drug Saf. 2015;6:20–31.
- Schaeffer JI, Haddad GG. Ventilatory response to moderate and severe hypoxia in adult dogs: role of endorphins. J Appl Physiol. 1988;65:1383–1388.
- Schlenker EH, Inamdar SR. Effects of naloxone on oxygen consumption and ventilation in awake golden Syrian hamsters. Physiol Behav. 1995;57:655–658.
- Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the Guideline Committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48–61.