
Abstract
Context: Inhalation of sulfur dioxide (SO2) affects the lungs and exposure to high concentrations can be lethal. The early pulmonary response after inhaled SO2 involves tissue injury, acute neutrophilic lung inflammation and airway hyperresponsiveness (AHR). In rats, long-term pulmonary fibrosis is evident 14 days post-exposure as indicated by analysis of collagen deposition in lung tissue. Early treatment with a single dose of dexamethasone (DEX,10 mg/kg) significantly attenuates the acute inflammatory response in airways. However, this single DEX-treatment is not sufficient for complete protection against SO2-induced injuries.
Methods: Female Sprague–Dawley rats exposed to SO2 (2200 ppm, nose-only exposure, 10 min) were given treatments (1, 5 and 23 h after SO2-exposure) with the anti-fibrotic and anti-inflammatory substance Pirfenidone (PFD, 200 mg/kg) or DEX (10 mg/kg) to evaluate whether the inflammatory response, AHR and lung fibrosis could be counteracted.
Results: Both treatment approaches significantly reduced the total leukocyte response in bronchoalveolar lavage fluid and suppressed pulmonary edema. In contrast to DEX-treatment, PFD-treatment reduced the methacholine-induced AHR to almost control levels and partially suppressed the acute mucosal damage whereas multiple DEX-treatment was the only treatment that reduced collagen formation in lung tissue.
Conclusions: To enable an accurate extrapolation of animal derived data to humans, a detailed understanding of the underlying mechanisms of the injury, and potential treatment options, is needed. The findings of the present study suggest that treatments with the capability to reduce both AHR, the inflammatory response, and fibrosis are needed to achieve a comprehensive mitigation of the acute lung injury caused by SO2.
Introduction
Sulfur dioxide (SO2) is extensively produced in numerous industrial applications and hence transported in high volumes as pressurized liquid which may be accidently released upon tanker disruption forming widespread clouds of highly concentrated gas. Inhalation to very high concentrations of SO2 can be immediately dangerous to life or health (IDLH, 100 ppm (NIOSH)) [Citation1]. The mechanisms by which SO2 damages the lungs and the long-term consequences of this injury are not fully understood, but it has been shown that SO2 can cause toxic effects both in the respiratory system (e.g., severe airway obstructions, bronchitis, sloughing of the mucosa of large and small airways) and in the cardiopulmonary system in animals and in humans [Citation2–14].
In a previous study using Sprague–Dawley rats [Citation15], the acute pulmonary response to inhaled SO2 was shown to involve tissue injury, acute neutrophilic lung inflammation and hyperreactivity of large airways. The acute response was followed by a long-term inflammation dominated by macrophages and eosinophils, and an upregulation of the pro-fibrotic cytokine TGF-β1. Pulmonary fibrosis was evident 14 days post-exposure as indicated by histopathological evaluation and increased collagen deposition in lung tissue. Treatment with a single dose of dexamethasone (DEX, 10 mg/kg) to counteract SO2-induced acute inflammation, AHR and long-term lung fibrosis was investigated showing significant down-modulation of the acute inflammatory response in airways. This treatment was, however, insufficient for complete protection against 1) the pulmonary toxicity (AHR and fibrosis), 2) animal body weight-loss and 3) cardiopulmonary effects (DEX-treatment did not reduce the increased heart-weight detected in the SO2-exposed animals). The reducing effects of DEX on AHR were inconsistent, indicating that hyperreactive airways may only partly be linked to acute cellular inflammation [Citation15].
The importance of early anti-inflammatory treatment of chemical-induced lung injury has been implicated in previous studies [Citation16–20]. An ideal treatment against SO2-induced lung injury includes protection against both acute and long-term effects. From the previous animal studies, it is clear that corticosteroids are generally beneficial in reducing adverse physiological effects induced by inflammatory responses, but also that alternative treatment approaches targeting other mechanisms such as pulmonary fibrosis in the pathogenesis are required for comprehensive protection [Citation15,Citation19,Citation20]. It should also be emphasized that the lung injuries caused by toxic compounds depend on the specific interactions (due to physicochemical properties, e.g., water solubility, pH and oxidation strength) between the chemical, and biological tissues, i.e., the mechanisms leading to inflammation and hyperreactive airways are expected to differ between toxic irritant chemicals.
To enable an accurate extrapolation of animal-derived data to humans, a detailed understanding of the underlying mechanisms of the injury may give important information about which treatments to test, with the aim to increase the therapeutic effect in humans exposed to high concentrations of toxic chemical gases. In this study, we employed a rat model of SO2-induced lung injury suitable for studying treatment approaches and evaluated two different drug treatment protocols, both with a potential protection against bronchoconstriction, cellular inflammation and pulmonary fibrosis. An animal ventilator and a forced oscillation method were used to evaluate respiratory mechanics and airway reactivity in response to various treatments, and together with the pulmonary physiology we also described the histopathology, cellular pulmonary inflammation and inflammatory mediators in bronchoalveolar lavage fluid (BALF) and serum.
Materials and methods
Animals
Female Sprague–Dawley rats (8–9 weeks old, Envigo RMS B.V, Netherlands) were used in this study. The care of the animals and the experimental protocols were approved by the regional ethics committee on animal experiments in accordance to Swedish law. Food and water were provided ad libitum. All rats were weighed immediately before the experimental start, and their health condition was monitored during the experimental period. The experiment was divided into two studies:
Study 1. Short-term 24 h post-exposure
The experiment terminated at 24 h (n = 6 rats per group). Animals were allocated into four different groups: 1) control animals were breathing room air (Control (211 ± 5 g), n = 6), 2) SO2-exposed animals given vehicle instead of treatment (SO2 (214 ± 3 g), n = 6), 3) SO2-exposed animals given dexamethasone (SO2+DEX (214 ± 6 g), n = 6) and 4) SO2-exposed animals given pirfenidone (SO2+PFD (214 ± 4 g), n = 6). Data regarding body weight, cells in BALF, inflammatory cytokines, respiratory mechanics and histopathology are shown for study 1.
Study 2. Long-term 14 days post-exposure
The experiment terminated at 14 days (n = 4–6 rats per group). Animals were allocated into four different groups: 1) control animals were breathing room air (Control (211 ± 5 g), n = 6), 2) SO2-exposed animals given vehicle instead of treatment (SO2 (210 ± 4 g), n = 4, (n = 2 (221 ± 1 g) were terminated before 14 days), 3) SO2-exposed animals given dexamethasone (SO2+DEX (213 ± 6 g), n = 6) and 4) SO2-exposed animals given pirfenidone (SO2 + PFD (212 ± 4 g), n = 5). Data regarding body weight and collagen deposition are shown for study 2.
SO2-exposure protocol
Animals were placed in individual nose-only containers (EMMS, UK) and coupled to an inhalation exposure tower (Battelle) providing equal and simultaneous exposure to SO2 (AGA gas, Sweden; compressed gas in gas cylinders: 10 mol-% SO2, 90 mol-% nitrogen). The compressed gas mixture was diluted with air to a final concentration of 2200 ppm. Rats were subjected to a single exposure of SO2–gas mixture for 10 min. The SO2 concentration in the inhalation tower was monitored and the experiments were conducted in a designated fume hood for toxic gas exposures [Citation15]. In the meantime, control animals were retained in their cages while breathing room air for 10 min (not placed in individual containers).
Interventions in study 1 and study 2
The dose of DEX (dexamethasone 21-phosphate disodium salt, Sigma-Aldrich, St. Louis, MO) was selected based on the results of previous studies [Citation15,Citation21,Citation22]. DEX was dissolved in NaCl (0.9% NaCl in water) and a volume of 500 μl was administered intraperitoneally (i.p.) (10 mg/kg) to the rats at three time-points (1 h, 5 h and 23 h) following exposure to SO2. This DEX-dose has also been used in previous studies with rats [Citation15,Citation22].
The dose of PFD (Pirfenidone (Esbriet®), Roche) was modified from Seifirad et al. [Citation23]. PFD was dissolved in NaCl and a volume of 1000 μl was administered as an i.p. treatment (200 mg/kg) at three time-points (1 h, 5 h and 23 h) following exposure to SO2.
The SO2-animals treated with vehicle received 500 μl NaCl i.p. The animals included in the 14-days study, received the same treatment schedule.
Respiratory mechanics
Animals in the 24 h group were weighed and anesthetized with pentobarbital sodium (50 mg/kg, i.p.). Rats were tracheostomized with a gauge cannula and mechanically ventilated with a small animal ventilator (flexiVent™, SCIREQ®) at a frequency of 1.5 Hz (90 breaths/min) and a tidal volume (VT) of 10 ml/kg bw. Rats were paralyzed with pancuronium (0.1 mg/kg bw, i.p.) and a positive end-expiratory pressure of 3 cm H2O was applied. In order to measure AHR, incremental doses of inhaled methacholine (MCh, acetyl-â-methylcholine chloride, Sigma-Aldrich) were given at 5-min intervals. The MCh, diluted in saline to a volume of 20 μl, was given during 10 s as an aerosol (Aeroneb™ PRO, SCIREQ®). Each dose of MCh (0, 5 and 25 mg/ml) was aerosolized without any interference with the ventilation pattern. A more detailed description of the method can be found in previously published papers [Citation15,Citation24].
Serum sampling
Animals were euthanized through exsanguination of the abdominal aorta during anesthesia and blood samples were collected at 24 h or 14 days post-exposure. The blood was centrifuged (15 min, 20 °C, 3000 rpm) and the serum was stored in −70°C until analysis.
Differential cell count in BALF
The lungs were lavaged six times via a tracheal tube with a total volume of 2 ml +23 ml (4 × 5 ml +3 ml) Ca2+/Mg2+ free Hanks’ balanced salt solution (HBSS, Sigma-Aldrich (St. Louis, MO)) and the BALF was centrifuged (10 min, 4 °C, 1500 rpm). After removing the supernatant, the cell pellet was resuspended in 1 ml PBS. The supernatant (the first 2 ml recovered after lavage) was stored in −70°C until analysis of inflammatory mediators. Leukocytes were counted manually in a hemacytometer and 20,000 cells were stained with May-Grünwald-Giemsa reagents (Merck Millipore, VWR International, Sweden), and differential cell counts of pulmonary inflammatory cells (macrophages, neutrophils, lymphocytes and eosinophils) were performed in duplicates using standard morphological criteria and counting 200 cells per slide. A more detailed description of the method can be found in a previously published paper [Citation15].
Inflammatory mediators in BALF and serum
Inflammatory mediators in BALF and serum were analyzed for the presence of IL-1â, IL-6 CINC-1 and MMP-9 using specific assay kits from R&D Systems, Inc. The inflammatory mediators MPO, PAI-1 and IL-18 were measured using specific assay kits from Nordic BioSites, Sweden. The amount of each analyte was analyzed with an ELISA reader (Thermo Scientific Multiskan FC, Thermo Fischer Scientific Oy, Vantaa, Finland) using the software program for the ELISA reader (SkanIt for Multiskan FC 3.1. Inc), referring to the standards added. The three isoforms of TGFβ (TGF-β1-3) were analyzed simultaneously in BALF using a multiplex kit (Bio-Plex Pro TGF-β 3-Plex Immunoassay) and detected with a Bio-Plex™ system (Bio-Rad).
Histopathologic analyses 24 hours post-exposure
Following lavage, the right lung lobe was fixed in 4% paraformaldehyde until paraffin embedding. After embedding, the tissue was cut into 3 μm thick sections and mounted on slides. To assess inflammatory cell infiltration, the sections were deparaffinized, dehydrated and stained with hematoxylin and eosin. Histopathological evaluations of stained sections were performed in a blinded manner using light microscopy for 4 randomly selected animals in each group (n = 4 rats/group). Specifically, scores of 0–4 (0 = normal histology, 4 = most severe injury) were assigned to each of the following characteristics: 1) Pulmonary congestion; 2) Bronchi (mucosal damage, leukocyte infiltration in the bronchi space and the regeneration of the epithelium); 3) Alveoli (leukocyte infiltration in the alveoli space); and 4) Interstitium (interstitial edema and leukocyte infiltration of the peribronchial and perivascular spaces, and interlobular septa).
Analysis of collagen deposition in lung tissue 14 days post-exposure
Following lavage, the left lung lobe was rinsed in HBSS, and frozen in liquid nitrogen and stored in −70°C until analysis. The collagen content of 500 mg lung tissue was measured by a spectrophotometric method, Sircol™ Collagen Assay kit for rats (Biocolor Ltd., Belfast, UK). The assay was performed in duplicate and the mean of two data was determined for each individual sample. A more detailed description of the method can be found in a previously published paper [Citation15].
Statistical analysis
Data are presented as means ± standard error of means (SEM). Statistical significance was assessed by parametric methods using a two-way analysis of variance (ANOVA) or when appropriate, Student’s unpaired t-test or one-way ANOVA followed by Bonferroni post hoc test to determine differences between the untreated SO2-group and the other groups (Control, SO2 + DEX and SO2 + PFD). A statistical result of p < .05 was considered significant. Statistical analysis was carried out and graphs were prepared with GraphPad Prism program (version 6.07 GraphPad software Inc., San Diego, CA).
Results
The observed health effects after SO2-exposure
Rats exposed to SO2 showed significant body weight-loss 24 h post-exposure (p < .001) (). They also displayed an increased number of total leukocytes, neutrophils and eosinophils in BALF compared to the control group (). A significant increase of MMP-9 was observed in BALF of SO2-exposed animals compared to the control group. In contrast, a reduction of MMP-9 was observed in serum of the SO2-exposed animals which differed significantly from the control group (). None of the other analyzed inflammatory mediators differed between the groups.
Figure 1. Measurement of body weight in treated (DEX: dexamethasone (10 mg/kg i.p) or PFD: Pirfenidone (200 mg/kg i.p)) and untreated rats (SO2) exposed to a 10-min exposure to 2200 ppm sulfur dioxide (SO2). Control animals were breathing room-air while the other rats were exposed to SO2. Statistical significant differences between initial weight and weight on termination day within each group are shown. (A) 24 h post-exposure (n = 6 rats per group) and (B) 14-days post-exposure (n = 5–6 rats per group). #Two animals in the 14-day SO2 group were excluded on day 13–14 from the study due to severe decline in general health status (last time-point n = 4) Values indicate means ± SEM, **p < .01, and ***p < .001.
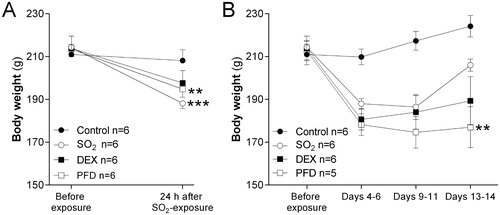
Figure 2. Total cell counts in bronchoalveolar lavage fluid (BALF) from rats exposed to 2200 ppm sulfur dioxide (SO2) at 24 h post-exposure, after treatment with dexamethasone (DEX, 10 mg/kg i.p) or pirfenidone (PFD, 200 mg/kg i.p) at 1 h, 5 h and 23 h. The numbers of (A) total leukocytes, macrophages and neutrophils and (B) eosinophils and lymphocytes are shown. Values indicate means ± SEM. Statistical significant differences compared to untreated SO2-group (vehicle) are shown, *p < .05, **p < .01 and ***p < .001 (n = 6 rats per group).
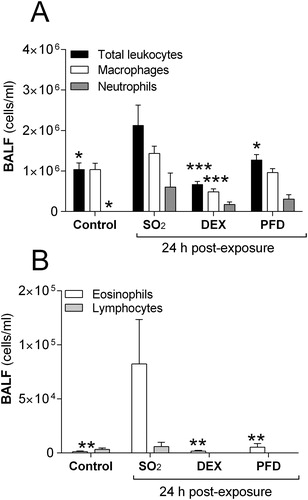
Table 1. Cytokines in bronchoalveolar lavage fluid (BALF) and serum, measured 24 h after a 10-minute exposure of female rats to 2200 ppm sulfur dioxide (SO2), with and without post-exposure treatment (1, 5 and 23 h after SO2-exposure).
Recordings of baseline respiratory parameters showed that hysteresivity (η) and tissue resistance (G) were the only baseline parameters significantly increased 24 h after the SO2-exposure (). Measurements of the MCh-induced changes in respiratory responses showed significant increases in respiratory resistance (RRS), Newtonian resistance (RN), tissue elastance (H), tissue resistance (G) and hysteresivity (η) in SO2-exposed animals compared to control animals ().
Figure 3. Respiratory mechanics in rats 24 h after exposure to 2200 ppm sulfur dioxide (SO2) and treatment (1 h, 5 h and 23 h) with dexamethasone (DEX, 10 mg/kg i.p) or pirfenidone (PFD, 200 mg/kg i.p). Measurements of methacholine (MCh)-induced (A) respiratory resistance, RRS (B) Newtonian resistance, RN (C) tissue resistance, G (D) respiratory elastance, ERS (E) tissue elastance, H and (F) hysteresivity (η) were performed using the Flexivent™. Values indicate means ± SEM. Statistical significant differences compared to untreated SO2-group are shown, *, #p < .05, **, # #, ¤¤p < .01, and ***, # # #, ¤¤ ¤p < .001 (*SO2 vs. control, #SO2 vs. PFD and ¤SO2 vs. DEX), (n = 6 rats per group).
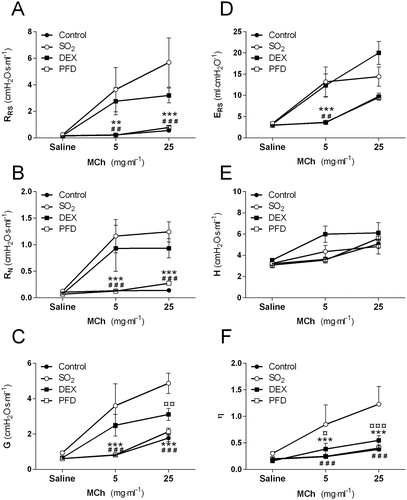
Table 2A. Acute effects of sulfur dioxide (SO2) at 24 h post-exposure. Measurement of baseline respiratory mechanics (without methacholine) in female rats exposed for 10 minutes to 2200 ppm SO2, with and without treatments (1, 5 and 23 h after SO2-exposure).
Based on previous results [Citation15] and results in this study, respiratory mechanics were only measured in a few animals (belonging to the 14-day group) before further analysis was canceled due to the fact that many animals died during anesthesia and surgery, even before being connected to the ventilator. The animals in the 14-day group that could be anesthetized and mechanically ventilated showed no MCh-induced AHR and had equivalent RRS response to control animals (data not shown). This reaction was not seen in animals belonging to the 24 h-group.
Histopathological analysis of the lung showed severe damage of the bronchial mucosa with epithelial degeneration, necrosis and erosion/ulceration 24 h post-exposure (, ). The epithelium was extensively disrupted, and in some animals there were no remaining epithelial cells. The large airways exhibited rich fibrin exudation and cellular infiltration but the intensity of the cellular exudate varied between rats, ranging from no infiltration to severe infiltration. The SO2-group at 24 h showed a marked cell infiltration in the large airways (bronchi) with numerous polymorphs, monocytes, macrophages but low numbers of lymphocytes in the exudates. The alveoli of the SO2-animals showed mild emphysema and leukocytes and there were also signs of mild to severe edema in the interstitium 24 h post-exposure (, ). After 14-days, there was an on-going fibrosis which was confirmed by quantitative measurement of collagen deposition in lung tissue (). Two animals were excluded from the study due to severe decline in general health status; one was terminated on day 11 and the other was terminated on day 13, both rats belonging to the 14-day SO2-group without treatment. The rationale for excluding these animals was that they lost too much weight according to the permission given by the regional ethics committee on animal experiments in accordance to Swedish law. The permission admits studies that induce a lung damage that could only be reconstituted by careful physiological measurements and avoid evoking an extreme lung damage in animals which could be life-threatening or deterioration in visible general functions. Symptoms such as body weight-loss, reduced mobility are expected to occur after exposure within the first days but be of a transitory nature.
Figure 4. Lung tissue sections from (A) healthy control and (B–D) rats 24 h post-exposure to 2200 ppm sulfur dioxide (SO2), (B) without treatment (vehicle) and with (C) dexamethasone (DEX, 10 mg/kg i.p)-treatment, or (D) pirfenidone (PFD, 200 mg/kg i.p)-treatment. Tissue sections were stained with hematoxylin-eosin and photos were taken at 20 x magnification using a light microscope. AL = airway lumen.
Table 2B. Analysis of collagen deposition in female rats at 14 days post-exposure for 10 minutes to 2200 ppm SO2, with and without treatments (1, 5 and 23 h after SO2-exposure).
Table 3. Scoring the main histopathology features in female rats 24 h after a 10-minute exposure to 2200 ppm sulfur dioxide (SO2), with and without post-exposure treatments (1, 5 and 23 h after SO2-exposure).
Multiple DEX-treatments
The initial animal body weight of the DEX-treatment group was not significantly different from the animal body weight at 24 h and 14 days post SO2-exposure (). The accumulation of leukocytes in BALF was significantly reduced after DEX-treatment compared to SO2-exposed rats (). Cell counts showed a decrease in the total number of macrophages and eosinophils, and non-significant decrease of neutrophils in BALF at 24 h post-exposure ().
The baseline alteration in lung configuration at 24 h, which showed heterogeneity after SO2-exposure (hysteresivity, η), was significantly decreased after DEX-treatment, reaching baseline hysteresivity similar to that of control animals (). Other baseline parameters such as the Newtonian resistance (RN) and tissue resistance (G) were significantly decreased after DEX-treatment compared to untreated SO2-exposed rats (). The respiratory recordings showed a significantly decreased MCh-induced hysteresivity (η) and tissue resistance (G) after DEX-treatment, but the other MCh-induced parameters compared to untreated SO2-exposed rats were not different ().
The histopathology evaluation of the lung tissue after DEX-treatment showed that the cellular response in the alveolar region and the interstitium was suppressed despite the severe damage to the bronchial mucosa of similar magnitude to that seen in SO2-animals without treatment 24 h post-exposure. The edema observed in the interstitium of the lung in the untreated SO2-group was undetectable after DEX-treatment at 24 h (, ). DEX-treatment was efficient in the reduction of long-term fibrosis 14 days post-exposure, as shown by quantitative measurement of collagen deposition in lung tissue ().
Multiple PFD-treatments
The animal body weight at 24 h and 14 days post-exposure in the PFD-treatment group was significantly lower than the initial body weight before exposure (p < .001, and p < .01 respectively, ).
The PFD-treatment decreased the total numbers of leukocytes in BALF in comparison to SO2-exposed rats without treatment at 24 h (). The number of eosinophils was significantly reduced while macrophages and neutrophils in BALF showed a non-significant decline after PFD-treatment ().
Compared to DEX-treatment, the alteration in baseline lung configuration, which showed heterogeneity after SO2-exposure, the hysteresivity (ç), was significantly decreased to a level similar to the level of control animals at 24 h (). In addition to hysteresivity, the baseline levels of Newtonian resistance (RN), respiratory resistance (RRS) and tissue resistance (G) were significantly declined after PFD-treatment (). Recordings of the MCh-induced changes in respiratory responses showed significant decreases of the respiratory resistance (RRS), Newtonian resistance (RN), tissue resistance (G) and hysteresivity (η) in the PFD-treated animals compared to untreated SO2-exposed animals at 24 h ().
The histopathology evaluation of the lung tissue at 24 h after PFD-treatment showed a tendency towards less mucosal damage in bronchi compared to SO2-animals without treatment, in addition to suppressed cellular response in alveoli region and the interstitium. The pulmonary edema observed in the SO2-group was undetectable also after PFD-treatment 24 h post-exposure (, ) and there was a tendency towards decreased collagen deposition in lung tissue from PFD-treated rats 14 days post-exposure ().
Discussion
Limited studies in animals have addressed the effects of a single inhaled exposure to high concentrations of SO2. The concentration of SO2 used in this study was considered to be relevant for a scenario of a chemical disaster e.g., at an industrial plant or during transportation, which can result in life-threatening injuries. Rats were subjected to a single exposure of SO2-gas for 10 min (367 ppm·h). The concentration used in this study was non-lethal and lower than a rat study performed by Cohen et al., who showed a 4-h LC50 of 1057 ppm (4227 ppm·h) and a BMCL05 (95% of the exposed mice are expected to survive) of 573 ppm SO2 (2292 ppm·h) [Citation6]. Fortunately, SO2 accidents are rare events but in case of a release of sufficient amounts, an exposure may cause both acute- and long-term health effects in humans [Citation15,Citation25]. It has been reported by us and others that the main acute injuries after SO2-exposure are located to larger bronchi and only minor changes are observed peripherally [Citation6,Citation7,Citation15]. In this study, we examined the protective effects of two different drug treatments in Sprague–Dawley rats after nose-only exposure to SO2, specifically addressing treatments that could provide protection against weight-loss, bronchoconstriction (AHR), cellular inflammation and pulmonary fibrosis.
We have previously demonstrated a rapid recruitment of inflammatory cells and release of inflammatory mediators in BALF and serum following SO2-exposure [Citation15]. These mediators could be used as possible biomarkers to identify the risk of inducing long-term lung injuries. From that study, it is also evident that SO2-induced acute inflammation can develop into prolonged respiratory symptoms similar to those that have been observed in the late-resolving phases of acute respiratory distress syndrome (ARDS) [Citation26] as well as symptoms associated with severe pathological airway obstruction in chronic obstructive pulmonary disease (COPD) patients [Citation27]. A concern with this study is that two out of six animals in the SO2 group had to be terminated before day 14, since those animals showed a severe decline in general health condition observed as major body weight-loss and air entrapment in the GI-tract, and thereby skewing the results at day 14. BALF, serum and lungs of those animals are not included in the analyses and for ethical reasons the investigation of lethal effects are not further studied. The underlying mechanism of how SO2 exerts its toxic effects is, however, unclear [Citation15].
A single dose of DEX administered early after chemical exposure seems to provide some protection against acute symptoms in animals, such as reduced number of inflammatory cells in BALF, diminished collagen deposition and MCh-induced AHR [Citation15], and here we show that multiple early treatments with DEX (within one day) also can protect against weight-loss and reduction in tissue resistance. In comparison with a previous study, these results show that even with multiple DEX-treatments, there is no significant improvement in protection against the AHR caused by SO2 in the central larger airways but a reduction in tissue resistance in peripheral airways. DEX does not have the complete ability to prevent the acute response after SO2-exposure such as AHR, but the insufficient therapeutic effect may to a large extent depend on the dose of DEX. Since DEX has been tested in other animal models with significant results, we used similar doses for easier comparisons of results [Citation15,Citation21–23]. Contrary to our findings, high-dose single treatment with DEX (100 mg/kg) on other chemical-induced lung injuries has proven ineffective, e.g., in phosgene-induced lung injury [Citation28].
Previously, we have shown that a single dose of DEX could reduce pulmonary fibrosis in lung tissue to some extent [Citation15]. In the present study, multiple treatment with DEX significantly counteracted also the long-term effects such as the pulmonary fibrosis shown 14 days post-exposure. Similar result of DEX-treatment has been shown to reduce the level of pulmonary fibrosis following inhalation exposure to chlorine in mice and following exposure to an alkylating nitrogen mustard analog (melphalan) in mice [Citation18,Citation19]. Altogether these exposure models support the notion that corticosteroids are generally beneficial, at least to some extent, in the protection against adverse physiological effects induced by inflammatory responses, but also that alternative treatment approaches targeting other mechanisms in the pathogenesis are required for complete elimination of adverse effects such as infiltration of leukocytes, AHR and pulmonary fibrosis. Based on clinical data it has previously been pointed out that the efficiency of corticosteroid treatment following human exposure to lung-damaging agents are inconclusive and that there is a lack of controlled studies following high-dose exposure to lung-damaging agents in humans [Citation29]. Although results from animal studies cannot be directly extrapolated to humans, these studies are of utmost importance to finding pathways and understanding the mechanism behind the complex interactions in the body.
PFD and Nintedanib are two approved medications used for the treatment of idiopathic pulmonary fibrosis (IPF). In this study PFD was used, as it has both anti-inflammatory and anti-fibrotic properties and acts as an important regulator of synthesis of cytokines, growth factors and collagen, and also attenuates pulmonary fibrosis by slowing functional decline and disease progression [Citation30–36]. However, the management of IPF with PFD-treatment does not cure the disease but slow down the natural course of the disease. IPF has some similarities with the acute effects of SO2-induced ALI, both appear to be driven by a mechanism that promote fibroblast recruitment and proliferation leading to a diffuse parenchymal lung disease, resulting in loss of lung function [Citation15,Citation34]. PFD-treatment has shown to have protective effects in other experimental lung disease-models induced by e.g., paraquat, bleomycin and allergen-induced asthma [Citation30,Citation37–39]. In addition to reducing pulmonary fibrosis, PFD-treatment has also been shown to attenuate fibrosis in the liver, heart and kidney [Citation34].
In the present study, it was found that PFD-treatment could reduce the AHR to levels similar to control-levels and suppressed the lung tissue inflammation and especially reduced the observed mucosal damage in larger airways, however, multiple PFD-treatment could not counteract the long-term effects such as the pulmonary fibrosis (14 days post-exposure) or protect against animal body weight-loss as a result of SO2-exposure. In the previous SO2-study, animals had on-going fibroblastic activity and fibrosis 14 days after exposure and after 28 days, animals were almost recovered. The findings in the animal model of long-term SO2-induced lung injury, however, show more resemblance with the late resolving phases in ARDS rather than IPF [Citation15,Citation21,Citation26,Citation40].
Combination therapy has proven to be successful in clinical studies and patients with advanced IPF had favorable outcomes after combined treatment with e.g., PFD and N-acetylcysteine (NAC) [Citation41]. PFD-treatment has also been proven to produce synergistic therapeutic effects together with inhalation of the antioxidant lecithinized superoxide dismutase in a bleomycin-induced pulmonary fibrosis mouse model [Citation42]. However, there are studies that provide evidence against the use of combination treatment, e.g., increased risks of death and hospitalization were observed in IPF-patients treated with a combination of prednisone, azathioprine, and NAC [Citation43]. The therapeutic effects of combining multiple treatments with NAC (200 mg/kg i.p) and DEX (10 mg/kg i.p) in the SO2 rat model described in the present study was not sufficient to reduce the SO2-induced lung injury any further than DEX alone (unpublished, data not shown). When given in combination, a protective effect on AHR and a significant reduction in inflammatory cells (neutrophils) has previously been observed in a Cl2-induced ALI mouse model [Citation20].
It must be emphasized that care should be taken when designing drug-treatment protocols. The method of administering treatments in this study has the advantage that it is simple and rapid but a limitation was that both DEX and PFD were administrated systemically via i.p. injection when there are oral and intravenous alternatives. DEX has been used systemically in previous studies and has proven to have a beneficial effect when given as an i.p injection within one hour after exposure in e.g., chlorine, endotoxin, or melphalan exposure models [Citation18,Citation19,Citation44]. A rapid absorption of the active substance is of importance and PFD was therefore given via the i.p route [Citation23] to enable comparisons with the effects of DEX-treatment. The injuries after exposure to toxic industrial chemicals are rapidly induced since the chemicals immediately react to the lung tissue and start an immune response [Citation45,Citation46], thus the most optimal solution was to treat SO2-induced ALI by i.p. administration. The rationale of using different time-points was that previous studies have shown that early treatment with a fast-acting drug is important to adequately treat chemical-induced ALI. The animals then received further maintenance doses after four hours (5 h time-point) and 23 hours similar to the recommendations in the medical guidelines for treatment of chemical-induced lung injury in humans.
Conclusions
Both treatment-protocols reduced the acute SO2-induced inflammatory cellular response in airways, but only PFD-treatment diminished the MCh-induced AHR while DEX was the only treatment that showed efficacy at reducing the long-term effects monitored 14 days post-exposure.
Due to lack of effective therapies that have the capability to stop the progression of chemical-induced ALI, there is a need for innovative treatment strategies and novel therapies. Few drugs may have the ability to completely stop the aggravating ALI and the therapeutic effects are highly dependent on the dose and timing of the treatment administered. It should also be emphasized that the lung injuries caused by toxic industrial chemicals are dependent on the specific chemical reactivity and that the mechanisms leading to inflammation and hyperreactive airways most likely differ depending on chemical agent. Given the different mechanisms, it cannot be assumed that one certain treatment protocol beneficially applies to all types of chemical-induced ALI. Collectively, taking the findings of the present study into consideration, we suggest treatments that have the capability to reduce both the AHR, the inflammatory response, and the fibrosis caused by SO2 or other industrial gases with pulmonary toxicity.
Acknowledgements
Karin Wallgren is gratefully acknowledged for invaluable help with the animal experiments and Dr. Ulrika Bergström and Dr. Åsa Gustafsson for thankful suggestions while writing the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Wang XB, Du JB, Cui H. Sulfur dioxide, a double-faced molecule in mammals. Life Sci. 2014;98:63–67.
- Meng Z, Qin G, Zhang B, et al. Oxidative damage of sulfur dioxide inhalation on lungs and hearts of mice. Environ Res. 2003;93:285–292.
- Meng Z. Oxidative damage of sulfur dioxide on various organs of mice: sulfur dioxide is a systemic oxidative damage agent. Inhal Toxicol. 2003;15:181–195.
- Woodford DM, Coutu RE, Gaensler EA. Obstructive lung disease from acute sulfur dioxide exposure. Respiration 1979;38:238–245.
- Charan NB, Myers CG, Lakshminarayan S, et al. Pulmonary injuries associated with acute sulfur dioxide inhalation. Am Rev Respir Dis. 1979;119:555–560.
- Cohen HJ, Drew RT, Johnson JL, et al. Molecular basis of the biological function of molybdenum: the relationship between sulfite oxidase and the acute toxicity of bisulfite and SO2. Proc Natl Acad Sci USA 1973;70:3655–3659.
- Stratmann U, Lehmann RR, Steinbach T, et al. Effect of sulfur dioxide inhalation on the respiratory tract of the rat. Zentralbl Hyg Umweltmed. 1991;192:324–335.
- Meng Z, Geng H, Bai J, et al. Blood pressure of rats lowered by sulfur dioxide and its derivatives. Inhal Toxicol. 2003;15:951–959.
- Rabinovitch S, Greyson ND, Weiser W, et al. Clinical and laboratory features of acute sulfur dioxide inhalation poisoning: two-year follow-up. Am Rev Respir Dis. 1989;139:556–558.
- Atkinson DA, Sim TC, Grant JA. Sodium metabisulfite and SO2 release: an under-recognized hazard among shrimp fishermen. Ann Allergy. 1993;71:563–566.
- Harkonen H, Nordman H, Korhonen O, et al. Long-term effects of exposure to sulfur dioxide. Lung function four years after a pyrite dust explosion. Am Rev Respir Dis. 1983;128:890–893.
- Huber AL, Loving TJ. Fatal asthma attack after inhaling sulfur fumes. JAMA 1991;266:2225
- Skalpe IO. Long-term effects of sulphur dioxide exposure in pulp mills. Br J Ind Med. 1964;21:69–73.
- Brooks SM, Weiss MA, Bernstein IL. Reactive airways dysfunction syndrome (RADS). Persistent asthma syndrome after high level irritant exposures. Chest. 1985;88:376–384.
- Wigenstam E, Elfsmark L, Bucht A, et al. Inhaled sulfur dioxide causes pulmonary and systemic inflammation leading to fibrotic respiratory disease in a rat model of chemical-induced lung injury. Toxicology 2016;368–369:28–36.
- Dinis-Oliveira RJ, Duarte JA, Remiao F, et al. Single high dose dexamethasone treatment decreases the pathological score and increases the survival rate of paraquat-intoxicated rats. Toxicology 2006;227:73–85.
- Wang J, Zhang L, Walther SM. Inhaled budesonide in experimental chlorine gas lung injury: influence of time interval between injury and treatment. Intensive Care Med. 2002;28:352–357.
- Jonasson S, Wigenstam E, Koch B, et al. Early treatment of chlorine-induced airway hyperresponsiveness and inflammation with corticosteroids. Toxicol Appl Pharmacol. 2013;271:168–174.
- Wigenstam E, Jonasson S, Koch B, et al. Corticosteroid treatment inhibits airway hyperresponsiveness and lung injury in a murine model of chemical-induced airway inflammation. Toxicology 2012;301:66–71.
- Wigenstam E, Koch B, Bucht A, et al. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology. 2015;328:40–47.
- Luo S, Pauluhn J, Trubel H, et al. Corticosteroids found ineffective for phosgene-induced acute lung injury in rats. Toxicol Lett. 2014;229:85–92.
- Wigenstam E, Elfsmark L, Koch B, et al. Acute respiratory changes and pulmonary inflammation involving a pathway of TGF-beta1 induction in a rat model of chlorine-induced lung injury. Toxicol Appl Pharmacol. 2016;309:44–54.
- Seifirad S, Keshavarz A, Taslimi S, et al. Effect of pirfenidone on pulmonary fibrosis due to paraquat poisoning in rats. Clin Toxicol. 2012;50:754–758.
- Gustafsson A, Jonasson S, Sandstrom T, et al. Genetic variation influences immune responses in sensitive rats following exposure to TiO2 nanoparticles. Toxicology. 2014;326:74–85.
- Bjornham O, Grahn H, von Schoenberg P, et al. The 2016 Al-Mishraq sulphur plant fire: source and health risk area estimation. Atmos Environ. 2017;169:287–296.
- Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 2016;140:345–350.
- Herfs M, Hubert P, Poirrier AL, et al. Proinflammatory cytokines induce bronchial hyperplasia and squamous metaplasia in smokers: implications for chronic obstructive pulmonary disease therapy. Am J Respir Cell Mol Biol. 2012;47:67–79.
- Liu F, Pauluhn J, Trubel H, et al. Single high-dose dexamethasone and sodium salicylate failed to attenuate phosgene-induced acute lung injury in rats. Toxicology. 2014;315:17–23.
- de Lange DW, Meulenbelt J. Do corticosteroids have a role in preventing or reducing acute toxic lung injury caused by inhalation of chemical agents?. Clin Toxicol. 2011;49:61–71.
- Knuppel L, Ishikawa Y, Aichler M, et al. A novel antifibrotic mechanism of nintedanib and pirfenidone. Inhibition of collagen fibril assembly. Am J Respir Cell Mol Biol. 2017;57:77–90.
- Lopez-de la Mora DA, Sanchez-Roque C, Montoya-Buelna M, et al. Role and new insights of pirfenidone in fibrotic diseases. Int J Med Sci. 2015;12:840–847.
- Fujimoto H, Kobayashi T, Azuma A. Idiopathic pulmonary fibrosis: treatment and prognosis. Clin Med Insights Circ Respir Pulm Med. 2015;9:179–185.
- Günther A, Korfei M, Mahavadi P, et al. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur Respir Rev. 2012;21:152–160.
- Schaefer CJ, Ruhrmund DW, Pan L, et al. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20:85–97.
- Yu W, Guo F, Song X. Effects and mechanisms of pirfenidone, prednisone and acetylcysteine on pulmonary fibrosis in rat idiopathic pulmonary fibrosis models. Pharm Biol. 2017;55:450–455.
- Robalo-Cordeiro C, Campos P, Carvalho L, et al. Idiopathic pulmonary fibrosis in the era of antifibrotic therapy: searching for new opportunities grounded in evidence. Rev Port Pneumol (2006). 2017;23:287–293.
- Ma J, Sun F, Chen B, et al. Tissue metabolic changes for effects of pirfenidone in rats of acute paraquat poisoning by GC-MS. Toxicol Ind Health. 2017;33:887–900.
- Mansoor JK, Decile KC, Giri SN, et al. Influence of pirfenidone on airway hyperresponsiveness and inflammation in a Brown-Norway rat model of asthma. Pulm Pharmacol Ther. 2007;20:660–668.
- Hirano A, Kanehiro A, Ono K, et al. Pirfenidone modulates airway responsiveness, inflammation, and remodeling after repeated challenge. Am J Respir Cell Mol Biol. 2006;35:366–377.
- Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163.
- Sakamoto S, Muramatsu Y, Satoh K, et al. Effectiveness of combined therapy with pirfenidone and inhaled N-acetylcysteine for advanced idiopathic pulmonary fibrosis: a case-control study. Respirology. 2015;20:445–452.
- Tanaka K, Azuma A, Miyazaki Y, et al. Effects of lecithinized superoxide dismutase and/or pirfenidone against bleomycin-induced pulmonary fibrosis. Chest. 2012;142:1011–1019.
- Raghu G, Anstrom KJ, King TE, Jr, et al. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366:1968–1977.
- Rocksen D, Lilliehook B, Larsson R, et al. Differential anti-inflammatory and anti-oxidative effects of dexamethasone and N-acetylcysteine in endotoxin-induced lung inflammation. Clin Exp Immunol. 2000;122:249–256.
- Hemström P, Larsson A, Elfsmark L, et al. L-alpha-phosphatidylglycerol chlorohydrins as potential biomarkers for chlorine gas exposure. Anal Chem. 2016;88:9972–9979.
- Elfsmark L, Ågren L, Akfur C, et al. 8-Isoprostane is an early biomarker for oxidative stress in chlorine-induced acute lung injury. Toxicol Lett. 2018;282:1–7.