Abstract
Introduction. Hydrofluoric acid (HF) is a weak inorganic acid used for etching and as rust remover. Systemic toxicity is manifested as ventricular dysrhythmias. The mechanisms for these dysrhythmias are not well elucidated. Case report. An 82-year-old woman ingested 8 ounces of 7% HF. Shortly after emergency department (ED) arrival, she became pulseless, developing recurrent ventricular dysrhythmias. She was defibrillated 17 times and received several doses of calcium, magnesium, and lidocaine. After three hours, she returned to sustained NSR. She was discharged home after four days. Discussion. The electrocardiographic findings in this patient demonstrate hypocalcemia, which has been implicated as the culprit in HF-induced arrhythmias. However, despite correction of the hypocalcemia, the ventricular arrhythmias persisted. The proposed mechanisms of systemic HF toxicity and the relevant literature are discussed. Conclusion. Ventricular dysrhythmias due to HF toxicity seem to be independent of either hypocalcemia or hyperkalemia. Systemic toxicity after ingestions may be delayed and precipitous.
Introduction
Hydrofluoric acid (HF) is a weak inorganic acid. Exposures to HF occur in the semiconductor industry, where it is used to etch glass and metal. In addition, household products marketed as rust removers commonly contain HF (Citation1,Citation2). The concentration of HF in household products is usually less than 10%, while industrial products may have concentrations as high as 70% (Citation3). Acute systemic toxicity from HF occurs through dermal exposure to highly concentrated solutions, and through ingestion (Citation1,Citation2,Citation4–11). Ingestion of the sodium salt form of HF (NaF), which was historically used as a pesticide, resulted in many deaths (Citation12–16). Death from both of these formulations is associated with refractory ventricular dysrhythmias.
The mechanisms by which fluoride induces these ventricular dysrhythmias are debated. Fluoride is the most electronegative element, and the fluoride (F-) anion readily binds physiologically important cations such as magnesium and calcium. Numerous reports of severe and fatal fluoride poisoning have demonstrated profound hypocalcemia, many with typical electrocardiographic (ECG) changes of QT prolongation (). Discussions of the management of fluoride toxicity have emphasized the importance of hypocalcemia and its prompt treatment to avoid cardiac complications (Citation1,Citation2,Citation11,Citation17–22). Hyperkalemia, which may be delayed in onset, has also been proposed as an etiology of fluoride-associated ventricular dysrhythmias (Citation4,Citation18,Citation23–27). Hyperkalemia has been documented in a number of cases of human poisoning with fluoride (Citation22,Citation27) and in dog studies of fluoride poisoning (Citation24,Citation25). In vitro studies of erythrocyte suspensions show that sodium fluoride induces potassium efflux from erythrocytes (Citation18,Citation23–26,Citation28). Due to uncertainty regarding the relative contributions of hypocalcemia and/or hyperkalemia to the development of ventricular dysrhythmias, appropriate therapies remain controversial.
Table 1. Initial potassium and calcium levels in patients with life-threatening toxicity associated with exposure to fluoride compounds
We report a case of a woman who experienced acute systemic fluoride toxicity following ingestion of a rust removal product containing 3% HF. This resulted in recurrent ventricular dysrhythmias from which she was successfully resuscitated. The significance of this patient's electrocardiographic and laboratory studies are discussed in the context of prevailing theories of the mechanisms of toxicity from hydrofluoric acid.
Case report
An 82-year-old woman was brought by ambulance to the Emergency Department (ED) six hours after ingesting approximately 8 ounces of Whisk© Brand rust stain remover (2.85-3% HF) in an attempt to harm herself. This product also contained water and 0.05% dentonium benzoate as a bittering agent. Before arrival the patient experienced epigastric pain and had one episode of non-bloody emesis.
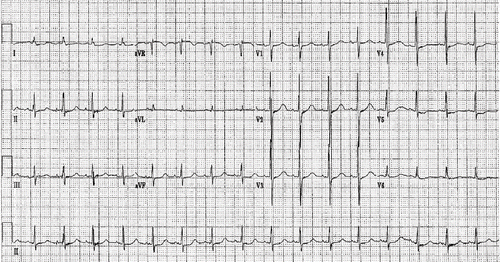
Vital signs upon arrival to the ED were blood pressure 129/82 mm Hg, pulse 108/minute, respiratory rate 24/minute and temperature 95.8° F. There were no oropharyngeal burns. The rest of the examination was unremarkable. The initial electrocardiogram (ECG), obtained within five minutes after her arrival to the ED, is shown in . It demonstrates normal sinus rhythm with a rate of 98 beats/minute; a QRS axis of +33; PR interval 134 msec (normal 120–200 msec), and QRS width 80 msec (normal 60–100 msec). The corrected QT interval was prolonged at 477 msec (normal ≤400 msec).
Fig. 1. Pre-arrest ECG.
Approximately 30 minutes after this first ECG during insertion of a urine foley catheter the patient developed pulseless ventricular tachycardia and ventricular fibrillation (). Normal sinus rhythm (NSR) was reestablished after defibrillation, but she lapsed back into the wide-complex rhythm. She was endotracheally intubated three minutes after the onset of ventricular tachycardia. Normal sinus rhythm was established after four defibrillation attempts (200 J, 300 J, 360 J, 360 J) delivered over 12 minutes. The subsequent period of NSR was sustained for one hour and forty minutes. A second ECG, done 41 minutes after the first, is depicted in .
Fig. 2. a) strip showing ventricular dysrhythmias and b) continuation of same strip with evidence of torsades de pointes.
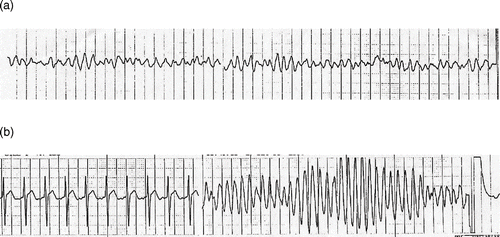
Fig. 3. Post-resuscitation ECG.
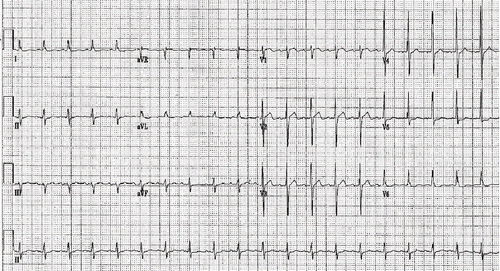
Pharmacologic interventions during this time were: intravenous (IV) boluses of three (10 cc) ampules of 10% CaCl2 solution (total 3 g); epinephrine 1 mg IV; atropine 1 mg IV, versed 2 mg IV, lidocaine 150 mg bolus and then infusion started at 2 mg/min. The ECG taken after this initial resuscitation is shown in . The corrected QT interval decreased to 408 msec.
The first set of laboratory tests, drawn after the initial episode of ventricular arrhythmia, was as follows: white blood cell count (WBC) 15 × 103/uL, hemoglobin 15 g/dL, hematocrit 44%, PLT 275 × 103/μL, sodium 142 mEq/L, potassium 4.2 mEq/L, chloride 110 mEq/L, bicarbonate 19 mEq/L, blood urea nitrogen (BUN) 20 mg/dL, creatinine 0.9 mg/dL, magnesium 1.7 mEq/L, anion gap 13. Arterial blood gas was performed after intubation on 100% FiO2 showed the following: pH 7.47, pCO2 24 mmHg, pO2 137 mmHg, BE 3.8, O2 sat 99.5%. Serum and urine tests for aspirin, acetaminophen, ethanol, and common drugs of abuse showed only the presence of benzodiazepines. The initial calcium level (drawn after IV treatment with 3 (10-cc or 1 gm each) ampules of 10% calcium chloride) was 11.4 mg/dl. A pre-resuscitation calcium level was not drawn.
The patient had recurrent episondes of polymorphic ventricular tachycardia (VT) over a period of 70 minutes, requiring an additional 13 DC shocks at 360 J and additional administrations of calcium and magnesium ( and ). Within 3.5 hours of her arrival she received a total of 8 grams of CaCl2, 3.5 grams of MgSO4, 1 mg each of epinephrine and atropine, and an IV bolus of lidocaine 150 mg followed by a continuous infusion at 2 mg/minute. Following one final electrical counter-shock at approximately three hours after presentation, she remained in normal sinus rhythm throughout the rest of her hospital stay. The lidocaine infusion was weaned and discontinued 12 hours after her initial cardiac arrest. She received approximately 1200 mg of lidocaine over the 12-hour period.
Serum and urine fluoride ion levels drawn eight hours after the initial cardiac arrest were measured using ion chromatography for serum and ion-specific electrode for urine specimens. The serum fluoride level was 2 mg/L (normal less than 0.2 mg/L), and the simultaneous urine fluoride was 72.46 mg/L (normal 0.2–3.2 mg/L). Serial calcium levels ranged from 8.1 mg/dl up to 19.5 mg/dl throughout the hospital stay. Repeated measurement of serum potassium showed levels that ranged from 4.2 mEq/L to a high of 5.2 mEq/L within the first eight hours from arrival; thereafter potassium levels were normal. Her first potassium level, sent after the first episode of ventricular arrhythmia, was 4.7 mEq/L.
She developed upper gastrointestinal bleeding on the second day that responded to initiation of a proton pump inhibitor. She was successfully extubated on the second hospital day and transferred to a psychiatric inpatient unit on hospital day 4 with intact neurologic function.
Discussion
Exposure to various fluoride-containing compounds has been associated with both hypocalcemia and hyperkalemia. Admission evaluation of our patient showed ECG changes consistent with hypocalcemia, but not hyperkalemia. The potassium levels were only slightly elevated (4.7–5.2 mEq/L). Administration of calcium resulted in a decrease in the QT interval and a period of sustained NSR. Despite normal to supra-normal calcium levels throughout resuscitation, repeated episodes of ventricular fibrillation (VF) and polymorphic VT recurred for hours.
Fluoride compounds disrupt many cellular processes. The toxicities of different fluoride compounds are likely related to their individual chemical properties, which may affect both accessibility to tissues as well as the potential to release free fluoride anion. Potassium fluoroborate and potassium hexafluorophosphate have no systemic toxicity, presumably due to tight binding of the fluoride anion (Citation20). In contrast, sodium fluoroacetate is a highly toxic compound that penetrates mitochondria and disrupts oxidative metabolism through inhibition of the Krebs cycle enzyme aconitase. (Citation29). Sulfuryl fluoride, an insecticide compound used in fumigation, causes rapid incapacitation, respiratory depression, and convulsions after inhalational exposure (Citation30, Citation31). Fluoride compounds also interfere with acetylcholinesterase and carbohydrate metabolism, but a contribution of these mechanisms to toxicity is not clear (Citation20).
Dysrhythmias and sudden cardiac death are well described following poisoning with HF and NaF, but the precise mechanisms underlying the cardiovascular toxicity of these compounds are not well-established. Severe and fatal poisonings by both NaF and HF have been associated with profound hypocalcemia in numerous published case reports (Citation1,Citation2,Citation5–11,Citation15,Citation16,Citation22,Citation34,Citation38,Citation39). In addition to direct binding and precipitation of free ionized calcium, potassium abnormalities have been described with sodium fluoride toxicity. These abnormalities are postulated to result from at least two other cellular actions of fluoride anion: 1) fluoride-mediated inactivation of the Na+/K+ ATPase, contributing to the accumulation of intracellular sodium and extracellular potassium; and 2) activation of a Na+/Ca++ ion exchanger that results in the intracellular accumulation of calcium, triggering a calcium-dependent potassium channel which in turn mediates potassium efflux (Citation4,Citation26). The addition of sodium fluoride to in vitro suspensions of red blood cells induces a delayed efflux of potassium from the erythrocytes (Citation18,Citation23). This efflux is more marked when propranolol is added to the red cells and is diminished by the presence of quinidine, observations attributed to the respective effects of these agents on calcium-dependent potassium channels in the red cell membrane (Citation24). A canine study demonstrated that concomitant administration of quinidine with sodium fluoride prevented ventricular dysrhythmias, but this study did not report potassium levels in the quinidine-treated animals (Citation24). Amiodarone attenuated the development of hyperkalemia due to sodium fluoride in a recent in vitro study using an erythrocyte model (Citation26).
Negative inotropic effects of sodium fluoride have also been demonstrated in animal models, but it is unclear whether this is a primary effect of the fluoride ion or a secondary effect from electrolyte derangements. Strubelt et al. demonstrated a concentration-dependent negative inotropic effect of NaF in isolated rat atria (Citation35). Plasma calcium declined in the presence of fluoride in this set of experiments, and supplementation of calcium to levels far above physiological levels still failed to reverse this toxic effect. Gaugl and Wooldridge demonstrated a decrease in CVP but no changes in cardiac output or contractility in a non-lethal canine model of NaF toxicity (Citation36). Peaked T-waves that suggested hyperkalemia were noted in the fluoride- treated animals, however neither of these studies reported potassium levels.
Most case reports and reviews attribute the toxicity of both HF and NaF to the fluoride anion per se and treat these two exposures as indistinguishable in their mechanisms of toxicity. However, our literature search showed that clinically consequential levels of hyperkalemia have been conclusively demonstrated only in cases of poisoning with sodium fluoride or sodium silicofluoride (Citation22,Citation27). Studies in canine models also utilized sodium fluoride (Citation24,Citation25). Three cases of HF-associated dermal injuries have documented or inferred elevations in serum potassium levels, but the association with cardiac dysfunction in each case is questionable. Burke (Citation37) reported a laboratory accident that resulted in dermal HF and possibly silver fluoroborate exposure. The patient developed hyperkalemia and acidosis but did not experience ventricular dysrhythmias. Takese et al. described a patient with fatal dermal exposure from HF, whose post-arrest electrolytes demonstrated both hyperkalemia and hypocalcemia. The patient's pre-arrest course is not reported (Citation34). One case report of HF attributed VF to hyperkalemia based on the demonstration of a wide complex tachycardia that preceded death, but in this case the only potassium level reported was normal (Citation4). summarizes the published human cases of toxicity from fluoride-containing compounds.
It is important to note that NaF and HF are separate compounds with unique chemical properties. While NaF is highly soluble and readily liberates the fluoride ion, HF is a weak acid that does not readily dissociate (Citation32). These biochemical differences may account for varying toxic effects of the two compounds. The available human literature and experimental studies in dogs suggest that significant hyperkalemia may complicate NaF poisoning, but there is at present little data to suggest that hyperkalemia is an important mechanism of myocardial toxicity in cases of poisoning with HF or other fluoride-containing compounds.
Although administration of calcium did not prevent dysrhythmias, our patient survived, as have numerous other reported patients who had documented normalization of calcium during resuscitation (Citation1,Citation6,Citation9–11,Citation22). In our patient, the inability to prevent the occurrence of VF and VT by careful correction of hypocalcemia suggests that HF, or possibly the fluoride anion itself, exerts some independent dysrhythmogenic effect unrelated to measurable disturbances of potassium and calcium. It is noteworthy that there was no laboratory or electrocardiographic demonstration of hyperkalemia in this case, which is consistent with the lack of published evidence of hyperkalemia with HF poisoning. It is possible that because this patient's serum electrolytes were not drawn until after her first cardiac arrest that we may have missed an episode of transient hyperkalemia, but the ECG was not suggestive of typical hyperkalemic changes and she did not receive any specific potassium-lowering treatments before the normal serum potassium level was drawn. Based on the limited but apparently normal serum electrolyte results, it is tempting to conclude that the recurrence of torsades de pointes despite aggressive calcium therapy was related to a direct toxic effect of HF or the fluoride ion on the myocardium.
Our case supports some value of calcium administration in patients poisoned with hydrofluoric acid. With regard to NaF poisoning, aggressive calcium administration also seems rational, as it can treat both hypocalcemia and hyperkalemia. As a therapy aimed specifically at preventing hyperkalemia, the reported ability of quinidine to prevent death in a dog model of fluoride poisoning (Citation24) suggests that this agent, as well as amiodarone, deserve further evaluation of their protective effects on the myocardium in fluoride poisoning. Experiments demonstrating the attenuation of hyperkalemia by insulin, amiodarone, or quinidine in in vitro erythrocyte suspensions are provocative, but more direct evidence of myocardial protection from these agents is necessary in order for them to be considered clinically as antidotes (Citation18,Citation23–26). Another important question is whether the use of bicarbonate for empirical treatment of hyperkalemia might exacerbate hypocalcemia due to the recognized effects of alkalinization on ionized serum calcium. One study in a rat model of sodium fluoride poisoning suggested that pre-alkalinization compared with pre-acidification led to higher serum fluoride levels, lower concentrations of fluoride anion in heart and skeletal muscle, and enhanced renal clearance of fluoride. The alkalinized animals lived twice as long (Citation33). However, further studies are needed to clarify the role of alkalinization in the setting of fluoride toxicity.
Conclusion
We believe that the recurrent ventricular arrhythmias in our patient with systemic HF toxicity occurred by a mechanism that was independent of hypocalcemia, as they occurred despite demonstrated normalization of serum calcium. Despite repeated episodes of VF and polymorphic VT, our patient did not manifest significant hyperkalemia. This prolonged toxicity may have been related to a direct toxic effect of HF or the fluoride ion on the myocardium. The delayed and sudden onset of life threatening symptoms in this case highlights the importance of intensive monitoring in asymptomatic patients after an HF ingestion.
Subsequent investigation of the literature showed that although hyperkalemia is likely associated with NaF poisoning, possibly due to release of potassium from erythrocytes. No patient with HF poisoning has been convincingly shown to develop hyperkalemia. The differences between the toxic effects of these two compounds may be related to differences in their biochemical properties, with regard to the availability of the fluoride anion or the compound itself.
References
- Yamaura K, Kao B, Iimori E, Urakami H, Takahashi S. Recurrent ventricular tachyarrhythmias associated with QT prolongation following hydrofluoric acid burns. J Toxicol Clin Toxicol 1997; 35(3)311–313
- Klasner AE, Scalzo AJ, Blume C, Johnson P. Marked hypocalcemia and ventricular fibrillation in two pediatric patients exposed to a fluoride-containing wheel cleaner. Ann Emerg Med 1996; 28(6)713–718
- Klauder JV, Shelanski L, Gabriel K. Industrial uses of compounds of fluorine and oxalic acid: Cutaneous reaction and calcium therapy. AMA Arch Ind Health 1955; 12(4)412–419
- McIvor ME. Delayed fatal hyperkalemia in a patient with acute fluoride intoxication. Ann Emerg Med 1987; 16(10)1165–1167
- Mayer TG, Gross PL. Fatal systemic fluorosis due to hydrofluoric acid burns. Ann Emerg Med 1985; 14(2)149–153
- Greco RJ, Hartford CE, Haith LR, Jr, Patton ML. Hydrofluoric acid-induced hypocalcemia. J Trauma 1988; 28(11)1593–1596
- Sanz-Gallen P, Nogue S, Munne P, Faraldo A. Hypocalcaemia and hypomagnesaemia due to hydrofluoric acid. Occup Med (Lond) 2001; 51(4)294–295
- Tepperman PB. Fatality due to acute systemic fluoride poisoning following a hydrofluoric acid skin burn. J Occup Med 1980; 22(10)691–692
- Chan BS, Duggin GG. Survival after a massive hydrofluoric acid ingestion. J Toxicol Clin Toxicol 1997; 35(3)307–309
- Bjornhagen V, Hojer J, Karlson-Stiber C, Selden AI, Sundbom M. Hydrofluoric acid-induced burns and life-threatening systemic poisoning-favorable outcome after hemodialysis. J Toxicol Clin Toxicol 2003; 41(6)855–860
- Stremski ES, Grande GA, Ling LJ. Survival following hydrofluoric acid ingestion. Ann Emerg Med 1992; 21(11)1396–1399
- Simpson E, Rao LGS, Evans RM, et al. Calcium metabolism in a fatal case of sodium fluoride poisoning. AnnClin Biochem 1980; 17: 10–14
- Maletz L. Report of a fatal case of fluoride poisoning. NEJM 1935; 22: 370–372
- Baltazar RF, Mower MM, Reider R, Funk M, Salomon J. Acute fluoride poisoning leading to fatal hyperkalemia. Chest 1980; 78(4)660–663
- Rabinowitch I. Acute fluoride poisoning. CMAJ 1945; 52: 345–349
- Abukurah AR, Moser AM, Jr, Baird CL, Randall RE, Jr, Setter JG, Blanke RV. Acute sodium fluoride poisoning. JAMA 1972; 222(7)816–817
- Kirkpatrick JJ, Enion DS, Burd DA. Hydrofluoric acid burns: A review. Burns 1995; 21(7)483–493
- McIvor ME, Cummings CC, Mower MM, Baltazar RF, Wenk RE, Lustgarten JA, Salomon J. The manipulation of potassium efflux during fluoride intoxication: implications for therapy. Toxicology 1985; 37(3–4)233–239
- Chan KM, Svancarek WP, Creer M. Fatality due to acute hydrofluoric acid exposure. J Toxicol Clin Toxicol 1987; 25(4)333–339
- Waldbott GL. Acute fluoride intoxication. Acta Med Scand. 1963; 174: S 400, 1–44
- Boink AB, Wemer J, Meulenbelt J, Vaessen HA, de Wildt DJ. The mechanism of fluoride-induced hypocalcaemia. Hum Exp Toxicol 1994; 13(3)149–155
- Yolken R, Konecny P, McCarthy P. Acute fluoride poisoning. Pediatrics 1976; 58(1)90–93
- McIvor ME, Cummings CC. Sodium fluoride produces a K+ efflux by increasing intracellular Ca2+ through Na+-Ca2+ exchange. Toxicol Lett 1987; 38(1–2)169–176
- Cummings CC, McIvor ME. Fluoride-induced hyperkalemia: the role of Ca2+-dependent K+ channels. Am J Emerg Med 1988; 6(1)1–3
- McIvor ME, Cummings CE, Mower MM, Wenk RE, Lustgarten JA, Baltazar RF, Salomon J. Sudden cardiac death from acute fluoride intoxication: the role of potassium. Ann Emerg Med 1987; 16(7)777–781
- Su M, Chu J, Howland MA, Nelson LS, Hoffman RS. Amiodarone attenuates fluoride-induced hyperkalemia in vitro. Acad Emerg Med 2003; 10(2)105–109
- McIvor M, Baltazar RF, Beltran J, Mower MM, Wenk R, Lustgarten J, Salomon J. Hyperkalemia and cardiac arrest from fluoride exposure during hemodialysis. Am J Cardiol 1983; 51(5)901–902
- Gardos G. The function of calcium in the potassium permeability of human erythrocytes. Biochim Biophys Acta 1958; 30(3)653–654
- Brockmann JL, McDowell AV, Leeds WG. Fatal poisoning with sodium fluoroacetate: Report of a case. J Am Med Assoc 1955; 159(16)1529–1532
- Nitschke KD, Albee RR, Mattsson JL, Miller RR. Incapacitation and treatment of rats exposed to a lethal dose of sulfuryl fluoride. Fundam Appl Toxicol 1986; 7(4)664–670
- Taxay EP. Vikane inhalation. J Occup Med 1966; 8(8)425–426
- Gutknecht J, Walter A. Hydrofluoric and nitric acid transport through lipid bilayer membranes. Biochim Biophys Acta 1981; 644(1)153–156
- Reynolds KE, Whitford GM, Pashley DH. Acute fluoride toxicity: The influence of acid-base status. Toxicol Appl Pharmacol 1978; 45(2)415–427
- Takase I, Kono K, Tamura A, et al. Fatality due to acute fluoride poisoning in the workplace. Legal Medicine 2004; 6: 197–200
- Strubelt O, Ivan H, Younes M. The pathophysiological profile of the acute cardiovascular toxicity of sodium fluoride. Toxicology 1982; 24: 313–323
- Gaugl JF, Wooldridge B. Cardiopulmonary response to sodium fluoride infusion in the dog. J Toxicol Environ Health 1983; 11: 765–782
- Burke WJ, Ulrich H, Phillips RE. Systemic fluoride poisoning resulting from a fluoride skin burn. J Occup Med 1973; 15(1)39–41
- Bordelon BM, Saffle JR, Morris SE. Systemic fluoride toxicity in a child with hydrofluoric acid burns: Case report. J Trauma 1993; 34(3)437–9
- Manoguerra AS, Neuman TS. Fatal poisoning from acute hydrofluoric acid ingestion. Am J Emerg Med 1986; 4(4)362–3