
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In order to improve our understanding of the origin and production processes of methylamine in the atmosphere and its contribution to the particulate WSON, simultaneous measurements of methylamine in size-segregated aerosols and the gas phase were conducted at a forested site in Yamanashi Prefecture, Japan, over one year. The concentration of the atmospheric methylamine that is the sum of methylamine in the gas phase, coarse-mode, and fine-mode particles was 0.962 ng-N m−3 on average. About 77% of the atmospheric methylamine was detected in the fine-mode particles and about 17% was in the gas phase. Similar seasonal variations were found in the fine-mode and gaseous methylamine including higher concentrations recorded in summer. The relationship between the concentrations of the gaseous methylamine and ammonia showed a significant positive correlation, suggesting that methylamine in the gas phase has similar sources with ammonia; for instance, the volatilization from soil and/or emission from forest biological activities. The concentration of the fine-mode methylamine showed a significant positive correlation with that of the gaseous methylamine, and the concentration ratio of the fine-mode methylamine to the sum of the fine-mode and gaseous methylamine that can be an indicator of the conversion of methylamine from the gas phase to the particulate phase showed a significant positive correlation with the fine-mode non-sea salt SO42- (nss-SO42-) concentration, suggesting that the gaseous methylamine is converted into the particulate methylamine through the acid-base reaction with sulfuric acid and/or nss-SO42-. Significant positive correlation was not obtained in the comparison of the concentration ratio of the fine-mode methylamine to the sum of the fine-mode and gaseous methylamine with the excessive amount of sulfuric acid for complete neutralization of ammonia, which implies the possibility that the sulfuric acid more preferentially reacts with the gaseous methylamine than ammonia.
1. Introduction
The atmospheric deposition of bioavailable nitrogen is an important input of the nutrients to terrestrial and marine ecosystems for the growth of organisms (Rastogi et al., Citation2011). Many previous studies about the bioavailable nitrogen in the atmosphere have focused on the water-soluble inorganic nitrogen (WSIN), such as NO3--N and NH4+-N, whereas the water-soluble organic nitrogen (WSON) has not received much attention. In recent studies, however, the WSON has been suggested to contribute about 10-30% of the particulate nitrogen (Cape et al. (Citation2011), and references therein).
Various potential sources for the particulate WSON including both primary and secondary processes have been suggested in previous studies (Cape et al., Citation2011, and references therein). The primary emissions, such as biomass burning (Mace et al., Citation2003; Zamora et al., Citation2011) and dust particles (Nakamura et al., Citation2006; Lesworth et al., Citation2010), have been reported as significant sources of the particulate WSON. On the other hand, the generation of the WSON in the fine particles from the gaseous WSON via gas-to-particle conversion processes has been also suggested (Matsumoto et al., Citation2017; Citation2018). Despite many efforts by these previous studies, the sources of the particulate WSON have not yet been sufficiently clarified.
Organic nitrogen compounds in the particulate WSON fraction have not been sufficiently identified, but amines and amino acids have been considered to be important components of the WSON fraction (Zhang and Anastasio, Citation2003; Ge et al., Citation2011). Previous studies have pointed out that the amines are much more effective than ammonia in terms of new particle formation processes (Barsanti et al., Citation2009; Smith et al., Citation2010; Zhang et al., Citation2012; Yao et al., Citation2018). Compared to ammonia, amines more preferentially react with sulfuric acid and contribute to new particle formation processes in the atmosphere (Yu et al., Citation2012; Zollner et al., Citation2012; Almeida et al., 2013; Berndt et al., Citation2014). The new particle formation by amines would have significant impacts on the global climate (Lee et al., Citation2019). The increase in the aerosol number in the atmosphere can affect the global climate through direct and indirect effects (IPCC, Citation2001; Boucher et al., 2013; Levy et al., Citation2013); the aerosols directly scatter and absorb sunlight and contribute to the cloud droplet formation through their ice nuclei and cloud condensation nuclei activities.
Methylamine (CH3NH2) is the most common compound in the aliphatic amine group, and some previous studies have detected methylamine in aerosols and the gas phase (Ge et al. (Citation2011), and references therein). In spite of the importance of amines in the new particle formation processes and atmospheric WSON chemistry, the origins and production processes of methylamine in the atmosphere have not yet been understood. Simultaneous measurement of methylamine in aerosols and the gas phase would improve our understanding of the origins and production processes of methylamine in the atmosphere, but has been almost never reported other than a few pioneering studies (Gibb et al., Citation1999; Akyüz, Citation2008). In the present study, the simultaneous measurements of methylamine and WSON in size-segregated aerosols and the gas phase were conducted at a forested site in Yamanashi Prefecture, Japan, over one year to improve our understanding of the origin and production processes of methylamine in the atmosphere, and its potential contribution to the WSON in aerosols.
2. Experiment
2.1. Sample collection
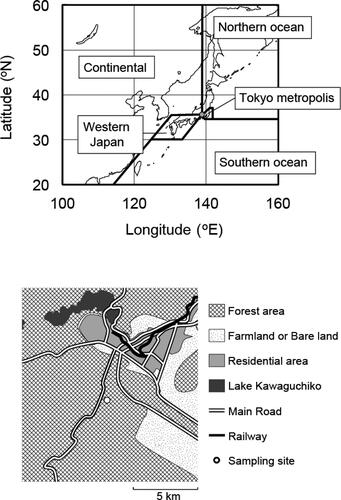
The aerosol and gas samples were collected at a forested site in Fujiyoshida, Yamanashi Prefecture, located about 90 km west of the Tokyo Metropolis. The sampling site was constructed at an open space in the experimental forest (red pine), with tree heights of 10-15 m, of the Mt. Fuji Research Institute, Yamanashi Prefectural Government, located at the northern foot of Mt. Fuji and about 5 km south of Lake Kawaguchiko. The residential areas with a population of about 66,000 was about 5 km away from the site, and there was no large stationary source around the site. The location of Fujiyoshida is shown in . Sample collections were conducted from 10 January 2018 to 9 January 2019, and the sample collection time of one sample was about 14 days. From 19 September to 4 October, the sample collection was interrupted by a collapse of the sampler due to a typhoon.
Figure 1. Location of Fujiyoshida (gray circle) and sections for calculating the passing frequency of air mass trajectories (upper panel), and the map of the surrounding area of the sampling site (lower panel).
Size-segregated aerosol samples with aerodynamic diameters smaller than 2.0 µm (fine particles) and of 2.0-10 µm (coarse particles) were collected on quartz fiber filters (QR100; Toyo Roshi Kaisha, Ltd.) using an NILU filter holder with a two-stage impactor (NL Series; Tokyo Dylec Corp.). Before the sample collections, the quartz fiber filters were heated at 850 °C for 4 h. Behind the collection filter of the fine particles, a 2% Na2CO3/1% glycerol-impregnated cellulose filter (5 A; Toyo Roshi Kaisha, Ltd.) and a 2% H3PO4/1% glycerol-impregnated cellulose filter (5 A; Toyo Roshi Kaisha, Ltd.) were placed in this order to collect the acid and basic gases, respectively. Sample collections were conducted at the flow rate of 20.0 L min−1. All of the filter samples were stored at −20 °C until chemically analyzed.
2.2. Chemical analysis
One-fourth of the aerosol sample filter was ultrasonically extracted for 30 min with 20 ml of ultrapure water in a glass vial. The whole of the basic gas sample filter was ultrasonically extracted for 30 min with 5 ml of ultrapure water in a glass vial. The extracts were then filtered using a PTFE membrane filter (0.45 μm pore size, 13HP045AN; Toyo Roshi Kaisha, Ltd.). The dissolved total organic carbon (DTOC) and total nitrogen (DTN) in the extract were measured using a total organic carbon (TOC)/total nitrogen (TN) analyzer (Model TOC-Vcsh/TNM-1; Shimadzu Corp.) that can detect the DTOC with an NDIR detector and the DTN with an ozone chemiluminescence detection after a thermo-catalytic oxidation process. The DTOC and DTN are considered to be the water-soluble organic carbon (WSOC) and water-soluble total nitrogen (WSTN), respectively. The ion species (Cl-, NO2-, NO3-, SO42-, Na+, NH4+, K+, Mg2+, Ca2+) in the extract were measured using ion chromatographs (DX-120; Dionex Corp. and ICS-1100; Thermo Fisher Scientific Inc.). Only the extract of the basic gas sample was analyzed after it was diluted 8-10 times by ultrapure water. The concentration of the total inorganic nitrogen (IN) was calculated from the sum of the NO2--N, NO3--N and NH4+-N concentrations, and the difference between the concentrations of the DTN and IN was considered to be the WSON.
The extract of the basic gas sample in 5 ml of ultrapure water, which was prepared for the analysis of the WSTN and ion species as already described, was used for the analysis of methylamine without dilution. For the analysis of methylamine in the aerosols, in addition, one-fourth of the filter that collected the aerosol sample was ultrasonically extracted for 30 min with 3 ml of ultrapure water in a glass vial, then the extract was filtered using the PTFE membrane filter. Methylamine in these extracts was quantified using the pre-column derivatization HPLC method described by Koop et al. (Citation1982) with some modifications; methylamine in 200 µL of the extract was reacted with 100 µL of 0.1 M phenylisothiocyanate in acetonitrile and 100 µL of 1 M triethylamine in acetonitrile at 40 °C for 20 min to form a phenylisothiocyanate derivative. After neutralization by adding 1.2 M HCl, the derivative was quantified using an HPLC system with an ODS column (Shim-pack VP-ODS; Shimadzu Corp.) and a UV-Vis detector (SPD-20A; Shimadzu Corp.).
In this study, the acid gas samples collected by 2% Na2CO3/1% glycerol-impregnated filters were not used because methylamine is a basic compound.
During the sample collections, operational blank tests were performed at given intervals, and the blank measurements were used to determine the detection limits of the WSON and methylamine. The detection limits were defined according to Matsumoto et al. (Citation2017), and the concentration below the detection limit was treated as zero in the following statistical analyses and discussion.
2.3. Meteorological data and trajectory analysis
Hourly data of ambient temperature, atmospheric pressure, relative humidity, wind speed, and sunshine duration at the Kawaguchiko Weather Station that is located about 5 km from the sampling site and those of solar radiation at the Kofu Local Meteorological Observatory that is about 30 km from the site were used for the following statistical analyses. The data of sunshine duration and solar radiation were summed up and the other data were averaged for each sampling period.
In order to clarify the origins of the air masses arriving at the sampling site, 5-day backward trajectories were calculated using the hybrid single-particle Lagrangian integrated trajectory (HYSPLIT) model. The trajectories were calculated at 3:00 and 15:00 UTC every day. The starting altitudes were set at 500 m above the ground surface. The passing frequency of the trajectories over the five regions shown in was calculated for each sample collection period by the following expression:
where PF(X) denotes the passing frequency of the trajectories over the region X, N(X) and N(T) denote the number of the trajectories that passed over the region X and total number of the trajectories, respectively, during each sample collection period.
3. Results and discussion
3.1. Concentrations of WSON and methylamine
A statistical summary of the concentrations of the WSON and methylamine and the nitrogen concentration ratios of methylamine to the WSON obtained in this study are shown in . Because the gaseous amine compounds can easily react with sulfuric acid and form thermodynamically stable aminium salt in the particulate phase, there would be a danger that the gaseous methylamine adsorbs on the aerosols collected on the filter during sample collection, which would lead to the overestimation of methylamine in the aerosols, especially in the fine-mode particles, and the underestimation of that in the gas phase.
Table 1. Concentrations of the WSON (unit; µg-N m−3), methylamine (MA) (ng-N m−3), and the nitrogen concentration ratios of MA/WSON (%).
The concentration of the atmospheric methylamine that is the sum of methylamine in the gas phase, coarse-mode, and fine-mode particles was 0.962 ng-N m−3 on average. About 77% of the atmospheric methylamine was detected in the fine-mode particles, and about 17% was in the gas phase. The concentration of methylamine in the coarse-mode particles was very low, so that it hardly contributed to the atmospheric methylamine concentration. Higher concentrations of methylamine in the fine particles than that in the coarse particles have been also reported by a pioneering study of Akyüz (Citation2008).
The concentration of methylamine in fine particles obtained in this study was significantly lower than those at many oceanic sites; for instance, in PM2.5 at Jeju Island in South Korea (a mean was 6.1 ng-N m−3) (Yang et al., Citation2004), although lower concentrations than this study were also obtained over the remote ocean; for instance, in PM1 at the Cape Verde over the North Atlantic Ocean (0.09 ng-N m−3) (van Pinxteren et al., Citation2019). In addition, the concentration of the fine-mode methylamine in this study was lower than those of dimethylamine in previous investigations at coastal sites; for instance, in PM1 at Crete Island over the Eastern Mediterranean (2.8 ng-N m−3) (Violaki and Mihalopoulos, Citation2010) and in PM2.5 over Tampa Bay, U.S.A. (9.6 ng-N m−3) (Calderón et al., Citation2007). Oceanic emissions would be important for methylamine and other amine compounds in the aerosols. On the other hand, similar levels of methylamine in fine particles to this study were found in PM1.1 at a Japanese urban site, Kobe (0.48-0.99 ng-N m−3) (Suzuki et al., Citation2001), although higher concentrations were found in PM2.5 at a Chinese urban site, Xi’an (6.3 ng-N m−3) (Ho et al., Citation2015), probably due to heavy anthropogenic emissions.
The concentration of methylamine in the gas phase obtained in this study was similar to those at an urban site in Atlanta, U.S.A. (0.13 ng-N m−3) (Hanson et al., Citation2011), a coastal site in Hawaii (0.15 ng-N m−3) (van Neste et al., Citation1987), and over the northwest Pacific Ocean (0.2 ng-N m−3) (Mopper and Zika, Citation1987), but clearly lower than those at urban/rural/coastal sites in Sweden (2.3-10 ng-N m−3) (Grönberg et al., Citation1992a, Citation1992b), a coastal site in Rhode Island, U.S.A. (0.73 ng-N m−3) (van Neste et al., Citation1987) and over the Arabian Sea (1.6-2.0 ng-N m−3) (Gibb et al., Citation1999).
The contribution of methylamine to the WSON was the highest in the fine particles, but was very low (< 1%) even in the fine particles. A similar result was reported for the nitrogen concentration ratio of dimethylamine to the WSON (0.9 ± 3.4%) in the PM1 particles obtained at a coastal site (Violaki and Mihalopoulos, Citation2010), even though the concentration of dimethylamine in their measurement was higher than that of methylamine in this study. On the other hand, Ho et al. (Citation2015) reported a low contribution (about 0.15%) of methylamine to the WSON from the measurements at an urban site in Xi’an, China.
Mutzel et al. (Citation2013) has pointed out that the ultrasonic extraction used in this study for the extraction of filter samples can lead to a formation of OH radicals and consequently to an oxidation of organic compounds in the solution. Therefore, it is possible that the measurements of the WSON and methylamine concentrations in this study were underestimated, although it is also possible that the measurements of the WSON and amines concentrations reported by many previous studies have been underestimated because the ultrasonic extraction has been commonly used in these studies.
3.2. Temporal trends in the concentrations of methylamine
Temporal variations in the concentrations of the coarse-mode, fine-mode, and gaseous methylamine are shown in . Similar seasonal variations were found in the fine-mode and gaseous methylamine; higher concentrations were recorded in the summer. This suggests that the emission of the gaseous methylamine into the atmosphere and subsequent conversion to the particulate methylamine would be enhanced in the warm season, which will be discussed in the section 3.3. Coarse-mode methylamine, on the other hand, showed higher concentrations from summer to early winter, which might be caused by a seasonality of direct emissions of particulate methylamine from the primary sources, such as plant defoliate, although the potential primary sources cannot be clarified in this study.
Figure 2. Temporal variations in the concentrations of the methylamine (MA) in the coarse-mode particles (a), fine-mode particles (b), and gas phase (c).
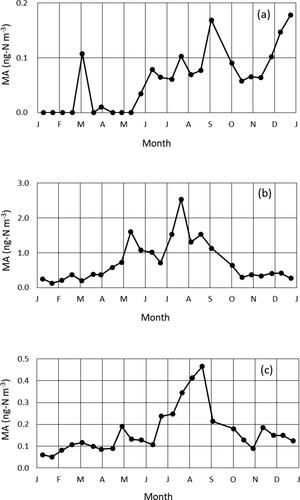
As with this study, enhanced concentrations of the fine-mode dimethylamine and diethylamine in summer were observed at a coastal site in Ireland due to high marine biological activity (Facchini et al., Citation2008). Contrary to these trends, Ho et al. (Citation2015) reported that the higher concentrations of methylamine in PM2.5 were found in the cold season from winter to spring at an urban site in Xi'an, China, due to enhanced atmospheric stability and anthropogenic emissions. Seasonal trend of the atmospheric methylamine would be largely dependent on local sources and meteorology.
3.3. Gas-to-particle conversion process of methylamine
Previous researches have pointed out many primary emission sources for methylamine; animal husbandry, fish processing, factories and power plants, sewage, biomass burning, food odor, tobacco smoke, terrestrial and marine biota (Ge et al. (Citation2011), and references therein). In order to clarify the source of methylamine in the gas phase, correlation analyses of the concentration of the gaseous methylamine with those of the particulate and gaseous species, meteorological parameters, and the passing frequency of the trajectories were conducted and summarized in . A strong positive correlation was found between the gaseous methylamine concentration and ambient air temperature (r = 0.71, p < 0.01). In addition, a significant positive correlation was also found in the relationship between the concentrations of the gaseous methylamine and ammonia (r = 0.56, p < 0.05). These results suggest that methylamine in the gas phase has sources similar to that of ammonia; for instance, the volatilization from soil and emission from forest biological activities, and the volatilization and/or emission of the gaseous methylamine is affected by the ambient air temperature. A strong positive correlation was also found between the gaseous and fine-mode methylamine concentrations (r = 0.71, p < 0.01), which would imply the gas-to-particle conversion from the gaseous methylamine to the fine-mode methylamine.
Table 2. Correlation coefficient (r) between the concentration of the gaseous methylamine and those of other particulate and gaseous species, meteorological parameters, and passing frequency of the air mass trajectories. Significance (s) is as follows; ***:p < 0.01, **:p < 0.05, *:p < 0.1, and -:p > 0.1.
Strong positive and negative correlations of the gaseous methylamine concentration with the passing frequency of the trajectories from the southern oceanic (r = 0.64, p < 0.01) and continental regions (r=-0.74, p < 0.01), respectively, were found as shown in . A negative correlation was also found between the fine-mode methylamine concentration and the passing frequency of the trajectories from the continental region (r=-0.69, p < 0.01). These results indicate that the long-range transport of methylamine from the Asian continental region to the site can be negligible. The influences of the air mass transport from the Tokyo metropolis and western Japanese areas to the site would be also small because less significant correlations were found between the gaseous methylamine concentration and the passing frequency of the trajectories from both regions (see ). On the other hand, a strong positive correlation of the gaseous methylamine concentration with the passing frequency of the trajectories from the southern oceanic regions was found, which would be consistent with previous reports that amine compounds have been ubiquitously found in the marine atmosphere (van Pinxteren et al. (Citation2019), and references therein). However, this positive correlation would be caused by the seasonal air mass alternation; the air masses from the southern oceanic region covered the site in the warm season, whereas the continental air masses from the Asian continent in the cold season. Indeed, a stronger positive correlation was found between the air temperature and the passing frequency of the trajectories from the southern oceanic region (r = 0.84, p < 0.01). In addition, the passing frequency of the trajectories from the northern oceanic region was not correlated with the methylamine concentration (see ). These results suggest that the influence of oceanic emissions of the gaseous methylamine on our measurements would be small and the local emission dependent on air temperature would be important for the atmospheric methylamine at the site, although the influence of the oceanic emission on our measurement cannot be fully excluded at the present stage.
In order to clarify the mechanism of the gas-to-particle conversion of methylamine, the concentration ratio of the fine-mode methylamine to the sum of the fine-mode and gaseous methylamine was compared with the concentrations of the fine-mode species and meteorological factors, which was summarized in . The ratios ranged within 0.63-0.92 and their average was 0.78, which was somewhat larger than previous reported values of about 0.6 (Akyüz, Citation2008). The ratio showed significant positive correlations with the concentration of the fine-mode nss-SO42- (r = 0.53, p < 0.1), ambient air temperature (r = 0.57, p < 0.05), and solar radiation (r = 0.58, p < 0.05). On the other hand, no significant correlation was obtained for the comparison of the ratio with the fine-mode NO3- concentration. A significant positive correlation of the ratio with the fine-mode nss-SO42- concentration indicates that the gaseous methylamine is converted to the particulate methylamine by the acid-base reaction with sulfuric acid. The reactions of the gaseous methylamine with the particulate ammonium sulfate and bisulfate can also be expected to form the particulate methylamine (Bzdek et al., Citation2010; Qiu et al., Citation2011). Significant positive correlations of the ratio with ambient air temperature and solar radiation can be attributed to the fact that the production of sulfuric acid and/or nss-SO42- is accelerated in the warm season due to photochemical reactions. A significant positive correlation of the ratio with the concentration of the fine-mode WSOC was also found (r = 0.56, p < 0.05), which can be attributed to the enhanced secondary production of the WSOC due to the photochemical reactions in the warm season.
Table 3. Correlation coefficient (r) between the concentration ratio of the fine-mode methylamine to the sum of the fine-mode and gaseous methylamine and the concentrations of the fine-mode species and meteorological parameters. Significance (s) is as follows; ***:p < 0.01, **:p < 0.05, *:p < 0.1, and -:p > 0.1.
In summary, the results from the present study suggest that the gaseous methylamine was volatilized from soil and/or emitted from forest biological activities, and subsequently converted to the fine-mode methylamine through the acid-base reaction with sulfuric acid and/or nss-SO42-, both of which would be accelerated in the warm season due to the high temperature and enhanced photochemical reactions. No positive correlation was obtained for the comparison of the concentration ratio of the fine-mode methylamine to the sum of the fine-mode and gaseous methylamine with the excessive amount of sulfuric acid for complete neutralization of ammonia that is the equivalent concentration difference between the fine-mode nss-SO42- and NH4+ ([nss-SO42-]-[NH4+]), suggesting that the presence of ammonia in the gas phase and/or ammonium in the aerosols may not prevent the gas-to-particle conversion of methylamine. This implies the possibility that the sulfuric acid more preferentially reacts with the gaseous methylamine than ammonia, which is consistent with the results from the laboratory experiments (Almeida et al., 2013; Glasoe et al., Citation2015).
4. Conclusion
Simultaneous measurements of methylamine and WSON in size-segregated aerosols and the gas phase were conducted at a forested site in Yamanashi Prefecture, Japan, over one year to improve our understanding of the origin and production processes of methylamine in the atmosphere, and its potential contribution to the WSON in aerosols.
The concentration of the atmospheric methylamine that is the sum of methylamine in the gas phase, coarse-mode, and fine-mode particles was 0.962 ng-N m−3 on average. About 77% of the atmospheric methylamine was detected in the fine-mode particles, and about 17% was in the gas phase. The contribution of methylamine to the WSON was the highest in the fine particles, but was very low (< 1%) even in the fine particles.
Correlation analyses suggested that the gaseous methylamine would be emitted from the sources similar to that of ammonia; for instance, the volatilization from soil and/or emission from forest biological activities, and subsequently converted to the particulate methylamine in the fine-mode range by the acid-base reaction with sulfuric acid and/or nss-SO42-. The gas-to-particle conversion reaction of methylamine may be not affected by ammonia in the gas phase and/or ammonium in the aerosols. Seasonal trends of the methylamine in the gas phase and aerosols indicated that the emission of the gaseous methylamine into the atmosphere and subsequent conversion to the particulate methylamine would be enhanced during the warm season due to the high temperature and enhanced photochemical reactions.
The present study suggested the importance of the local emission from the forest ecosystem for discussing the atmospheric methylamine, although the influence of the regional scale air mass alternation on the temporal trend in the methylamine concentrations cannot be fully excluded.
Acknowledgment
We would like to acknowledge Dr. T. Nakano, M. Ishikawa, K. Sakata, Y. Watanabe, T. Ogawa, R. Kumagai, S. Kodama for their kind support of the sample collections and chemical analyses, and the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and/or READY website (http://www.ready.noaa.gov) used in this publication.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Akyüz, M. 2008. Simultaneous determination of aliphatic and aromatic amines in ambient air and airborne particulate matters by gas chromatography-mass spectrometry. Atmos. Environ. 42, 3809–3819. doi:https://doi.org/10.1016/j.atmosenv.2007.12.057
- Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O. and co-authors. 2013. Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere. Nature 502, 359–363. doi:https://doi.org/10.1038/nature12663
- Barsanti, K., McMurry, P. and Smith, J. 2009. The potential contribution of organic salts to new particle growth. Atmos. Chem. Phys. 9, 2949–2957. doi:https://doi.org/10.5194/acp-9-2949-2009
- Berndt, T., Sipila, M., Stratmann, F., Petaja, T., Vanhanen, J. and co-authors. 2014. Enhancement of atmospheric H2SO4/H2O nucleation: organic oxidation products versus amines. Atmos. Chem. Phys. 14, 751–764. doi:https://doi.org/10.5194/acp-14-751-2014
- Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G. and co-authors. 2013.,. Clouds and Aerosols. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (eds. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex, and P. M. Midgley). Cambridge University Press, Cambridge, UK and New York, NY, pp. 571–657.
- Bzdek, B. R., Ridge, D. P. and Johnston, M. V. 2010. Amine exchange into ammonium bisulfate and ammonium nitrate nuclei. Atmos. Chem. Phys. 10, 3495–3503. doi:https://doi.org/10.5194/acp-10-3495-2010
- Calderón, S. M., Poor, N. D. and Campbell, S. W. 2007. Estimation of the particle and gas scavenging contributions to wet deposition of organic nitrogen. Atmos. Environ. 41, 4281–4290. doi:https://doi.org/10.1016/j.atmosenv.2006.06.067
- Cape, J. N., Cornell, S. E., Jickells, T. D. and Nemitz, E. 2011. Organic nitrogen in the atmosphere-where does it come from? A review of sources and methods. Atmos. Res. 102, 30–48. doi:https://doi.org/10.1016/j.atmosres.2011.07.009
- Facchini, M. C., Decesari, S., Rinaldi, M., Carbone, C., Finessi, E. and co-authors. 2008. Important source of marine secondary organic aerosol from biogenic amines. Environ. Sci. Technol. 42, 9116–9121. doi:https://doi.org/10.1021/es8018385
- Ge, X. L., Wexler, A. S. and Clegg, S. L. 2011. Atmospheric amines - part I. A review. Atmos. Environ. 45, 524–546. doi:https://doi.org/10.1016/j.atmosenv.2010.10.012
- Gibb, S. W., Mantoura, R. F. C. and Liss, P. S. 1999. Ocean-atmosphere exchange and atmospheric speciation of ammonia and methylamines in the region of the NW Arabian Sea. Global Biogeochem. Cycles 13, 161–178. doi:https://doi.org/10.1029/98GB00743
- Glasoe, W. A., Volz, K., Panta, B., Freshour, N., Bachman, R. and co-authors. 2015. Sulfuric acid nucleation: an experimental study of the effect of seven bases. J. Geophys. Res. Atmos. 120, 1933–1950. doi:https://doi.org/10.1002/2014JD022730
- Grönberg, L., Lovkvist, P. and Jönsson, Å.,J. 1992b. Determination of aliphatic amines in air by membrane enrichment directly coupled to a gas chromatograph. Cromatographia 33, 77–82. doi:https://doi.org/10.1007/BF02276856
- Grönberg, L., Lovkvist, P. and Jönsson, Å. J. 1992a. Measurement of aliphatic amines in ambient air and rainwater. Chemosphere 24, 1533–1540. doi:https://doi.org/10.1016/0045-6535(92)90273-T
- Hanson, D. R., McMurry, P. H., Jiang, J., Tanner, D. and Huey, L. 2011. Ambient pressure proton transfer mass spectrometry: detection of amines and ammonia. Environ. Sci. Technol. 45, 8881–8888. doi:https://doi.org/10.1021/es201819a
- Ho, K. F., Ho, S. S. H., Huang, R. J., Liu, S. X., Cao, J. J. and co-authors. 2015. Characteristics of water-soluble organic nitrogen in fine particulate matter in the continental area of China. Atmos. Environ. 106, 252–261. doi:https://doi.org/10.1016/j.atmosenv.2015.02.010
- IPCC. 2001. Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change (eds. J. T. Houghton, Y. Ding, D. J. Griggs, M. Noguer, P. J. van der Linden, X. Dai, K. Maskell, and C. A. Johnson). Cambridge University Press, Cambridge, UK and New York, NY, pp. 289–348.
- Koop, D. R., Morgan, E. T., Tarr, G. E. and Coon, M. J. 1982. Purification and characterization of a unique isozyme of cytochrome P-450 from liver microsomes of ethanol-treated rabbits. J. Biol. Chem. 257, 8472–8480. doi:https://doi.org/10.1016/S0021-9258(18)34356-4
- Lee, S.-H., Gordon, H., Yu, H., Lehtipalo, K., Haley, R. and co-authors. 2019. New particle formation in the atmosphere: From molecular clusters to global climate. J. Geophys. Res. Atmos. 124, 7098–7146. doi:https://doi.org/10.1029/2018JD029356
- Lesworth, T., Baker, A. and Jickells, T. 2010. Aerosol organic nitrogen over the remote Atlantic Ocean. Atmos. Environ. 44, 1887–1893. doi:https://doi.org/10.1016/j.atmosenv.2010.02.021
- Levy, H. I. I., Horowitz, L. W., Schwarzkopf, M. D., Ming, Y., Golaz, J.-C. and co-authors. 2013. The roles of aerosol direct and indirect effects in past and future climate change. J. Geophys. Res. Atmos. 118, 4521–4532. doi:https://doi.org/10.1002/jgrd.50192
- Mace, K., Artaxo, P. and Duce, R. 2003. Water-soluble organic nitrogen in Amazon Basin aerosols during the dry (biomass burning) and wet seasons. J. Geophys. Res. 108, 4512. doi:https://doi.org/10.1029/2003JD003557
- Matsumoto, K., Takusagawa, F., Suzuki, H. and Horiuchi, K. 2018. Water-soluble organic nitrogen in the aerosols and rainwater at an urban site in Japan: Implications for the nitrogen composition in the atmospheric deposition. Atmos. Environ. 191, 267–272. doi:https://doi.org/10.1016/j.atmosenv.2018.07.056
- Matsumoto, K., Yamamoto, Y., Nishizawa, K., Kaneyasu, N., Irino, T. and co-authors. 2017. Origin of the water-soluble organic nitrogen in the maritime aerosol. Atmos. Environ. 167, 97–103. doi:https://doi.org/10.1016/j.atmosenv.2017.07.050
- Mopper, K. and Zika, R. G. 1987. Free amino-acids in marine rains - evidence for oxidation and potential role in nitrogen cycling. Nature 325, 246–249. doi:https://doi.org/10.1038/325246a0
- Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O. and Herrmann, H. 2013. An improved method for the quantification of SOA bound peroxides. Atmos. Environ. 67, 365–369. doi:https://doi.org/10.1016/j.atmosenv.2012.11.012
- Nakamura, T., Ogawa, H., Maripi, D. and Uematsu, M. 2006. Contribution of water soluble organic nitrogen to total nitrogen in marine aerosols over the East China Sea and western North Pacific. Atmos. Environ. 40, 7259–7264. doi:https://doi.org/10.1016/j.atmosenv.2006.06.026
- Qiu, C., Wang, L., Lal, V., Khalizov, A. F. and Zhang, R. 2011. Heterogeneous reactions of alkylamines with ammonium sulfate and ammonium bisulfate. Environ. Sci. Technol. 45, 4748–4755. doi:https://doi.org/10.1021/es1043112
- Rastogi, N., Zhang, X. L., Edgerton, E. S., Ingall, E. and Weber, R. J. 2011. Filterable water soluble organic nitrogen in fine particles over the southeastern USA during summer. Atmos. Environ. 45, 6040–6047. doi:https://doi.org/10.1016/j.atmosenv.2011.07.045
- Smith, J. N., Barsanti, K. C., Friedli, H. R., Ehn, M., Kulmala, M. and co-authors. 2010. Observations of aminium salts in atmospheric nanoparticles and possible climatic implications. Proc. Natl. Acad. Sci. USA 107, 6634–6639. doi:https://doi.org/10.1073/pnas.0912127107
- Suzuki, Y., Kawakami, M. and Akasaka, K. 2001. 1H NMR application for characterizing water-soluble organic compounds in urban atmospheric particles. Environ. Sci. Technol. 35, 2656–2664. doi:https://doi.org/10.1021/es001861a
- Van Neste, A., Duce, R. A. and Lee, C. 1987. Methylamines in the marine atmosphere. Geophys. Res. Lett. 14, 711–714. doi:https://doi.org/10.1029/GL014i007p00711
- van Pinxteren, M., Fomba, K. W., Van Pinxteren, D., Triesch, N., Hoffmann, E. H. and co-authors. 2019. Aliphatic amines at the Cape Verde Atmospheric Observatory: Abundance, origins and sea-air fluxes. Atmos. Environ. 203, 183–195. doi:https://doi.org/10.1016/j.atmosenv.2019.02.011
- Violaki, K. and Mihalopoulos, N. 2010. Water-soluble organic nitrogen (WSON) in size-segregated atmospheric particles over the Eastern Mediterranean. Atmos. Environ. 44, 4339–4345. doi:https://doi.org/10.1016/j.atmosenv.2010.07.056
- Yang, H., Xu, J. H., Wu, W. S., Wan, C. H. and Yu, J. Z. 2004. Chemical characterization of water-soluble organic aerosols at Jeju Island collected during ACE-Asia. Environ. Chem. 1, 13–17. doi:https://doi.org/10.1071/EN04006
- Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C. and co-authors. 2018. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 361, 278–281. doi:https://doi.org/10.1126/science.aao4839
- Yu, H., McGraw, R. and Lee, S.-H. 2012. Effects of amines on formation of sub-3 nm particles and their subsequent growth. Geophys. Res. Lett. 39, L02807. doi:https://doi.org/10.1029/2011GL050099
- Zamora, L. M., Prospero, J. M. and Hansell, D. A. 2011. Organic nitrogen in aerosols and precipitation at Barbados and Miami: Implications regarding sources, transport and deposition to the western subtropical North Atlantic. J. Geophys. Res. 116, D20309. doi:https://doi.org/10.1029/2011JD015660
- Zhang, Q. and Anastasio, C. 2003. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmos. Environ. 37, 2247–2258. doi:https://doi.org/10.1016/S1352-2310(03)00127-4
- Zhang, G., Bi, X., Chan, L. Y., Li, L., Wang, X. and co-authors. 2012. Enhanced trimethylamine-containing particles during fog events detected by single particle aerosol mass spectrometry in urban Guangzhou. China. Atmos. Environ, 55, 121–126. doi:https://doi.org/10.1016/j.atmosenv.2012.03.038
- Zollner, J. H., Glasoe, W. A., Panta, B., Carlson, K. K., McMurry, P. H. and co-authors. 2012. Sulfuric acid nucleation: power dependencies, variation with relative humidity, and effect of bases. Atmos. Chem. Phys. 12, 4399–4411. doi:https://doi.org/10.5194/acp-12-4399-2012