
ABSTRACT
Background: The anti–interleukin-6 receptor antibody tocilizumab is approved for subcutaneous injection using a prefilled syringe (PFS). We report results from a bioequivalence study in healthy subjects and a user-handling study in patients with rheumatoid arthritis (RA) using an autoinjector (AI) for tocilizumab.
Methods: A randomized crossover study in healthy subjects (N = 161) examined the bioequivalence, safety, and tolerability of tocilizumab after a single subcutaneous injection by AI versus PFS. A nonrandomized observational, real-life human factors study in RA patients (N = 54) assessed user (RA patients, caregivers, health care providers) ability to administer tocilizumab effectively by AI.
Results: Bioequivalence criteria for tocilizumab AI versus PFS were met for key pharmacokinetic parameters. Safety was comparable between devices and consistent with the established tocilizumab profile. In the real-life human factors study, the proportion of users who successfully performed all essential tasks required to operate the AI to deliver the full dose was 92.3% at first assessment and 98.1% at second assessment, with no safety concerns.
Conclusions: Tocilizumab administration by AI was bioequivalent to administration by PFS. Intended users were successful in performing the tasks required to administer tocilizumab by AI. No new safety signals were observed in either study.
Clinical Trial Registration: NCT02678988, NCT02682823
1. Introduction
Tocilizumab is a humanized monoclonal antibody against interleukin-6 receptor-α, indicated for the treatment of patients with rheumatoid arthritis (RA), giant cell arteritis (GCA), polyarticular juvenile idiopathic arthritis (pJIA), systemic JIA (sJIA), and cytokine release syndrome induced after the administration of chimeric antigen receptor T-cell therapy [Citation1]. In many regions of the world, tocilizumab administered via subcutaneous (SC) injection using a prefilled syringe (PFS) is approved for the treatment of patients with RA, GCA, sJIA, and pJIA [Citation1].
In recent years, disposable autoinjectors (AIs) have come into use and are designed to be easy for patients to self-administer [Citation2]. AIs are in development or are marketed for several drug products [Citation2–Citation5], and evidence is emerging to suggest patient preference for administration with an AI over administration with a PFS [Citation6,Citation7]. Among the potential benefits of AIs are improved convenience and ease of use (and, consequently, patient compliance), reduced risk for dosage error, reduced anxiety from ‘needle phobia,’ and integrated needle safety [Citation3,Citation8]. Herein, we report the results from a bioequivalence study in healthy subjects that was designed to evaluate the bioequivalence, safety, and tolerability of tocilizumab after a single SC injection from an AI compared with the currently marketed PFS. In addition, data from a real-life human factors study in patients with RA are presented in which user ability to administer tocilizumab with the AI was assessed to ensure that the target population would be able to use the device effectively. Together, these studies provide evidence for effective use of the AI as an alternative delivery device to the PFS for approved indications of SC tocilizumab.
2. Subjects and methods
2.1. Ethics
Both studies were conducted in accordance with the principles of the Declaration of Helsinki and in compliance with the International Conference on Harmonisation E6 Guideline for Good Clinical Practice (Committee for Proprietary Medicinal Products guideline CPMP/ICH/135/95). The clinical study protocols and the informed consent forms were reviewed and approved by an institutional review board in the United States. All participants provided written informed consent before initiation of any study-related procedures.
2.2. Bioequivalence study (NCT02678988)
2.2.1. Study design
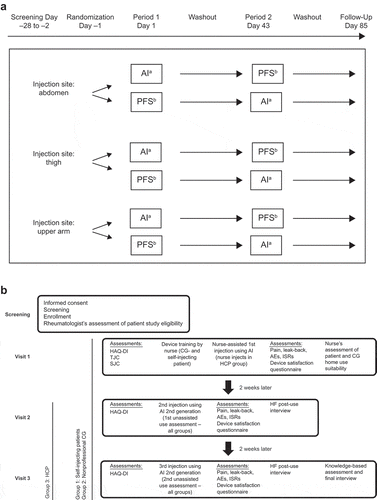
This was an open-label, randomized, 2-period, crossover study in which healthy subjects received a single 162-mg SC dose of tocilizumab by PFS (marketed dose and delivery system) and a single 162-mg SC dose of tocilizumab by AI (same marketed dose; AI-1000 G2 device). The study included a screening period (up to 4 weeks), a 6-week washout interval between doses, and a 6-week follow-up period, for a total duration of up to 16 weeks. Based on an earlier bioequivalence study with a previous version of an AI, a geometric mean ratio (AI/PFS) of 1.1 and a coefficient of variation of 39% was assumed to estimate the sample size. A sample size of 150 subjects was estimated to provide adequate power to meet the bioequivalence criteria; given an expected dropout rate of 20%, a sample size of 188 subjects was planned. Eligible subjects were randomly assigned (1:1) to 1 of the 2 possible devices in the first period, with a crossover to the alternative device in the second period of the study, and to 1 of 3 approved injection sites (abdomen, thigh, or upper arm) for SC administration of tocilizumab (). The injection was administered by the investigational staff at the same body site (approximate location) and the same side of the body for both treatment periods. The PFS (incorporating an ISO 9626–compliant, 27-gauge, half-inch needle) was assembled with a needle-safety device that extended a rigid plastic shield on completion of the injection. The AI was a mechanical, spring-driven device that incorporated a PFS with an ISO–compliant, 26-gauge, thin-wall, half-inch needle. Because of the design of the AI, needle protrusion was within a range of 4.5 to 8 mm. As soon as the user pressed the activation button, the AI automatically inserted the needle into the skin and injected the product. The AI featured a transparent window through which the user could observe the colored plunger moving inside the syringe as a visual indicator of injection progress. A needle cover extended over the needle on completion of the injection.
Figure 1. Study design. (a) Bioequivalence. (b) Real-life human factors study. AE, adverse event; AI, autoinjector; CG, caregiver; gen, generation; HAQ-DI, Health Assessment Questionnaire–Disability Index; HCP, health care professional; HF, human factors; ISR, injection site reaction; PFS, prefilled syringe; SC, subcutaneous; SJC, swollen joint count; TJC, tender joint count. a,bTocilizumab 162 mg/0.9 mL SC using AI or PFS.
Users of the PFS and the AI were instructed to let the device warm up to room temperature, 30 minutes for the PFS and 45 minutes for the AI. Users of both devices were instructed to perform the injection at a 90° angle. Users of the PFS were not given a specific injection time, but users of the AI were instructed to observe the visual indicator to determine when the injection was complete and were informed that this might take up to 10 seconds. Users of the PFS were instructed to pinch the injection site while inserting the needle and to let go of the pinch while injecting the product. Users of the AI were instructed to pinch the site during the injection.
2.2.2. Subject eligibility criteria
The bioequivalence study included healthy men and women aged 18–65 years inclusive. Healthy subjects were included to allow a clear interpretation of the study results and to limit confounding factors resulting from changes in disease state and/or concomitant medication use. Among the key inclusion criteria were non-childbearing potential (use of contraception [men]; postmenopausal or surgically sterile [women]); no evidence of active or chronic disease based on detailed medical and surgical history and physical and laboratory testing; intact normal skin (free from obscuring tattoos, pigmentation, or lesions) at injection site; and body weight <150 kg. Key exclusion criteria were any current or history of recurrent infection, clinically significant disease, malignant disease (with the exception of localized skin or cervical carcinoma that had been excised and cured), or immunodeficiency; dependence on or positive test result for any substance of abuse; smoking >10 cigarettes/day or equivalent; positive screening result for hepatitis B, hepatitis C, or human immunodeficiency virus; any clinically relevant abnormality in laboratory test results or vital signs; requirement for any concomitant medication during the study period (except hormone replacement therapy); and any other condition likely to render the subject unsuitable for the study.
2.2.3. Study outcomes
The primary outcome of the study was evaluation of bioequivalence after single-dose SC administration of 162 mg tocilizumab by AI or PFS based on criteria recommended by various health authorities [Citation9,Citation10]. Secondary end points were safety and tolerability of tocilizumab administered using AI or PFS.
2.2.4. Analyses
Tocilizumab concentrations were analyzed up to 42 days after dose, with blood samples for pharmacokinetic (PK) analysis taken on the dosing day (0 and 8 hours after dose) and on days 1–5, 7, 9, 12, 15, 18, 21, 28, 35, and 42 of both dosing periods as well as the day 85 follow-up. The day 42 sample after the first dose corresponded with the first (predose) sample of period 2, and the day 42 sample after the second dose corresponded with the day 85 follow-up sample. Additional blood samples were taken on the last day of follow-up (approximately 6 weeks after the second dose). Tocilizumab in serum samples was analyzed using a validated (F. Hoffmann-La Roche Ltd., unpublished data) enzyme-linked immunosorbent assay method. PK parameters included maximum observed serum concentration (Cmax), area under the serum concentration-time curve (AUC) from time 0 to the time of the last measurable concentration (AUClast), AUC from time 0 to infinity (AUCinf), time of occurrence of Cmax (Tmax), time of last measurable plasma concentration (Tlast), and apparent elimination rate constant (Kel). PK parameters were calculated using noncompartmental analysis from the serum concentration-time data for tocilizumab. To test for bioequivalence, the ratios of the least squares (LS) geometric means for Cmax, AUClast, and AUCinf were compared with equivalence bounds of 0.80 to 1.25. The PK population consisted of all subjects who received both scheduled doses in periods 1 and 2; however, subjects who did not receive the full dose of study drug in both periods and subjects with more than 3 nonevaluable samples in any period were not included in the population. Safety and tolerability assessments consisted of adverse events (AEs), clinical and laboratory tests, vital signs, 12-lead electrocardiography, physical examination, and pain measurements. AEs investigated included anaphylaxis and serious hypersensitivity reactions, injection site reactions, infections, and device-related AEs. The safety population consisted of all randomly assigned subjects who received at least 1 dose of study drug.
2.3. Real-life human factors study (NCT02682823)
2.3.1. Study design
This was a nonrandomized, open-label, observational, phase 4 study to test device safety and effectiveness during use and to test human factors impacting the administration of tocilizumab by AI in patients with RA who previously received tocilizumab by the commercially available PFS. The planned sample size was 15 participants per user type (for a total of 45 participants), following the recommendations of the US Food and Drug Administration on applying human factors to medical devices [Citation11]. The study included 3 groups of patients who received 162 mg tocilizumab injected SC using the AI device at 3 visits (day 1, day 14, and day 28) (). Group 1 self-injected tocilizumab, group 2 received injections from nonprofessional caregivers, and group 3 received injections from qualified health care professional (HCPs). For groups 1 and 2, visit 1 consisted of comprehensive device training and injection executed by the subject or caregiver with the support and guidance of an HCP trainer. No training was provided to HCPs in group 3; patients received injections from the study nurse at visit 1. For all 3 groups, visits 2 and 3 were conducted for evaluation of use and performance.
The clinical settings in which injections were conducted were designed to match those of actual homes, with consideration of lighting, acoustics, and climate.
2.3.2. Patient eligibility criteria
The real-life human factors study included men and women who had RA (per American College of Rheumatology [ACR] 1987 criteria) [Citation12] for ≥6 months, were ≥18 years of age, and were receiving tocilizumab SC using the commercially available PFS. To facilitate a realistic usage scenario, any caregiver or HCP who was already acquainted with or supporting a patient enrolled into group 2 or 3, respectively, could be paired with that patient for the duration of the study. Key inclusion criteria included non-childbearing potential or continued use of current contraception; receiving 162 mg tocilizumab by PFS for ≥8 weeks and suitable to continue treatment at the currently prescribed dose regimen; suitable for AI treatment at home; left-hand dominance in ≥2 self-injecting patients; and paired caregivers and professionally qualified HCPs who were ≥18 years of age and were able and willing to administer injections. Key exclusion criteria were any serious medical condition or abnormality in clinical laboratory tests that precluded safe participation in and completion of the study; functional RA status class IV (per ACR criteria) [Citation13]; any condition interfering with pain evaluation; pregnancy or breastfeeding; participation in any previous tocilizumab study involving an AI, or previous use of the AI-1000 G1 or AI-1000 G2 in any human factors study.
2.3.3. Study outcomes
The primary purposes of this study were to demonstrate that the intended users could safely and effectively perform critical and essential tasks and to investigate the root cause for use errors and/or use-related difficulties. As such, the primary end point was the percentage of users who correctly performed all essential tasks during visit 2 (day 14) and visit 3 (day 28). Essential tasks had to be performed sequentially as follows: 1. Open carton, remove device and associated documents. 2. Remove cap. 3. Start injection by depressing needle-shield at injection site and pressing activation button. 4. Hold AI until the complete dose has been delivered.
Additional outcomes were AEs, including injection site reactions and AEs related to device use; injection site pain (according to a 100-mm visual analog scale [VAS] and a 6-point categorical score); injection site leak-back; and device satisfaction (assessed using a questionnaire).
2.3.4. Analyses
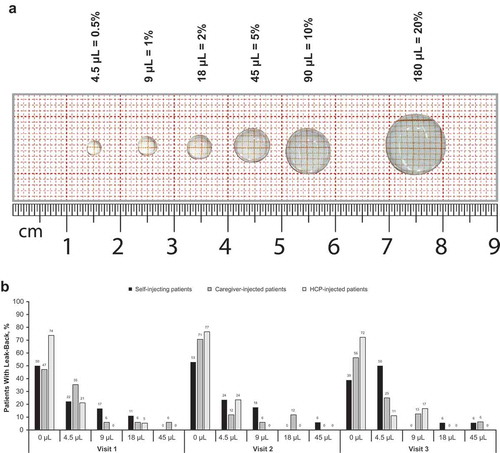
During visits 2 and 3, the investigator observed the injection process and recorded the completion of essential tasks, time to injection, hold time, and any usage errors or close calls; this was followed by a postinjection interview to capture root cause for any usage errors or close calls and any additional subject feedback. The Health Assessment Questionnaire–Disability Index (HAQ-DI) score was collected at baseline (visit 1), visit 2, and visit 3. This was used as a surrogate indicator for hand impairment to assess whether it influenced the patient’s ability to successfully use the AI. Data were collected on patient satisfaction with the AI and any injection pain experienced by the patient when using the AI. Safety outcome measures assessed the nature, frequency, and severity of AEs and injection site reactions throughout the study. Pain assessment was performed immediately after the injection had been completed and again 15 minutes later. The continuous assessment used a 100-mm horizontal VAS, from ‘no pain’ to ‘unbearable pain.’ The categorical assessment used a 6-point scoring method (0 = no pain, 1 = minimal but tolerable pain, 2 = mild but tolerable pain, 3 = moderate but tolerable pain, 4 = moderately severe but tolerable pain, and 5 = severe and intolerable pain). The volume of leak-back at each visit was estimated by comparing the droplet on the skin surface with a series of droplet standards on a life-scale photograph and selecting the droplet volume that corresponded most closely (). Clinical ease of use (satisfaction with the AI) was assessed with a device satisfaction questionnaire collected at visits 1 to 3. The questionnaire consisted of a set of statements for which users expressed their level of agreement, on a 5-point Likert-type scale, ranging from ‘strongly disagree’ (0) to ‘strongly agree’ (4). The all-participants population consisted of all patients and subjects who signed the informed consent form. The safety population consisted of all patients who signed the informed consent form, received ≥1 dose of the study medication, and had ≥1 safety assessment.
Figure 2. Leak back in the real-life human factors study. (a) Assessment.a (b) Outcomes.b HCP, health care professional. aNot to scale. bSelf-injecting patients: visit 1, n = 18; visit 2, n = 17; visit 3, n = 18. Caregiver-injected patients: visit 1, n = 17; visit 2, n = 17; visit 3, n = 16. HCP-injected patients: visit 1, n = 19; visit 2, n = 17; visit 3, n = 18.
3. Results
3.1. Bioequivalence study
3.1.1. Subject disposition and demographics
In total, 188 subjects were randomly assigned to a treatment sequence and received the study drug (safety population), and 161 had sufficient available data to be included in the PK analyses. Of the 27 subjects excluded from the PK population, 23 had not received study drug in both treatment periods and 4 did not have sufficient evaluable PK samples in a given treatment period.
Baseline demographics are shown in ; no meaningful differences were observed between the groups. Subjects’ mean age was 37.6 years, and most were male (82.4%) and white (71.3%).
Table 1. Baseline demographics of participants enrolled in the bioequivalence and real-life human factors studies.
3.1.2. Pharmacokinetics
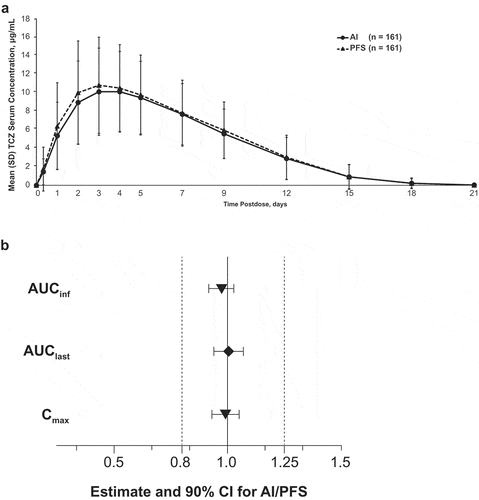
Mean tocilizumab serum concentrations were comparable after dosing with an AI or a PFS (, ). There were no subjects in whom Ctrough represented >5% of Cmax and no outliers. Mean Tmax was 88.9 hours with the AI and 85.4 hours with the PFS, and mean Kel was 0.0208 L/h with the PFS and 0.0206 L/h with the AI. Mean peak (Cmax) and overall systemic exposure (AUClast and AUCinf) to tocilizumab after AI administration were comparable to those after PFS administration, and bioequivalence criteria were met for Cmax, AUClast, and AUCinf (). The geometric mean ratio for AUClast was 1.00, and that for Cmax and AUCinf was 0.99 and 0.97, respectively. The 90% confidence intervals were included within the predefined bioequivalence range (0.80–1.25) (). For all administration sites (abdomen, thigh, upper arm), tocilizumab concentration-time profiles and associated variability were comparable for both devices. Although a bioequivalence analysis based on administration site was not preplanned, post hoc analysis indicated that mean Cmax, AUClast, and AUCinf were comparable for all 3 administration sites for both devices, and the 90% CIs of the geometric mean ratios were included in the equivalence range of 0.80 to 1.25 ().
Table 2. Summary statistics of tocilizumab serum PK parameters by administration device.
Table 3. Statistical analysis of tocilizumab bioequivalence by administration device.
Table 4. Tocilizumab bioequivalence by administration site.
Figure 3. Analysis of tocilizumab pharmacokinetic parameters by administration device in the bioequivalence study. (a) Mean serum concentration–time profiles. (b) Bioequivalence. AI, autoinjector; AUCinf, area under the serum concentration–time curve from time 0 to infinity; AUClast, area under the serum concentration–time curve from time 0 to the time of the last measurable concentration; CI, confidence interval; Cmax, maximum concentration; PFS, prefilled syringe; SD, standard deviation; TCZ, tocilizumab. Analyses were performed on log-transformed parameters with treatment, period, sequence, and subject nested within sequence as fixed effects. Dashed vertical lines represent the predefined bioequivalence range.
3.1.3. Safety and tolerability
In total, 149 AEs were reported by 92 subjects (48.9%); 72 were considered related to tocilizumab (). Most AEs were grade 1 in intensity, and none were grade ≥4; almost all AEs (145/149) had resolved at follow-up. No deaths or serious AEs were reported during the study, and there were no cases of anaphylaxis. Hypersensitivity AEs occurred in 7 patients (after administration by PFS in 3 patients and AI in 3 patients); all were nonserious grade 1 AEs that resolved without treatment or sequelae.
Table 5. Safety in the bioequivalence and real-life human factors studies.
The only AE that occurred in ≥5% of the study population was injection site reaction (36 subjects [19.1%]). Reactions following tocilizumab SC administration by AI and PFS occurred to a similar extent; all were grade 1 in intensity and resolved without treatment or sequelae. The most common symptoms were pain, erythema, and swelling. Most subjects experienced no injection site pain (categorical score 0; VAS <5 mm on 100-mm scale), and the degree and time course of pain was similar for both devices. Subjects experiencing injection site pain had categorical pain scores generally limited to minimal or mild but tolerable pain (scores of 1 or 2 on the 6-point categorical pain assessment), and the highest VAS scores observed were similar for both devices (range, 30–45 mm on 100-mm scale). The overall safety profile was comparable for both devices and was consistent with the well-established safety profile for tocilizumab.
3.2. Real-life human factors study
3.2.1. Patients
The all-participants population included 55 patients. One patient in the self-injecting group withdrew before receiving study drug; thus, the safety population consisted of 18 patients who self-injected tocilizumab, 17 who received injections from nonprofessional caregivers, and 19 who received injections from HCPs. The median treatment duration was 29 days (range, 1−36 days). Baseline demographics are shown in Supplementary Table S1; there were no notable differences between patients who self-injected and those who received injection from a caregiver or an HCP (not shown). The mean age of patients was 52.1 years, and most were female (72.2%) and white (92.6%). Four patients (7.4%) were left-handed.
3.2.2. Primary end point (overall success rate)
In total, including patients, caregivers, and HCPs, 92.3% and 98.1% of users at visits 2 and 3, respectively, performed all essential tasks required to deliver the full dose of tocilizumab.
3.2.3. Additional end points
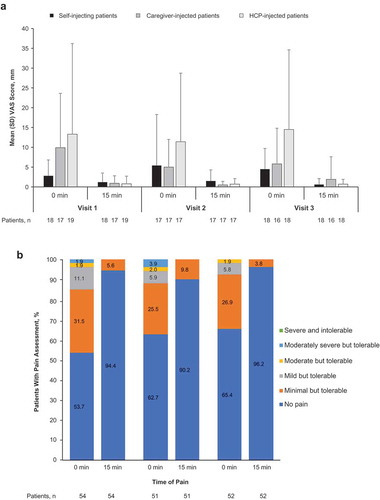
The HAQ-DI was assessed as a means to benchmark disease activity and to be used as a surrogate measure of hand impairment and was not intended to monitor treatment efficacy. The median HAQ-DI score at baseline was 0.8750, and there was little change in HAQ-DI at visits 2 and 3 (median, 0.8125 at both visits). Safety outcomes are described in . AEs occurred in 11 patients overall (20.4%). None of the AEs reported in this study were considered related to tocilizumab or to the AI. Two local injection site reactions were reported: 1 in the self-injecting patient group (bruising, grade 1) and 1 in the caregiver-injected group (pruritus, grade 2); both events resolved without sequelae. The only AE occurring in >1 patient was arthropod bites (n = 2; 3.7%). All AEs were grade 1 or 2; no AEs grade ≥3 were reported; no deaths or serious AEs occurred during the study. One patient in the caregiver-injected group withdrew from the study because of an AE of upper respiratory tract infection. This AE was not serious, was not considered by the investigator to be related to tocilizumab, and resolved without sequelae. VAS pain scores are shown in . At visit 1, immediately after injection, mean VAS scores were higher in the caregiver-injected and the HCP-injected patients than in the self-injecting patients; however, after 15 minutes, VAS scores were similar across all 3 study groups. Overall, VAS scores after the second and third injections were similar to those after the first injection. Categorical pain scores are shown in . After each injection, >50% of patients reported no pain immediately after injection, and >90% of patients reported no pain after 15 minutes. Most patients who felt pain reported that it was minimal and tolerable; no patients reported intolerable pain. No leak-back at the injection site was recorded for >50% of injections, regardless of study visit (). Minimal leak-back (4.5 μL or 0.5% of the total injection volume) was reported for ~25% of injections, and the volume of leak-back never exceeded 45 μL (5% of the total injection volume) in any group. Overall, most users were satisfied with the device. After the third injection, all self-injecting patients and HCPs and all but 1 caregiver agreed or strongly agreed that they were able to tell when the injection was completed and felt confident that they injected successfully. After the third injection, 92.3% of all patients agreed or strongly agreed that they would continue to use the AI, if possible.
Figure 4. Summary of patient assessment of injection pain. (a) 100-mm VAS score by injection group and (b) categorical pain scores in all patients at 0 and 15 minutes after injection in the real-life human factors study. HCP, health care professional; SD, standard deviation; VAS, visual analog scale.
4. Discussion
Convenience of use plays an important role in increasing patient compliance. An autoinjector as an alternative device for delivery of a subcutaneous injection was developed with this goal in mind. The potential comparability data package contains 3 components: analytical characterization, nonclinical assessment, and clinical assessment [Citation14]. The current manuscript summarizes the clinical assessment aspect for development of an injectable drug-device combination of tocilizumab with an AI. Data indicate that tocilizumab 162-mg administered SC using the AI was bioequivalent to the same dose administered using the marketed PFS and that RA patients, caregivers, and HCPs were all able to successfully use the AI in the treatment environment.
It is critical to establish bioequivalence of a new device – to ascertain that there are no significant differences in the rate and extent to which the active moiety becomes available at the site of drug action when administered at the same molar dose under similar conditions – to ensure the same therapeutic effect [Citation15,Citation16]. For SC injection, the needle has to pass through the skin (epidermis and dermis) to deliver the medicine into the SC tissue but should not penetrate any deeper into the underlying muscle fascia because this could result in an intramuscular injection. The PFS and the AI were both designed to ensure SC injection, but slight differences in injection depth and injection speed with the 2 devices could theoretically have affected the rate of absorption, injection pain, or leak-back. These factors were therefore robustly assessed in a bioequivalence and a real-life human factors study.
The study design for bioequivalence was rigorous. An important aspect in the design of a bioequivalence study is to account for all variables affecting the PK of the study drug. For tocilizumab, that meant not only accounting for degradation of the drug by the major clearance pathway but also monitoring the pharmacodynamic-driven effect on PK. Although the half-life of tocilizumab is long, as expected for a monoclonal antibody, a crossover trial design was chosen to allow intra-subject comparisons. A washout period of 6 weeks was used to account for any potential residual systemic tocilizumab following the first dose and to allow for the pharmacodynamic effects of tocilizumab to return to baseline, in particular IL-6 receptor levels. Tocilizumab undergoes target-mediated drug disposition (TMDD) [Citation17], which involves binding the drug to its specific target, IL-6 receptor, and contributes to overall clearance of the drug, especially at the low levels anticipated in the terminal phase after a single dose. Given that this has an impact on the serum levels of tocilizumab, designing a crossover trial for a drug with this mechanism of clearance must account not only for the half-life of the antibody but also for any effects of TMDD. With the exception of 1 subject (tocilizumab concentration, 0.135µg/mL), no one else had measurable concentrations of tocilizumab at the start of the second period, indicating an adequate washout period. Levels of soluble IL-6R (sIL-6R) were similar at baseline, end of period 1 (corresponding with start of dosing period 2), and end of period 2 (Supplementary Table S1), confirming that an adequate washout period was used from a pharmacodynamics perspective.
Because subcutaneous tocilizumab is approved for injection at 3 distinct body sites, bioequivalence was also evaluated between the AI and the PFS at each of these injection sites (abdomen, thigh, upper arm). Although this was a post hoc analysis, statistical analysis confirmed that the exposure of tocilizumab from the AI was comparable to that from the PFS at each site. The incidence of ISRs (19.1%) was higher in this bioequivalence study than in phase 3 clinical trials in which patients with RA received tocilizumab injections using the same PFS device either weekly (ISR incidence, 10.1%) or every other week (ISR incidence, 7.1%) over a period of 24 weeks [Citation1]. In this bioequivalence study, all injections were administered by a health care professional, which might have been the reason for the higher ISR incidence because the most common ISR symptom was injection site pain.
In addition to bioequivalence, the current data were intended to demonstrate usability of the new delivery device; for patients with RA who may have weakened hand grip or reduced dexterity, the ability to successfully administer treatment is an essential component of effective therapy. Human factor studies are an essential tool to evaluate safe and appropriate use by assessing functionality and correct procedure and highlighting potential sources of error [Citation18,Citation19]. In recent years, several such studies have been published reporting the usability and safety of AI devices for various patient populations, including those with RA [Citation2,Citation20–Citation22]. The results of the real-life human factors study indicated that most patients, caregivers, and HCPs were able to successfully use the AI and had confidence in it. This study was designed to demonstrate that the AI could be used safely and effectively by the intended users (patients, caregivers, and HCPs) in a clinical treatment environment that was configured to meet the conditions expected during anticipated usage. Patients enrolled into this study were representative of the overall population of patients with RA who would receive SC tocilizumab. By visit 3, the overall success rate at performing all essential tasks was 98.1%; of all patients who agreed that they would continue to use the AI, if possible, it was 92.3%. However, it must be noted that the study was not designed to assess patient preference, and some users may still prefer the experience of injecting with a PFS rather than an AI. More than half of all injections were completed without any leak-back, and minimal leak-back (0.5% of the total injection volume) was reported for ~25% of injections. The volume of leak-back never exceeded 5% of the total injection volume in any group, which would have no clinical consequence, especially given that tocilizumab is dosed frequently (weekly or every other week) for the treatment of RA and that users were observed performing SC injections by a human factors expert and premature withdrawals were recorded as use errors. Although some patients reported pain immediately after the injection, categorical scores after 15 minutes were 0 or 1, suggesting that the pain experienced was likely associated with the needle insertion or injection process. The tolerability of this treatment regimen was positive; no AEs were considered related to tocilizumab or to the AI, and only 2 local injection site reactions were reported.
This analysis had several limitations that should be considered. Given that the real-life human factors study was not randomized and that there was an element of choice, the self-injecting group enrolled more quickly than the other groups because of patient preference. Patients in the HCP-injected group had a numerically greater HAQ-DI at baseline, though this was likely due to chance because there was a high level of variability between patients regarding RA activity. In addition, no accepted and clearly defined measure for hand impairment was used in this study, so HAQ-DI was used as a proxy measure.
5. Expert Opinion
Criteria for demonstrating bioequivalence are well established and provide robust and reliable measures of comparability. However, given the lack of validated measures of the ease of use of new devices for patients, we used the Health Assessment Questionnaire–Disability Index as a proxy measure for hand impairment in our study of patient user handling of an autoinjector for subcutaneous administration of tocilizumab.
The goal of this research was to investigate an autoinjector as an alternative delivery device to the existing prefilled syringe for subcutaneous administration of tocilizumab. Our results show that patients can successfully use an autoinjector or a prefilled syringe for subcutaneous administration of tocilizumab according to their personal preference. The main challenge was in meeting rigorous statistical criteria in a controlled setting. A new delivery device should be tested in a controlled setting in the most sensitive and relevant patient population to establish comparability with existing devices. The evidence generated from this research was provided along with additional evidence to the United States Food and Drug Administration and the European Medicines Agency and formed the basis of license approval of the autoinjector for subcutaneous administration of tocilizumab from these authorities. Patient convenience is key to patient compliance, and providing alternative delivery devices to meet patients’ needs improves health care, especially for patients with limited physical ability. Novel delivery devices for these patients offer increased independence, which is a goal for drug delivery research that must be continually examined.
6. Conclusions
Single-dose SC administration of 162-mg tocilizumab with an AI was bioequivalent to administration with the currently marketed PFS. Intended users of the AI were successful in performing the tasks required to administer doses of tocilizumab. No new safety signals were observed with tocilizumab in either study. Given the positive results from both studies, tocilizumab administered via the AI is expected to result in clinical outcomes similar to those for the existing PFS.
Declaration of interest
All authors are employees of F. Hoffmann-La Roche Ltd. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, approved the final draft to be published, and agree be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conception and design of study: N Mallalieu, C Mela, S Fettner, F Wildenhahn, M Tavanti, C Wells, W Douglass
Analysis or interpretation of data: N Mallalieu, C Mela, S Fettner, F Wildenhahn, C Wells
Data availability statement
Qualified researchers may request access to data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
EODD-2018-ST-0249_Supplementary_Table_S1.docx
Download MS Word (24 KB)Acknowledgments
The authors thank Caroline Kreuzer for supporting the bioanalysis of pharmacokinetic and pharmacodynamic samples. The first draft of the manuscript was prepared by the authors with professional writing and editorial assistance provided by Sally-Anne Mitchell, PhD, and Sara Duggan, PhD, on behalf of F. Hoffmann-La Roche Ltd.
Supplementary materials
Supplemental data for this article can be accessed here
Additional information
Funding
References
- ACTEMRA® (tocilizumab) injection, for intravenous or subcutaneous use. South San Francisco, CA: Genentech, Inc.; 2018.
- Lange J, Richard P, Bradley N. Usability of a new disposable autoinjector platform device: results of a formative study conducted with a broad user population. Med Devices. 2015;8:255–264.
- Callis Duffin K, Bukhalo M, Bobonich MA, et al. Usability of a novel disposable autoinjector device for ixekizumab: results from a qualitative study and an open-label clinical trial, including patient-reported experience. Med Devices. 2016;9:361–369.
- Bayas A, Ouallet JC, Kallmann B, et al. Adherence to, and effectiveness of, subcutaneous interferon beta-1a administered by RebiSmart® in patients with relapsing multiple sclerosis: results of the 1-year, observational SMART study. Expert Opin Drug Deliv. 2015;12:1239–1250.
- Paul C, Lacour JP, Tedremets L, et al. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29:1082–1090.
- Berteau C, Schwarzenbach F, Donazzolo Y, et al. Evaluation of performance, safety, subject acceptance, and compliance of a disposable autoinjector for subcutaneous injections in healthy volunteers. Patient Prefer Adherence. 2010;4:379–388.
- Limmroth V, Gerbershagen K. Single-use autoinjector for once-weekly intramuscular injection of IFNbeta-1a. Expert Opin Drug Deliv. 2014;11:1969–1978.
- Hupperts R, Becker V, Friedrich J, et al. Multiple sclerosis patients treated with intramuscular IFN-beta-1a autoinjector in a real-world setting: prospective evaluation of treatment persistence, adherence, quality of life and satisfaction. Expert Opin Drug Deliv. 2015;12:15–25.
- Guidance for Industry: statistical approaches to establishing bioequivalence. Rockville, MD: US Department of Health and Human Services; 2001 Accessed 2018 Nov 5. https://www.fda.gov/downloads/drugs/guidances/ucm070244.pdf
- Agency EM. Guideline on the investigation of bioequivalence. London, UK: European Medicines Agency Website; 2010. https://www.ema.europa.eu/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf. cited 2018 Nov 5
- Guidance for Industry and Food and Drug Administration Staff: applying human factors and usability engineering to medical devices. Rockville, MD: US Department of Health and Human Services; 2016 Accessed 2018 Nov 5. https://www.fda.gov/downloads/medicaldevices/./ucm259760.pdf
- Arnett FC, Edworthy SM, Bloch DA, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324.
- Hochberg MC, Chang RW, Dwosh I, et al. The American College of Rheumatology 1991 revised criteria for the classification of global functional status in rheumatoid arthritis. Arthritis Rheum. 1992;35:498–502.
- Li Z, Easton R. Practical considerations in clinical strategy to support the development of injectable drug-device combination products for biologics. MAbs. 2018;10:18–33.
- Guidance for Industry: bioavailability and bioequivalence studies submitted in new drug applications or investigtional new drug applications—general considerations. Rockville, MD: US Department of Health and Human Services;2014. Accessed 2018 Nov 5. https://www.gpo.gov/fdsys/pkg/FR-2014-03-18/pdf/2014-05849.pdf
- Chow SC. Bioavailability and bioequivalence in drug development. Wiley Interdiscipl Rev Computat Stat. 2014;6:304–312.
- Frey N, Grange S, Woodworth T. Population pharmacokinetic analysis of tocilizumab in patients with rheumatoid arthritis. J Clin Pharmacol. 2010;50:754–766.
- Borsci S, Buckle P, Hanna GB. Why you need to include human factors in clinical and empirical studies of in vitro point of care devices? Review and future perspectives. Expert Rev Med Dev. 2016;13:405–416.
- Domanska B, Stumpp O, Poon S, et al. Using patient feedback to optimize the design of a certolizumab pegol electromechanical self-injection device: insights from human factors studies. Adv Ther. 2018;35:100–115.
- Schiff M, Koo J, Jin E, et al. Usability and acceptability of the abatacept pre-filled autoinjector for the subcutaneous treatment of rheumatoid arthritis. Adv Ther. 2016;33:199–213.
- Allaert FA, Schueller R, Mabile F, et al. Human factors study of ZENEO® (needle-free autoinjector) and comparison of different user instruction formats. Panminerva Med. 2018;60:52–59.
- Brand-Schieber E, Munjal S, Kumar R, et al. Human factors validation study of 3 mg sumatriptan autoinjector, for migraine patients. Med Devices. 2016;9:131–137.