
ABSTRACT
Introduction
Idiosyncratic drug-induced liver injury (IDILI) causes morbidity and mortality in patients and leads to curtailed use of efficacious pharmaceuticals. Unlike intrinsically toxic reactions, which depend on dose, IDILI occurs in a minority of patients at therapeutic doses. Much remains unknown about causal links among drug exposure, a mode of action, and liver injury. Consequently, numerous hypotheses about IDILI pathogenesis have arisen.
Areas covered
Pharmacokinetic and toxicodynamic characteristics underlying current hypotheses of IDILI etiology are discussed and illustrated graphically.
Expert opinion
Hypotheses to explain IDILI etiology all involve alterations in pharmacokinetics, which lead to plasma drug concentrations that rise above a threshold for toxicity, or in toxicodynamics, which result in a lowering of the toxicity threshold. Altered pharmacokinetics arise, for example, from changes in drug metabolism or from transporter polymorphisms. A lowered toxicity threshold can arise from drug-induced mitochondrial injury, accumulation of toxic endogenous factors or harmful immune responses. Newly developed, interactive freeware (DemoTox-PK; https://bit.ly/DemoTox-PK) allows the user to visualize how such alterations might lead to a toxic reaction. The illustrations presented provide a framework for conceptualizing idiosyncratic reactions and could serve as a stimulus for future discussion, education, and research into modes of action of IDILI.
1. Introduction
Adverse drug reactions (ADRs) remain an important contributor to mortality and morbidity. The liver is a major target of these reactions. Many drug candidates that are intrinsically hepatotoxic are eliminated from development during preclinical toxicity testing. Nevertheless, several hundred marketed drugs have been linked to liver injury, and the number continues to grow [Citation1]. Drug-induced liver injury (DILI) is an important cause of failure in clinical trials and a major reason for issuing warnings or withdrawal of drugs from the market [Citation2].
More than half of acute liver failure cases in the U.S. occurs from DILI. Most of these cases arise from overdose of acetaminophen (APAP). APAP-induced liver injury is an example of intrinsic drug toxicity, for which reactions leading to injury are clearly dose-dependent and usually occur in overdose situations. About 12% of acute liver failure cases are due to idiosyncratic reactions to other drugs or to herbal dietary supplements [Citation3]. Idiosyncratic DILI (IDILI) is best defined as an hepatotoxic reaction to a drug that occurs in a minority of patients during drug therapy [Citation4]. The fraction of patients who experience IDILI from a specific drug is typically quite small. Idiosyncratic toxicity is distinguished from intrinsic drug toxicity in that, although idiosyncratic reactions likely depend on dose within each particular patient, they are not as obviously dose-dependent across a population due to marked differences in susceptibility among individuals. Although the fraction of acute liver failure cases due to IDILI may seem small, these reactions are particularly insidious because they occur at therapeutic drug doses, often without warning, and are difficult to diagnose [Citation5]. This applies also to liver injury arising from the intake of herbal dietary supplements, which are subject to less regulation and typically less toxicity evaluation than pharmaceuticals and for which liver injury cases are underreported and on the rise [Citation3,Citation6,Citation7].
IDILI liability is sometimes discovered prior to or during clinical trials, leading to abandonment of further development of the offending drug. However, because of the rarity with which these adverse reactions occur, IDILI is often not identified until the drug has been on the market and many people have been exposed. Accordingly, the IDILI liability of newer drugs might not reveal itself immediately. Withdrawal from the market by order of the FDA often accompanies the discovery of IDILI association, but such action can depend on the indication of use for the drug; for example, an anticancer drug with IDILI liability might retain FDA approval because its life-saving potential outweighs the risk of hepatotoxicity. There are ongoing efforts to develop cell-based assays and quantitative biological models for the preclinical identification of drug candidates likely to cause IDILI in order to prevent offending drugs from reaching the marketplace. Genetic and transcriptomic analyses of patient characteristics associated with susceptibility have provided suggestions as to mechanisms of action and potential predictive strategies [Citation8]. However, no tests have emerged so far that have gained universal use by pharmaceutical companies and that can predict IDILI liability consistently across drug classes and with high accuracy.
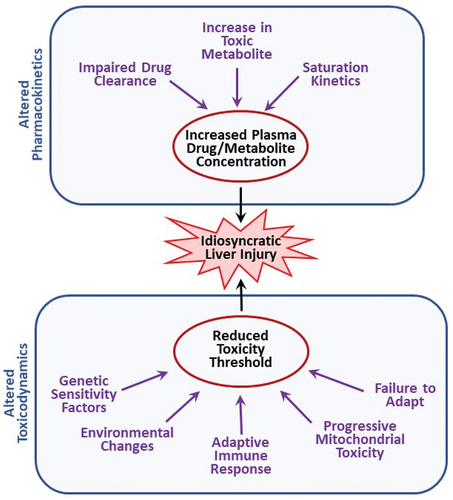
Idiosyncratic reactions depend on (1) the characteristics of the offending drug and (2) characteristics of the individual. Examples of drug characteristics that tend to associate with IDILI include drug lipophilicity, therapeutic dose of the drug (drugs given at larger doses tend to have greater IDILI liability) and structural features that can lead to reactive metabolites or to activation of cell death pathways [Citation9]. Individual characteristics can encompass any genetic or environmental factors that increase susceptibility to an adverse reaction. As diagramed in , these can either affect the disposition of a drug, leading to altered pharmacokinetics, or cause quantitative or qualitative changes in its mechanism(s) of toxicity (i.e. toxicodynamics), which would be reflected in an altered threshold for toxicity.
Figure 1. Potential modes of action of idiosyncratic adverse drug reactions. Idiosyncratic hepatotoxicity can arise either from altered drug disposition resulting in greater than normal exposure to a drug or its toxic metabolite(s) or from a change in toxicodynamics that leads to a reduction in the threshold for toxicity.
Much remains unknown about the etiology of IDILI or how liver injury relates to drug pharmacokinetics or toxicodynamics in affected patients. Indeed, inciting factors that precipitate IDILI reactions are numerous and often not identified. Nevertheless, there are features that appear commonly in IDILI cases. IDILI reactions from most drugs occur during maintenance drug therapy (see below), are typically rare and are delayed in onset from a few days to several months after commencing drug therapy. Although some IDILI cases result in patient death, in most cases withdrawal of drug treatment results in patient recovery when injury is not too severe [Citation10]. For many drugs, reinstitution of drug therapy leads to reoccurrence of the reaction.
Because IDILI reactions remain poorly understood and difficult to study, several hypotheses to explain their etiology have emerged. In this review, the likely pharmacokinetic and toxicodynamic implications of these hypotheses are discussed, and graphical illustrations of likely pharmacokinetic changes associated with various modes of action of IDILI are presented. It is hoped that these illustrations will aid in conceptualizing IADRs, especially for those not expert in pharmacokinetics, bolster discussion and research into modes of action and aid in the teaching of pharmacokinetic and pharmacodynamic principles related to IDILI.
2. Kinetics of maintenance drug therapy
For most drugs that cause IDILI, liver injury occurs amid repeated drug administration in the context of maintenance therapy. The goal of maintenance therapy is to provide drug effectiveness over a prolonged period of time. From a pharmacokinetic perspective, this translates into maintaining plasma concentration that is therapeutic but below the threshold concentration at which toxicity occurs. This is accomplished by repeated administration of a drug at a constant dose and interval between drug administrations (). Initially, the concentration of drug in plasma or blood is low, but it then increases with each administration until a steady state (plateau) concentration is achieved. Although idiosyncratic hepatotoxicity for most drugs occurs during maintenance therapy after a steady state has been achieved, a notable exception involves certain antibiotics, which can cause IDILI without reaching a steady-state plasma concentration.
Figure 2. Drug plasma concentration during maintenance therapy. When drugs are taken over an extended period, the usual goal is to maintain plasma drug concentrations above the minimum effective concentration but below the threshold for toxicity. This is accomplished by administering the drug repeatedly at the same dose and dosing interval. Initially, the concentration of drug in blood is small but increases with each dose until the amount of drug eliminated during a dosing interval equals the next dose. A plateau (steady state) in the concentration of drug in plasma is thereby achieved. Solid line depicts drug concentration in plasma; blue dashed line indicates the plasma concentration below which the drug is ineffective; red dashed line represents the plasma concentration above which toxicity occurs (i.e. the threshold for toxicity).
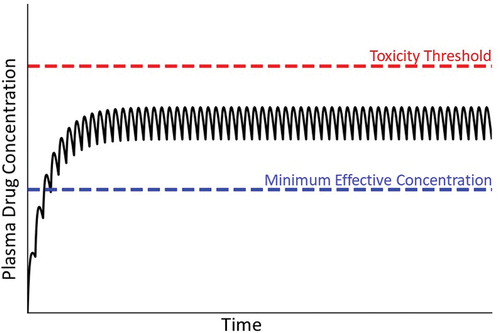
Attaining a steady-state plasma concentration requires that the drug is eliminated from the body as a first-order process (i.e. elimination rate is proportional to drug concentration). When a steady state is achieved, the amount of drug eliminated during the dosing interval equals the drug dose. In this treatise, we assume that at steady state the drug concentration in the target organ is proportional to plasma drug concentration. Conceptually, toxicity occurs when plasma concentration rises above a toxicity threshold (depicted as a red dashed line in ). When drug elimination is first-order, as dose increases the steady state drug concentration increases proportionately. In overdose situations, this increase in steady state drug concentration can eventually result in intrinsic toxicity, that is, as the steady state plasma concentration increases to exceed the threshold for toxicity, injury occurs (). However, even at a therapeutic dosing regimen the steady state plasma concentration can differ among individuals due to genetic and/or environmental factors and can sometimes exceed the threshold for toxicity in a small subset of patients, leading to an idiosyncratic reaction.
2.1. Importance of first-order kinetics in achieving a steady state
Intrinsic toxicity usually occurs in overdose situations, in which first-order drug elimination can transition to zero-order kinetics. Most small molecule pharmaceuticals are eliminated from the body by metabolism. Enzyme kinetics of drug metabolism reactions can be complex, but important principles of simpler, Michaelis–Menten kinetics, such as enzyme saturation, still apply [Citation11]. According to Michaelis–Menten kinetics, the rate of production of a metabolite from a parent drug (reaction velocity, V) is directly proportional to drug concentration when the drug concentration is much less than the enzyme’s Km (Michaelis constant; the concentration of drug at 50% maximal velocity [Vmax]) ()). For a drug that is eliminated by metabolism, this condition defines first-order kinetics and is necessary for a steady state drug concentration to occur during maintenance therapy. Fortunately, the Km of most drug-metabolizing enzymes is in the mM range, whereas therapeutic drug concentrations are in the µM range, so that a steady state concentration is readily achieved.
Figure 3. Michaelis-Menten kinetics and drug concentration in plasma over time under the condition of zero-order kinetics. A. Michaelis-Menten kinetics. As the concentration of drug increases above zero, the velocity of the reaction (V, metabolism rate) increases. When the drug concentration is substantially smaller than Km (the drug concentration at half-maximal V), first-order kinetics apply (i.e. metabolism rate is proportional to drug concentration). At concentrations much larger than Km (i.e. at Vmax), zero-order kinetics occur (i.e. metabolism rate is constant and independent of drug concentration). At concentrations near Km, a transition from first- to zero-order will occur in vivo as plasma drug concentration increases, because first-order kinetics no longer apply. B. Transitional kinetics. When approaching saturation kinetics during maintenance therapy, the dose of drug taken during the dosing interval exceeds the ability to eliminate (e.g. metabolize) the dose within the dosing interval. This results in increasing plasma concentration of drug with time and a failure to attain a steady state (plateau). Ultimately, plasma drug concentration exceeds the threshold for toxicity, and an adverse reaction occurs.
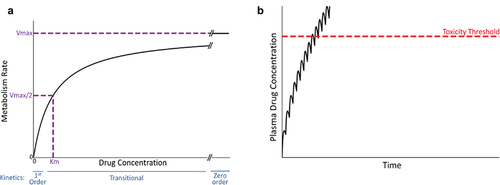
When drug concentration markedly exceeds Km, the amount of enzyme available rather than the drug substrate concentration limits the rate at which metabolism of a drug occurs. Under this ‘saturation’ condition, the rate of metabolism becomes constant–i.e. obeys zero-order kinetics (see )). Under these conditions, rather than achieving a steady state plateau during repeated drug administration, plasma concentration continually increases, at some point in time exceeding the toxicity threshold ()). If drug concentration is near Km, plasma drug concentration can appear to approach a steady state early after the onset of maintenance therapy only to transition to zero-order kinetics associated with a steep, linear rise in plasma concentration. Similar principles of zero-order kinetics apply to drug elimination by transporters in the liver and kidney.
In theory, the kinetics depicted in ) can occur, for example, in individuals with a drug metabolism polymorphism that results in a Km near the therapeutic drug concentration in the liver. Indeed, many polymorphisms in cytochromes P450 (CYPs) and other drug metabolizing enzymes have been identified that affect metabolic activity; however, effects on substrate affinity (Km) and the relationship of polymorphisms to toxicity are less well studied [Citation12]. Other possibilities include allosteric modifications of drug metabolizing enzymes and conditions in which cofactors for metabolism become limiting. Such cofactors could include O2 or NADPH for phase 1 CYPs; glutathione (GSH), glucuronic acid or sulfate for phase 2 enzymes; and ATP for transporters. Under these limiting conditions, elimination of the drug would appear to obey transitional and ultimately zero-order kinetics, and maintenance therapy would lead to a scenario similar to that pictured in ), a rise in plasma drug concentration that can exceed the toxicity threshold. For CYPs, polymorphisms that affect binding of cofactors have been identified, and in theory these too could effect a transition to zero-order kinetics [Citation12].
Green tea extracts (GTEs) are an example of an herbal supplement that might obey zero-order kinetics. A minority of people who consume GTEs experience hepatocellular injury linked to the catechin components in GTEs [Citation13]. Saturation of drug metabolizing enzymes or efflux transporters is thought to play a role in the kinetics of GTE components and in their hepatotoxicity, but more study is needed to support this contention [Citation13]. The incidence of liver injury from green tea preparations is 5% with latency of 10 days to seven months, and in some cases rechallenge following cessation of dosing resulted in return of liver injury [Citation14,Citation15]. Fasting is a clear risk factor for toxicity, and genetic susceptibility is presumed to be a factor in some patients. The dose-relatedness of GTE toxicity is not entirely clear, but toxicity tends to occur with higher GTE intake and/or longer consumption periods. GTE toxicity is likely an example of intrinsic toxicity, although individual susceptibility factors could play important roles.
The time required to reach the threshold for toxicity under transition from first-order to zero-order kinetics can vary depending on the proximity of initial drug concentration to the Km of the metabolizing enzyme(s). Accordingly, the toxicity can be somewhat delayed in onset. Cessation of drug administration would, of course, lead to a reduction in plasma concentration as remaining drug is metabolized and the concentration ultimately falls below the toxicity threshold. If drug therapy is subsequently resumed at the same dosing regimen in a sensitive individual, reoccurrence of toxicity associated with rising plasma concentration would be expected. Accordingly, a toxic reaction arising from transitional pharmacokinetics can demonstrate characteristics of an idiosyncratic reaction. That said, to our knowledge, toxicities due to transitional/saturation kinetics have only occurred as intrinsic toxicity in drug overdose situations and not during usual maintenance therapy.
3. Conditions under which an IDILI reaction can occur
As noted above, much uncertainty and controversy exist surrounding IDILI reactions; as a result, numerous theories as to the etiology of IDILI have arisen. What follows are brief descriptions of inciting factors or conditions that plausibly result in IDILI reactions. The scenarios described represent various hypothesized modes of action of IDILI in the context of maintenance drug therapy. Some DILI reactions that depend minimally on characteristics of the individual and which are more appropriately classified as ‘intrinsic toxicity’ (e.g. drug–diet interactions) can be misconstrued as an idiosyncratic reaction if the inciting cause remains undiscovered; accordingly, this category is included in the discussion.
For each hypothesis, examples are provided of drugs for which some evidence exists that the drug acts by a particular mode of action. Importantly, for most IDILI-associated drugs mode-of-action evidence in humans is lacking. Where human studies have provided evidence for a particular IDILI hypothesis, the evidence is associative and does not prove cause-and-effect. For several drugs, evidence from studies in animals or in vitro points to a potential mechanism, and we allude to such evidence in the review. Worth emphasizing is that for essentially no drug is there sufficient evidence to corroborate fully a specific mechanism of IDILI. A final caveat is that some drugs might cause liver injury by more than one mechanism.
For each of the IDILI hypotheses described, a graphical illustration highlighting the most probable associated alterations in pharmacokinetics or toxicodynamics is presented. Toxicodynamic alterations are reflected in an altered drug toxicity threshold, that is, the plasma concentration above which toxicity occurs. This threshold is often considered to be constant (as in ), but it can differ among individuals, and even within an individual it can change with time. Since toxicity is influenced by both toxicity threshold and steady state plasma concentration, and these are in turn influenced by genetic and environmental factors, several different scenarios underpinning IDILI need to be considered. Finally, even in intrinsic reactions there can be an element of ‘idiosyncrasy’ due to differences in individual susceptibility. These factors can blur the lines between intrinsic and idiosyncratic DILI [Citation16,Citation17]. A defining quality of idiosyncratic reactions is that they occur during normal drug therapy; accordingly, the sections below describe conditions under which toxicity can occur within the context of a therapeutic dosing regimen.
3.1. Steady state plasma drug concentration elevates above toxicity threshold
Therapeutic drug treatment regimens can lead to greater than usual steady state drug concentrations under conditions of reduced drug elimination. This can occur, for example, when drug metabolizing enzymes or export transporters responsible for drug elimination are inhibited or reduced in expression. For example, a pediatric patient undergoing standard dosing with the thrombopoietin receptor agonist, eltrombopag, suffered acute liver failure [Citation18]. Her liver toxicity was associated with markedly elevated plasma drug concentration. Genetic analysis revealed allelic variations in drug metabolizing enzymes (CYP2C8, UDP glucuronyltransferase 1A1) and the transporter, ABCG2.
Diseases and genetic polymorphisms in hepatic transporter proteins have the potential to alter drug clearance and affect plasma drug concentrations. Tolvaptan is a vasopressin V2-receptor antagonist used to slow decline in renal function in patients with polycystic kidney disease, and in some patients it causes liver injury classified as hepatocellular or mixed. Tolvaptan and its metabolites are secreted into the bile by transporters, and studies in a rodent model of polycystic kidney disease indicated enhanced liver exposure to the drug due to impaired expression of transporters and reduced biliary drug excretion [Citation19,Citation20]. Additionally, specific transporter polymorphisms have been identified that impair hepatocellular uptake of statins, thereby increasing systemic drug exposure. There is speculation that this contributes to the hepatocellular or cholestatic hepatitis associated with statins [Citation21]. However, although many drugs have been shown to interact with transporters, and many transporter polymorphisms have been identified, studies definitively linking transporter polymorphisms causally to IDILI are lacking.
Notably, several dietary supplements have been identified as inhibitors of CYPs [Citation22,Citation23]. For example, usnic acid is a nutritional supplement used for weight loss. It causes hepatocellular IDILI but is also a potent inhibitor of several CYP isoforms [Citation24]. As such, it has the potential to elevate plasma and liver concentrations of drugs that are eliminated by CYPs. If such drug-supplement interaction results in toxicity, it could be construed as an idiosyncratic reaction to some drugs metabolized by CYPs. Importantly, patients often do not report their consumption of herbal supplements to clinicians, and so the inciting cause can remain unknown.
Finally, reduced drug elimination associated with elevated drug concentration would be expected under conditions in which cofactors required for drug metabolism become limiting (e.g. limitations in O2 for oxidative drug metabolism; deficiency in glucuronic acid, GSH, or sulfur donors for conjugation). For example, hypoxia can slow the clearance of numerous drugs eliminated by oxidative metabolism [Citation25–27]. However, more research is needed to link cofactor limitations and consequent reductions in drug clearance to hepatotoxicity of drugs, especially in clinical situations.
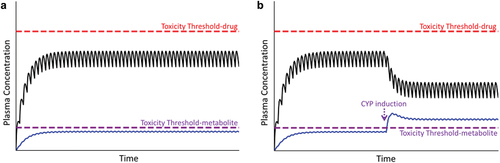
The result of such conditions is illustrated in , in which the plasma drug concentration during usual maintenance therapy exceeds the threshold for toxicity (compare with ). If a patient’s metabolism/transport capabilities are compromised prior to the initiation of drug therapy, toxicity should occur early after drug administration ()). However, if these conditions develop sometime during the course of drug therapy, a new steady state will be attained that exceeds the toxicity threshold later during the course of therapy ()).
Figure 4. Plasma drug concentration over time under conditions in which the drug concentration exceeds the toxicity threshold. Panel A represents a situation in which the patient’s metabolism or transport capabilities are compromised before the start of therapy; in this case, the plasma drug concentration can exceed the threshold for toxicity, resulting in an early onset of injury (violet dashed line). Panel B represents a decrease in the patient’s drug metabolism or excretion during maintenance therapy. In this case, plasma drug concentration rises and has the potential to exceed the toxicity threshold. A new, elevated steady state plasma concentration will ultimately be established (not shown).
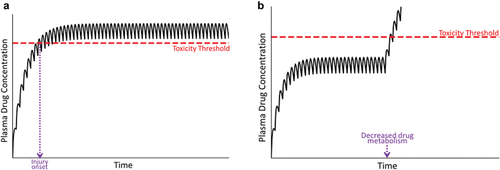
This situation could demonstrate all of the characteristics of an idiosyncratic reaction listed above, namely, the toxicity occurs at therapeutic doses, can be delayed in onset, could lead to recovery from injury upon drug withdrawal (i.e. when the plasma concentration decreases below the toxicity threshold), and reinstitution of drug therapy would see recurrence of the toxicity. If the cause of the elevated plasma drug concentration was known, for example, an identified drug–drug interaction, this situation would probably not be considered to be idiosyncratic; however, if the inciting factor(s) was not known, for example, an unrecognized dietary supplement interaction, it would likely be construed as ‘idiosyncratic.’
3.2. Production of toxic metabolite(s) is elevated
As mentioned above, DILI tends to be associated with drug lipophilicity, which is a characteristic of drugs that are metabolized by enzymes such as CYPs. Indeed, database reviews revealed that a majority (61%) of drugs that have caused DILI are substrates for CYPs [Citation28,Citation29]. Although such association does not prove cause and effect, for some drugs a toxic drug metabolite appears to mediate injury to liver and other organs. Such metabolites can be reactive intermediates produced during CYP-mediated metabolism but can form for some drugs from conjugation with glutathione or other endogenous molecules [Citation30,Citation31]. If a metabolite is responsible for liver injury from a drug, then the relevant threshold for toxicity will be determined by the metabolite rather than by the parent drug (). Under normal therapeutic situations, concentrations of the toxic metabolite remain subthreshold, and no toxicity occurs ()). However, if for genetic or environmental reasons conversion of drug to the toxic metabolite is more efficient than normal, the toxicity threshold of the metabolite can be exceeded, predisposing the liver to injury. If induction of the relevant metabolic enzyme(s) occurs during drug therapy, then an enhanced production of the toxic metabolite can occur, leading to a sudden and unexpected toxic response during maintenance therapy that is delayed in onset and may be construed as an idiosyncratic event ()).
Figure 5. Plasma concentration of parent drug and its metabolite over time. Toxicity thresholds are shown for both parent compound (red dashed line) and metabolite (violet dashed line). Panel A represents the desired therapeutic condition in which the plasma concentration of parent drug as well as its metabolite(s) (blue line) remain below their respective toxicity thresholds. Panel B represents the condition in which the concentration of the metabolite exceeds its toxicity threshold, in this case due to induction of CYP during maintenance therapy, prompting an adverse reaction.
Evidence from genome-wide association studies suggests a relationship between DILI and polymorphisms in several drug metabolizing enzymes or drug transporters, but such associations do not prove causality, and many of the studies are in need of replication [Citation32]. The causal role of reactive drug metabolites in IDILI is difficult to study, but several animal models have been developed that point to their potential importance in causing injury, and cell culture systems are being used to explore mechanisms and better tests to predict toxicity [Citation33]. For example, diphenylhydantoin (DPH, phenytoin) and carbamazepine (CBZ) are antiepileptic drugs associated with human IDILI classified as hepatocellular, mixed, or cholestatic. In vitro studies revealed that both of these drugs are metabolized by CYPs to reactive metabolites capable of binding to cellular proteins. Repeated administration of DPH to mice for 5 days resulted in pronounced hepatocellular necrosis [Citation34] that was attenuated by a CYP inhibitor, suggesting that the toxicity was mediated by a reactive metabolite. Similarly, repeated administration of CBZ to rats for several days resulted in liver injury accompanied by elevated hepatic CYP3A4 expression and activity [Citation35]. A CYP3A inhibitor suppressed the injury. The development of liver injury was associated with a decline in hepatic GSH concentration, and depletion of GSH prior to drug administration exacerbated the hepatotoxicity, presumably because inactivation of a reactive metabolite was reduced. Notably, in both rodents and humans, CBZ and DPH require repeated administration for DILI to occur, and both drugs induce CYPs in rodents and humans [Citation36–38]. In humans, CBZ can induce its own metabolism [Citation38], raising the possibility that increased expression and activity of CYPs that result in reactive CBZ metabolites contribute to IDILI from this drug. These observations suggest that autoinduction of metabolism to a toxic metabolite occurs during repeated administration of some drugs and plays a role in liver injury from these drugs.
These metabolic attributes can explain why such drugs cause liver injury but not why IDILI from CBZ and DPH occurs in a minority of patients who are particularly susceptible. It could be that metabolic activation or detoxification differences in certain individuals render them more susceptible to injury. Indeed, polymorphisms in CYP genes affect autoinduction and the elimination of DPH in people [Citation36]. Alternatively, CBZ and DPH have been administered in combination with other drugs, such as phenobarbital, which is well recognized as a CYP inducer [Citation39,Citation40].
Notably, results of in vitro and animal studies showed that some dietary supplements can induce drug metabolizing enzymes and thereby have the potential to enhance formation of reactive metabolites; however, the clinical significance of such induction as it relates to IDILI remains largely unproven [Citation23,Citation24,Citation41–43]. Thus, drug metabolism polymorphisms and/or coexposure to agents that induce bioactivating enzymes or reduce activities of enzymes that detoxify harmful metabolites could contribute to IDILI and could explain why IDILI from these drugs occurs infrequently. Depending on the severity of injury, withdrawal of drug would eliminate production of toxic metabolite and could therefore result in recovery; reinstitution of drug therapy might or might not lead to recurrence of injury, depending on whether exposure to the inducing agent remained.
3.3. Genetic sensitivity factors reduce the threshold for toxicity
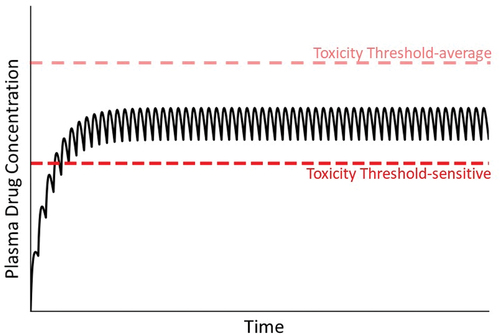
In addition to the pharmacokinetic alterations discussed above, ‘toxicodynamic’ conditions can lead to IDILI. These would appear as a decrease in the threshold for toxicity (compare with ). For example, a genetic deficiency in a cytoprotective mechanism could lower the threshold for toxicity. A loss-of-function polymorphism in antioxidant enzymes or in other genes needed for induction of cellular protectants would fall into this category.
Figure 6. Concentration of drug in plasma during maintenance therapy under condition in which a patient’s toxicity threshold is low relative to the majority of patients. As mentioned in the text, a variety of factors can lead to a reduced toxicity threshold for an individual. Without prior knowledge of this, normal maintenance therapy could lead to concentrations of drug in plasma that exceed the lower-than-normal toxicity threshold (compare to ).
As an example, the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2), controls the expression of several antioxidant enzymes. In a mouse model of 1-bromopropane (1-BP)-induced liver injury, Nrf2 null mice given 1-BP had more liver necrosis and lipid peroxidation than wild-type mice [Citation44]. 1-BP exposure increased expression of enzymes involved in antioxidant defense, including heme oxygenase-1 (HO-1), NAD(P)H: quinone oxidoreductase 1 (NQO1), glutathione reductase and enzymes involved in glutathione biosynthesis in wild type but not Nrf2-null mice, suggesting that a compromised antioxidant defense rendered Nrf2 mice susceptible to 1-BP hepatotoxicity. Human patients carrying a specific GG genotype in the gene encoding HO-1 were found to be at greater risk of hepatocellular injury from antitubercular drugs than those with the AA genotype [Citation45]. Similarly, manganese superoxide dismutase (MnSOD) is an enzyme that protects against oxidant damage, and human subjects with a mutant allele in the gene encoding MnSOD had an increased risk of hepatocellular injury from antitubercular and other drugs [Citation46]. Impairment of antioxidant defense would be expected to lower the toxicity threshold for drugs that act through a mechanism involving oxidative damage.
A genetic difference that favors activation of cell death pathways can increase sensitivity to drug-induced injury and could result in a reduced toxicity threshold. In this regard, genetic defects in apoptotic or necrotic pathway constituents, such as signaling kinases, phosphatases, cell death receptors, or transcription factors that control expression of these proteins could be important [Citation47,Citation48]. As an example, intracellular signaling involving signal transducer and activator of transcription 1 (STAT1) can lead to death of hepatocytes, and STAT1 activation contributes to cell death from certain IDILI-associated drugs in vitro [Citation48]. Recently, a single nucleotide polymorphism (SNP) in the gene encoding STAT1 was found to be associated with severe hepatocellular injury from crizotinib in non-small cell lung cancer patients [Citation49].
Since there is no routine evaluation of such genetic sensitivity factors, they escape detection in patients, so that injury resulting from them would appear as unexplained and idiosyncratic.
3.4. Drug causes a progressive decrease in toxicity threshold
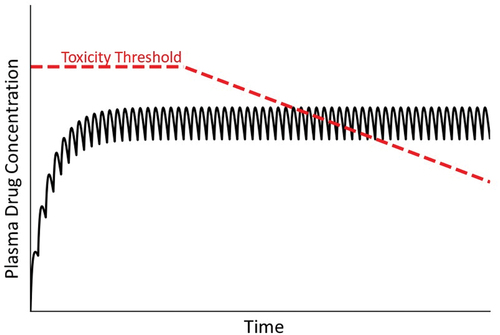
Several drugs that cause IDILI in people also cause mitochondrial injury in hepatocytes in vitro [Citation50]. Mitochondrial injury can arise from mitochondrial oxidative stress (e.g. from anticancer quinones, fibrates), inhibition of respiration (e.g. barbiturates, thiazolidinediones), uncoupling of oxidative phosphorylation (e.g. nitrophenols, nonsteroidal anti-inflammatory drugs), or inhibition of mitochondrial protein synthesis or DNA replication (e.g. antiretroviral drugs) [Citation51]. Such observations have led to the hypothesis that progressive injury to mitochondria leads ultimately to hepatocellular dysfunction and death. This idea is consistent with a delayed injury onset during maintenance therapy but cannot explain why IDILI is typically rare, unless some individuals have mitochondria that are inherently susceptible to drug-induced dysfunction (). Alternatively, factors such as environmental exposures or genetic deficiencies that cause mitochondrial dysfunction might render some individuals more susceptible to injury from drugs that cause toxicity by mechanisms independent of, but influenced by, compromised mitochondria. This would appear as a lower threshold for hepatocellular injury in these susceptible individuals. If mitochondrial impairment is either reversible or can be overcome by replication of hepatocytes, withdrawal of drug could presumably lead to recovery. Thus, there seem to be circumstances under which mitochondrial toxicity could lead to IDILI.
Figure 7. Drug concentration in plasma over time under condition in which the toxicity threshold decreases progressively. As mentioned in the text, mitochondrial dysfunction and other factors can lead to a gradual decrease in the toxicity threshold within an individual. As such, at some time the threshold can fall below the steady state plasma concentration, leading to overt injury.
One consequence of mitochondrial toxicity is lactic acidosis (increased concentration of lactic acid in blood). This arises from inhibition of mitochondrial pyruvate dehydrogenase, which is the rate-limiting step in aerobic oxidation of pyruvate, and from inhibition of mitochondrial respiration that leads to increased glycolysis. Lactic acidosis has been reported in patients undergoing therapy with one of several biguanide drugs (metformin, phenformin, and buformin) which cause IDILI that can present as hepatocellular, mixed, or cholestatic [Citation52,Citation53]. The incidence of liver injury within this class is related to the severity of mitochondrial toxicity in vitro [Citation54,Citation55].
Some nucleoside analog reverse transcriptase inhibitor (NRTI) antiretroviral drugs might also cause IDILI through mitochondrial toxicity. In vitro, these drugs inhibit mitochondrial DNA (mtDNA) polymerase-gamma (pol g) [Citation56], leading to depletion of mtDNA. mtDNA encodes components of the electron transport chain, and its depletion can lead to disruption of mitochondrial oxidative function. Some patients receiving NRTI therapy developed significant mtDNA depletion, lactic acidosis, and liver injury [Citation57–59]. Abnormal mitochondrial structure has been observed in skeletal muscle and heart in rodents treated for 5 weeks with zidovudine (AZT) [Citation60,Citation61], a NRTI drug associated with IDILI.
Tamoxifen is an antiestrogen used commonly for the treatment of estrogen receptor-positive breast cancer, but IDILI is a major concern in patients receiving tamoxifen. Patients can present with cholestatic, mixed, or hepatocellular injury. Tamoxifen has multiple effects on mitochondria, resulting in depressed phosphorylation and reduced ATP production. This mitochondrial toxicity likely contributes to tamoxifen-induced liver injury [Citation62].
As mentioned above, one possibility for the rare occurrence of IDILI reactions is that some individuals have mitochondria that make them particularly susceptible to drug-induced mitochondrial damage. In a small cohort of patients with valproic acid-induced liver injury, nearly half had mutations in the gene that encodes mtDNA pol g [Citation63]. In addition, flutamide, a drug associated with idiosyncratic hepatocellular injury, is metabolized to a reactive metabolite that inhibited respiration in platelets from patients in a manner dependent on their mtDNA haplotype [Citation64]. Furthermore, alterations in mitochondrial function underlie the mechanism by which flutamide caused hepatotoxicity in mice [Citation65]. Thus, mitochondrial damage can occur from numerous drugs by different mechanisms, and for several drugs evidence exists that mitochondrial dysfunction is associated with IDILI.
Although this section has emphasized mitochondrial injury, slowly developing stress can occur from other mechanisms, such as the accumulation of endogenous factors that ultimately reach toxic concentrations. For example, fasiglifam (aka, TAK-875) is a G-protein receptor 40 agonist that failed in phase III clinical trials because of cases of severe IDILI [Citation66]. In cultured hepatocytes, TAK-875 inhibited several transporters, including the bile salt export pump (BSEP) that transports bile acids out of hepatocytes and into the bile [Citation67–69]. TAK-875 given to dogs for 2 weeks resulted in increased serum bile acids over the treatment period and late-developing inflammation and liver injury [Citation70]. In rats, TAK-875-induced elevations in serum alanine aminotransferase (ALT) and total bilirubin, which were enhanced by BSEP knockdown [Citation71]. Although mechanisms of TAK-875 hepatotoxicity appear to be complex, accumulative stress from altered bile acid homeostasis seems to be an important contributor [Citation69].
Generally, the onset of liver injury from the drugs mentioned in this section occurs several weeks to months after the beginning of maintenance therapy [Citation72]. These examples illustrate that slowly developing cellular stress from a variety of mechanisms could lead to reduction in toxicity threshold and result in IDILI.
3.5. Environmental changes result in a threshold for toxicity that varies with time
We typically think of toxicity thresholds for drugs as invariant within an individual. However, due to changes in environment, this is unlikely to be true [Citation16]. For example, many factors including dietary alterations (e.g. fasting) and exposure to chemicals can lower tissue concentrations of GSH that protect against injury from reactive oxygen species (ROS) produced during exposure to some drugs. Studies in experimental animals showed that several drugs that cause IDILI become hepatotoxic under conditions of GSH depletion. Azathioprine is an immunosuppressive drug used to treat inflammatory bowel disease and autoimmune diseases, but hepatotoxicity in a small percentage of patients limits its usefulness. Azathioprine given to mice resulted in liver injury associated with increased ROS production [Citation73]. In cultured hepatocytes, GSH depletion prior to azathioprine exposure enhanced hepatocellular killing [Citation74]. Together, these results support the possibility that dietary alterations or exposure to environmental chemicals which lower GSH concentrations could render the liver more sensitive to drugs for which hepatotoxicity is mediated by ROS. This increased sensitivity would be reflected in a lowered and possibly variable threshold for toxicity.
People all experience episodic bouts of acute inflammation due to microbial infections or various other stresses, and such inflammatory episodes can lead to toxicity thresholds that vary with time (). Interestingly, antibiotics are the most common cause of IDILI and are used to combat infections that are associated with inflammation [Citation1]. Hepatic inflammation can occur from microbial infection but also from conditions that enhance permeability of the gastrointestinal tract to bacterial products, such as endotoxin (lipopolysaccharide; LPS) that can prompt hepatic inflammation. In rodent models, exposure to noninjurious doses of LPS led to pronounced liver injury and inflammation upon cotreatment with otherwise nontoxic doses of any of several drugs that cause human IDILI [Citation75]. Tumor necrosis factor-alpha (TNFα) and other inflammatory mediators underlie the drug–inflammation interaction in these animal models, and results in vitro suggest that inflammatory cytokines interact with hepatocytes stressed by drug exposure, leading to hepatocellular death [Citation4,Citation48,Citation76]. These animal models indicate clearly that interaction of IDILI-associated drugs with an acute inflammatory response can result in severe liver injury. This can be viewed as a lowering of toxicity threshold by the acute inflammatory response. In animals, exposure to virus that targets the liver can also interact with drugs to enhance liver injury [Citation77]. Similarly, Reye syndrome in children involves life-threatening liver injury that is always preceded by a viral infection. A link with aspirin administration has been reported, although controversy regarding cause and effect remains [Citation78].
Figure 8. Concentration of drug in plasma over time under condition of a variable toxicity threshold. As mentioned in the text, the toxicity threshold might not be constant but rather subject to a variety of factors that could change it over time. In this illustration, the variable toxicity threshold falls below the steady state concentration of drug in plasma, at which time an IDILI reaction occurs.
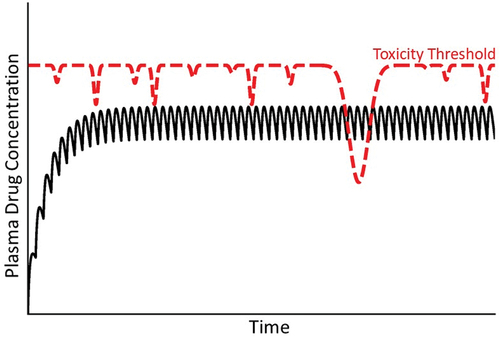
Thus, variable exposures to numerous environmental agents or dietary changes likely lead to variations in toxicity threshold, as illustrated in , and consequently to episodic toxicity that would be variable in time of onset, difficult to predict, sometimes reversible upon drug withdrawal and categorized as idiosyncratic.
3.6. Drug exposure activates a damaging adaptive immune response, qualitatively changing the toxicity threshold
A popular hypothesis regarding IDILI etiology is that a drug or its reactive metabolite acts as a hapten to activate the adaptive immune system. Liver immune tolerance normally prevents adaptive immune stimulation from causing injury, but escape from such tolerance in the face of immune system activation could hypothetically result in immune-mediated liver damage [Citation79]. Adaptive immune reactions typically require an initial sensitizing exposure to a drug followed by reexposure (drug challenge) that prompts proliferation and activation of lymphocytes and other immune cells, stimulates production of immune/inflammatory mediators and provokes hepatocellular damage. Continued dosing with a drug during maintenance therapy can provide both the sensitizing and challenge exposures. In this scenario, the original toxicity threshold from a nonimmune mechanism would be replaced by a new threshold dictated by the activated immune response. Conceptually, the threshold would be qualitatively different and likely to be markedly lower than in the absence of an immune response, as a rapid shift from a usual toxic mechanism to one driven by a damaging immune response occurs ().
Figure 9. Concentration of drug in plasma over time under conditions of an adaptive immune response. In this illustration, a patient’s usual toxicity threshold is above the concentration of drug in plasma. When a damaging adaptive immune response ensues, a new toxicity threshold applies that falls rapidly below the steady state plasma concentration. The normal toxicity threshold (red dashed line) is established by intrinsic properties of the drug, whereas the lower toxicity threshold (violet dashed line) is controlled by a different mechanism driven by the adaptive immune response.
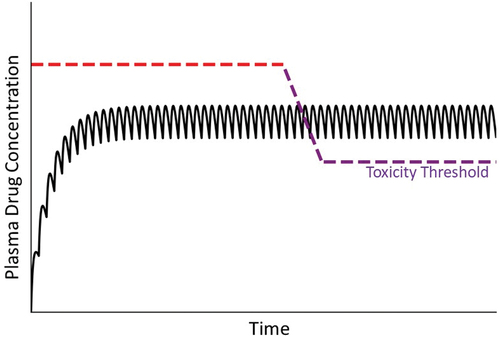
An adaptive immune response can require several weeks to develop, which can explain the delayed onset and suddenly developing injury that characterizes many IDILI responses. Drug withdrawal often halts the immune response, but upon reexposure to the offending drug, the immune system once again becomes activated and can result in injury. Inasmuch as immune responses can be triggered by very small exposures to antigens, the patient could be expected to be susceptible to very small, therapeutic drug doses, and this would be reflected in a markedly lowered toxicity threshold ().
Many drugs elicit immune responses. Most of these do not cause damage, and those that do cause injury only in susceptible individuals [Citation80]. For several drugs, patients who developed IDILI had a specific human leukocyte antigen (HLA) polymorphism, supporting an adaptive immune mode of action. For example, virtually all patients suffering cholestatic IDILI from the antibiotic flucloxacillin had the HLA B*5701 allelic polymorphism [Citation81]. CD8 + T-cells isolated from flucloxacillin-sensitized mice and then stimulated with the drug proliferated, secreted interferon-gamma and killed hepatocytes in vitro [Citation82]. High prevalence of a different HLA polymorphism (HLA-B*35:01) was found in liver injury cases associated with green tea consumption [Citation83]. Such results support the adaptive immunity hypothesis of IDILI and raise the possibility that changes in toxicity threshold related to an immune response underlie some cases of IDILI.
3.7. Patient fails to adapt to modest, drug-induced injury, thereby maintaining suprathreshold plasma concentration
In a study involving healthy human volunteers, acetaminophen given at the maximum recommended daily dose for 2 weeks resulted in modest increases in serum ALT that returned to normal in most individuals, despite continued drug treatment [Citation84]. There was considerable variability in the magnitude of the ALT increase among individuals, suggesting a degree of idiosyncrasy even with a drug that causes intrinsic hepatotoxicity. Although acetaminophen hepatotoxicity is clearly intrinsic in nature, this observation led to a novel, ‘failure-to-adapt’ hypothesis about the etiology of idiosyncratic reactions. According to the failure-to-adapt hypothesis of IDILI [Citation85], susceptible people fail to adapt to modest, drug-related injury, and clinically significant liver injury then ensues.
Other drugs too cause small increases in liver enzymes in serum that return to normal despite continued administration of the drug, suggesting modest liver injury to which the patient normally adapts even with continued drug exposure. A potential example is the antituberculosis agent, isoniazid, which is well known to cause hepatocellular IDILI in people. Between 10% and 20% of patients treated with isoniazid develop modest increases in serum ALT that resolve in most people despite continued maintenance therapy; however, in a smaller fraction of patients (i.e. <2%) progression to serious liver injury occurs [Citation86–88], suggesting that some people fail to adapt to the modest injury. No correlation was found between isoniazid plasma concentration and liver injury, suggesting that the adaptation defect was not pharmacokinetic in nature [Citation86].
A similar scenario occurred with troglitazone in clinical studies, in that some patients on maintenance therapy had rather large increases in serum ALT that resolved even with continued drug therapy, whereas others progressed to liver failure, usually with an hepatocellular pattern of injury [Citation89]. Idiosyncratic hepatotoxicity from tacrine might also involve adaptation. Tacrine causes large elevations in serum ALT in a minority of patients during maintenance therapy. Upon discontinuation of the drug and subsequent rechallenge with it, some people who had the initial ALT elevations had a reduced ALT response, consistent with adaptation to the initial injury [Citation90].
The failure-to-adapt hypothesis is illustrated in , which depicts initial intersection of the toxicity threshold with plasma drug concentration, during which time modest liver injury occurs, followed by an increase in toxicity threshold, reflecting an adaptation response that leads to disappearance of injury. In contrast, a susceptible individual who cannot adapt suffers overt, clinically significant liver injury (i.e. the steady state drug concentration remains above the toxicity threshold). The mechanisms by which such adaptation occurs are unclear but could relate to protective immune tolerance or to replicative liver repair [Citation75,Citation85], deficiencies in which could lead to failure to adapt to modest injury and consequent progression to serious injury displaying the usual characteristics of IDILI.
Figure 10. Drug concentration in plasma under condition in which a patient either adapts or fails to adapt to minor, drug-induced injury. Many drugs cause a minor degree of liver injury (depicted as an intersection of plasma drug concentration line with a low toxicity threshold [red dashed line]) to which most patients respond with adaptation (pictured as an elevation in toxicity threshold [green dashed line]) during continued drug treatment. Some patients do not adapt, so that plasma drug concentration remains above the toxicity threshold (red dashed line), resulting in continued and worsening liver damage.
![Figure 10. Drug concentration in plasma under condition in which a patient either adapts or fails to adapt to minor, drug-induced injury. Many drugs cause a minor degree of liver injury (depicted as an intersection of plasma drug concentration line with a low toxicity threshold [red dashed line]) to which most patients respond with adaptation (pictured as an elevation in toxicity threshold [green dashed line]) during continued drug treatment. Some patients do not adapt, so that plasma drug concentration remains above the toxicity threshold (red dashed line), resulting in continued and worsening liver damage.](/cms/asset/db1dfa5b-59c4-49f8-9335-2612e82d6202/iemt_a_2113379_f0010_oc.jpg)
4. Expert opinion
Much remains to be understood about the etiologies of IDILI. For almost all IDILI-associated drugs, definitive cause-and-effect evidence linking drug exposure, mechanism of action and liver dysfunction is incomplete. Consequently, much speculation and many hypotheses have emerged to explain IDILI pathogenesis, and it is likely that IDILI can occur by several modes of action depending on the drug and the individual. Any tenable hypothesis should be able to explain why IDILI occurs unpredictably at therapeutic drug doses, typically in a small subset of patients on maintenance therapy, why it is often delayed relative to the onset of drug exposure and that for many drugs ceasing exposure leads to recovery whereas resumption of exposure leads to reoccurrence of injury. For the most part, the scenarios depicted above account for these characteristics.
We attempted to provide examples of drugs which might act by each of the modes of action that are discussed. It should be emphasized that, for almost all drugs that cause IDILI, attributing a mechanism involves considerable uncertainty because evidence for a particular mechanism is almost always incomplete. Another caveat is that clinical diagnosis of DILI is a diagnosis of exclusion and always entails a degree of uncertainty. The preferred assessment of causality in patients uses the Roussel Uclaf Causality Assessment Method (RUCAM), which was updated in 2016 [Citation91], but even RUCAM provides only an estimated probability that a case of liver injury emanated from a drug rather than from some other cause. When available, clinical examples in this treatise employed RUCAM in the diagnosis. A final contributor to uncertainty is that a drug might cause toxicity by more than one mechanism (e.g. TAK-875) [Citation69]. Although not specifically illustrated above, pharmacokinetic and toxicodynamic modes of action might occur together and act in concert in some situations, as exemplified by the multiple determinant hypothesis of IDILI [Citation92,Citation93].
As emphasized in this review, IDILI can be conceptualized as having pharmacokinetic or toxicodynamic origins that are reflected in enhanced drug or drug metabolite concentrations or in lowered threshold for toxicity, respectively. We have developed freeware that allows the user to manipulate input variables to illustrate these concepts graphically (see DemoTox-PK at https://bit.ly/DemoTox-PK). It is important to keep in mind that a threshold for toxicity is not necessarily constant but can vary with time within an individual, leading to temporal fluctuations in susceptibility to intoxication.
It is hoped that the conceptualizations outlined herein will provide a framework for thinking about IDILI origins and events that can transpire in a patient suffering from idiosyncratic toxicity. The concepts may be useful in guiding research inquiries as well as for teaching principles of toxicokinetics related to adverse drug reactions.
Article highlights
Adverse drug reactions, and specifically idiosyncratic, drug-induced liver injury (IDILI), remain an important cause of morbidity, mortality, and withdrawal of efficacious drugs from the market.
Enhanced sensitivity of individuals underlies IDILI reactions.
Causes of IDILI remain poorly understood, and consequently several hypotheses to explain IDILI etiology have emerged.
Each of these hypotheses entails pharmacokinetic or toxicodynamic characteristics that confer susceptibility to IDILI.
Graphical illustration of these characteristics can provide conceptual frameworks for considering events that can transpire in a patient suffering from idiosyncratic toxicity and for understanding IDILI mechanisms.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Björnsson ES. Drug-induced liver injury: an overview over the most critical compounds. Arch Toxicol. 2015 Mar;89(3):327–334.
- Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005 Jun;4(6):489–499.
- Rao A, Rule JA, Hameed B, et al. Secular trends in severe idiosyncratic drug-induced liver injury in north america: an update from the acute liver failure study group registry. Am J Gastroenterol. 2022 Apr 1;117(4):617–626.
- Roth RA, Maiuri AR, Ganey PE. Idiosyncratic drug-induced liver injury: is drug-cytokine interaction the linchpin? J Pharmacol Exp Ther. 2017 Feb;360(2):461–470.
- Hassan A, Fontana RJ. The diagnosis and management of idiosyncratic drug-induced liver injury. Liver Int. 2019 Jan;39(1):31–41.
- Bunchorntavakul C, Reddy KR. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2013 Jan;37(1):3–17.
- Navarro VJ, Khan I, Björnsson E, et al. Liver injury from herbal and dietary supplements. Hepatology. 2017 Jan;65(1):363–373.
- Koido M, Kawakami E, Fukumura J, et al. Polygenic architecture informs potential vulnerability to drug-induced liver injury. Nat Med. 2020 Oct 1;26(10):1541–1548.
- Chen M, Borlak J, Tong W. A model to predict severity of drug-induced liver injury in humans. Hepatology. 2016;64(3):931–940.
- Medina-Caliz I, Robles-Diaz M, Garcia-Muñoz B, et al. Definition and risk factors for chronicity following acute idiosyncratic drug-induced liver injury. J Hepatol. 2016 Sep;65(3):532–542.
- Korzekwa K. Enzyme kinetics of oxidative metabolism: cytochromes P450. Methods Mol Biol. 2014;1113:149–166.
- Hart SN, Zhong XB. P450 oxidoreductase: genetic polymorphisms and implications for drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2008 Apr;4(4):439–452.
- Oketch-Rabah HA, Roe AL, Rider CV, et al. United States Pharmacopeia (USP) comprehensive review of the hepatotoxicity of green tea extracts. Toxicol Rep. 2020;7:386–402.
- Hu J, Webster D, Cao J, et al. The safety of green tea and green tea extract consumption in adults - Results of a systematic review. Regul Toxicol Pharmacol. 2018 Jun;95:412–433.
- Dostal AM, Samavat H, Bedell S, et al. The safety of green tea extract supplementation in postmenopausal women at risk for breast cancer: results of the Minnesota Green Tea Trial. Food Chem Toxicol. 2015 Sep;83:26–35.
- Roth RA, Ganey PE. Intrinsic versus idiosyncratic drug-induced hepatotoxicity–two villains or one? J Pharmacol Exp Ther. 2010 Mar;332(3):692–697.
- Senior JR. What is idiosyncratic hepatotoxicity? What is it not? Hepatology. 2008 Jun;47(6):1813–1815.
- Marano M, Serafinelli J, Cairoli S, et al. Eltrombopag-induced acute liver failure in a pediatric patient: a pharmacokinetic and pharmacogenetic analysis. Ther Drug Monit. 2018 Aug;40(4):386–388.
- Bezençon J, Beaudoin JJ, Ito K, et al. Altered expression and function of hepatic transporters in a rodent model of polycystic kidney disease. Drug Metab Dispos. 2019 Aug;47(8):899–906.
- Beaudoin JJ, Bezençon J, Cao Y, et al. Altered hepatobiliary disposition of tolvaptan and selected tolvaptan metabolites in a rodent model of polycystic kidney disease. Drug Metab Dispos. 2019 Feb;47(2):155–163.
- Pan G. Roles of hepatic drug transporters in drug disposition and liver toxicity. Adv Exp Med Biol. 2019;1141:293–340.
- Delgoda R, Westlake AC. Herbal interactions involving cytochrome p450 enzymes: a mini review. Toxicol Rev. 2004;23(4):239–249.
- Wanwimolruk S, Prachayasittikul V. Cytochrome P450 enzyme mediated herbal drug interactions (Part 1). EXCLI J. 2014;13:347–391.
- Foti RS, Dickmann LJ, Davis JA, et al. Metabolism and related human risk factors for hepatic damage by usnic acid containing nutritional supplements. Xenobiotica. 2008 Mar;38(3):264–280.
- Roth RA Jr., Rubin RJ. Role of blood flow in carbon monoxide- and hypoxic hypoxia-induced alterations in hexobarbital metabolism in rats. Drug Metab Dispos. 1976 Sep-Oct;4(5):460–467.
- Zhou X, Nian Y, Qiao Y, et al. Hypoxia plays a key role in the pharmacokinetic changes of drugs at high altitude. Curr Drug Metab. 2018;19(11):960–969.
- Hui L, Rong W, Zheng-Ping J, et al. Effects of high altitude exposure on physiology and pharmacokinetics. Curr Drug Metab. 2016;17(6):559–565.
- Teschke R, Danan G. Idiosyncratic drug induced liver injury, cytochrome P450, metabolic risk factors and lipophilicity: highlights and controversies. Int J Mol Sci. 2021;22(7):3441.
- Yu K, Geng X, Chen M, et al. High daily dose and being a substrate of cytochrome p450 enzymes are two important predictors of drug-induced liver injury. Drug Metab Dispos. 2014;42(4):744–750.
- Monks TJ, Anders MW, Dekant W, et al. Glutathione conjugate mediated toxicities. Toxicol Appl Pharmacol. 1990 Oct;106(1):1–19.
- Boelsterli UA. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 2003 Nov 1;192(3):307–322.
- Daly AK. Are polymorphisms in genes relevant to drug disposition predictors of susceptibility to drug-induced liver injury? Pharm Res. 2017 Aug 1;34(8):1564–1569.
- Yokoi T, Oda S. Models of idiosyncratic drug-induced liver injury. Annu Rev Pharmacol Toxicol. 2021;61(1):247–268.
- Sasaki E, Matsuo K, Iida A, et al. A novel mouse model for phenytoin-induced liver injury: involvement of immune-related factors and P450-mediated metabolism. Toxicol Sci. 2013 Nov;136(1):250–263.
- Iida A, Sasaki E, Yano A, et al. Carbamazepine-induced liver injury requires CYP3A-mediated metabolism and glutathione depletion in rats. Drug Metab Dispos. 2015 Jul;43(7):958–968.
- Chaudhry AS, Urban TJ, Lamba JK, et al. CYP2C9*1B promoter polymorphisms, in linkage with CYP2C19*2, affect phenytoin autoinduction of clearance and maintenance dose. J Pharmacol Exp Ther. 2010 Feb;332(2):599–611.
- Fleishaker JC, Pearson LK, Peters GR. Phenytoin causes a rapid increase in 6 beta-hydroxycortisol urinary excretion in humans–a putative measure of CYP3A induction. J Pharm Sci. 1995 Mar;84(3):292–294.
- Oscarson M, Zanger UM, Rifki OF, et al. Transcriptional profiling of genes induced in the livers of patients treated with carbamazepine. Clin Pharmacol Ther. 2006 Nov;80(5):440–456.
- Waxman DJ, Azaroff L. Phenobarbital induction of cytochrome P-450 gene expression. Biochem J. 1992 Feb 1;281(Pt 3):577–592.
- Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol. 2006 Mar;61(3):246–255.
- Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006 Mar;20(2):187–210.
- Brewer CT, Chen T. Hepatotoxicity of herbal supplements mediated by modulation of cytochrome P450. Int J Mol Sci. 2017 Nov 8;18(11):2353.
- Ryu SD, Chung WG. Induction of the procarcinogen-activating CYP1A2 by a herbal dietary supplement in rats and humans. Food Chem Toxicol. 2003 Jun;41(6):861–866.
- Liu F, Ichihara S, Valentine WM, et al. Increased susceptibility of Nrf2-null mice to 1-bromopropane-induced hepatotoxicity. Toxicol Sci. 2010 Jun;115(2):596–606.
- Yang M, Zhang H, Tao B, et al. Possible association of HMOX1 and NQO1 polymorphisms with anti-tuberculosis drug-induced liver injury: a matched case-control study. J Clin Pharm Ther. 2019 Aug;44(4):534–542.
- Huang YS, Su WJ, Huang YH, et al. Genetic polymorphisms of manganese superoxide dismutase,NAD(P)H:quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J Hepatol. 2007 Jul;47(1):128–134.
- Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 2008 Apr;22(4):954–965.
- Maiuri AR, Breier AB, Gora LF, et al. Cytotoxic synergy between cytokines and NSAIDs associated with idiosyncratic hepatotoxicity is driven by mitogen-activated protein kinases. Toxicol Sci. 2015 Aug;146(2):265–280.
- Xin S, Fang W, Li J, et al. Impact of STAT1 polymorphisms on crizotinib-induced hepatotoxicity in ALK-positive non-small cell lung cancer patients. J Cancer Res Clin Oncol. 2021 Mar;147(3):725–737.
- Pessayre D, Fromenty B, Berson A, et al. Central role of mitochondria in drug-induced liver injury. Drug Metab Rev. 2012 Feb;44(1):34–87.
- Chan K, Truong D, Shangari N, et al. Drug-induced mitochondrial toxicity. Expert Opin Drug Metab Toxicol. 2005 Dec;1(4):655–669.
- Luft D, Schmülling RM, Eggstein M. Lactic acidosis in biguanide-treated diabetics: a review of 330 cases. Diabetologia. 1978 Feb;14(2):75–87.
- Gan SC, Barr J, Arieff AI, et al. Biguanide-associated lactic acidosis. Case report and review of the literature. Arch Internal Med. 1992 Nov;152(11):2333–2336.
- Dykens JA, Jamieson J, Marroquin L, et al. Biguanide-induced mitochondrial dysfunction yields increased lactate production and cytotoxicity of aerobically-poised HepG2 cells and human hepatocytes in vitro. Toxicol Appl Pharmacol. 2008 Dec 1;233(2):203–210.
- Will Y, Shields JE, Wallace KB. Drug-induced mitochondrial toxicity in the geriatric population: challenges and future directions. Biology (Basel). 2019 May 11;8(2):32.
- Gardner K, Hall PA, Chinnery PF, et al. HIV treatment and associated mitochondrial pathology: review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014 Jul;42(5):811–822.
- Senise JF, Castelo A, Martinez M. Current treatment strategies, complications and considerations for the use of HIV antiretroviral therapy during pregnancy. AIDS Rev. 2011 Oct-Dec;13(4):198–213.
- Montessori V, Press N, Harris M, et al. Adverse effects of antiretroviral therapy for HIV infection. CMAJ = Journal de l’Association Medicale Canadienne. 2004 Jan 20;170(2):229–238.
- Montessori V, Harris M, Montaner JS. Hepatotoxicity of nucleoside reverse transcriptase inhibitors. Semin Liver Dis. 2003 May;23(2):167–172.
- Lewis W, Gonzalez B, Chomyn A, et al. Zidovudine induces molecular, biochemical, and ultrastructural changes in rat skeletal muscle mitochondria. J Clin Invest. 1992 Apr;89(4):1354–1360.
- Lewis W, Papoian T, Gonzalez B, et al. Mitochondrial ultrastructural and molecular changes induced by zidovudine in rat hearts. Lab Invest. 1991 Aug;65(2):228–236.
- Ribeiro MP, Santos AE, Custódio JB. Mitochondria: the gateway for tamoxifen-induced liver injury. Toxicology. 2014 Sep 2;323:10–18.
- Stewart JD, Horvath R, Baruffini E, et al. Polymerase γ gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010 Nov;52(5):1791–1796.
- Ball AL, Bloch KM, Rainbow L, et al. Assessment of the impact of mitochondrial genotype upon drug-induced mitochondrial dysfunction in platelets derived from healthy volunteers. Arch Toxicol. 2021 Apr;95(4):1335–1347.
- Kashimshetty R, Desai VG, Kale VM, et al. Underlying mitochondrial dysfunction triggers flutamide-induced oxidative liver injury in a mouse model of idiosyncratic drug toxicity. Toxicol Appl Pharmacol. 2009 Jul 15;238(2):150–159.
- Shavadia JS, Sharma A, Gu X, et al. Determination of fasiglifam-induced liver toxicity: insights from the data monitoring committee of the fasiglifam clinical trials program. Clin Trial. 2019;16(3):253–262.
- Li X, Zhong K, Guo Z, et al. Fasiglifam (TAK-875) inhibits hepatobiliary transporters: a possible factor contributing to fasiglifam-induced liver injury. Drug Metab Dispos. 2015 Nov;43(11):1751–1759.
- Chalasani N, Bonkovsky HL, Fontana R, et al. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015 Jun;148(7):1340–52 e7.
- Otieno MA, Snoeys J, Lam W, et al. Fasiglifam (TAK-875): mechanistic investigation and retrospective identification of hazards for drug induced liver injury. Toxicol Sci. 2018 Jun 1;163(2):374–384.
- Wolenski FS, Zhu AZX, Johnson M, et al. Fasiglifam (TAK-875) alters bile acid homeostasis in rats and dogs: a potential cause of drug induced liver injury. Toxicol Sci. 2017 May 1;157(1):50–61.
- Li Y, Evers R, Hafey MJ, et al. Use of a bile salt export pump knockdown rat susceptibility model to interrogate mechanism of drug-induced liver toxicity. Toxicol Sci. 2019 Jul 1;170(1):180–198.
- Hoofnagle JH, Björnsson ES. Drug-induced liver injury - types and phenotypes. N Engl J Med. 2019;381(3):264–273.
- Matsuo K, Sasaki E, Higuchi S, et al. Involvement of oxidative stress and immune- and inflammation-related factors in azathioprine-induced liver injury. Toxicol Lett. 2014 Jan 13;224(2):215–224.
- Lee AU, Farrell GC. Mechanism of azathioprine-induced injury to hepatocytes: roles of glutathione depletion and mitochondrial injury. J Hepatol. 2001 Dec;35(6):756–764.
- Roth RA, Ganey PE. What have we learned from animal models of idiosyncratic, drug-induced liver injury? Expert Opin Drug Metab Toxicol. 2020 Jun;16(6):475–491.
- Shaw PJ, Ganey PE, Roth RA. Idiosyncratic drug-induced liver injury and the role of inflammatory stress with an emphasis on an animal model of trovafloxacin hepatotoxicity. Toxicol Sci. 2010 Nov;118(1):7–18.
- Maddox JF, Amuzie CJ, Li M, et al. Bacterial- and viral-induced inflammation increases sensitivity to acetaminophen hepatotoxicity. J Toxicol Environ Health Part A. 2010;73(1):58–73.
- Schrör K. Aspirin and Reye syndrome: a review of the evidence. Paediatr Drugs. 2007;9(3):195–204.
- Uetrecht J. Mechanisms of idiosyncratic drug-induced liver injury. Adv Pharmacol. 2019;85:133–163.
- Kim SH, Naisbitt DJ. Update on advances in research on idiosyncratic drug-induced liver injury. Allergy Asthma Immunol Res. 2016 Jan;8(1):3–11.
- Daly AK, Day CP. Genetic association studies in drug-induced liver injury. Drug Metab Rev. 2012 Feb;44(1):116–126.
- Nattrass R, Faulkner L, Vocanson M, et al. Activation of flucloxacillin-specific CD8+ T-cells with the potential to promote hepatocyte cytotoxicity in a mouse model. Toxicol Sci. 2015 Jul;146(1):146–156.
- Hoofnagle JH, Bonkovsky HL, Phillips EJ, et al. HLA-B*35:01 and green tea-induced liver injury. Hepatology. 2021;73(6):2484–2493.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. Jama. 2006 Jul 5;296(1):87–93.
- Dara L, Liu ZX, Kaplowitz N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2016 Feb;36(2):158–165.
- Mitchell JR, Long MW, Thorgeirsson UP, et al. Acetylation rates and monthly liver function tests during one year of isoniazid preventive therapy. Chest. 1975 Aug;68(2):181–190.
- Mitchell JR, Zimmerman HJ, Ishak KG, et al. Isoniazid liver injury: clinical spectrum, pathology, and probable pathogenesis. Ann Intern Med. 1976 Feb;84(2):181–192.
- Metushi I, Uetrecht J, Phillips E. Mechanism of isoniazid-induced hepatotoxicity: then and now. Br J Clin Pharmacol. 2016 Jun;81(6):1030–1036.
- Watkins PB. Idiosyncratic liver injury: challenges and approaches. Toxicol Pathol. 2005;33(1):1–5.
- Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. Jama. 1994 Apr 6;271(13):992–998.
- Danan G, Teschke R. RUCAM in drug and herb induced liver injury: the update. Int J Mol Sci. 2015;17(1):14.
- Ulrich RG. Idiosyncratic toxicity: a convergence of risk factors. Annu Rev Med. 2007;58:17–34.
- Li AP. A review of the common properties of drugs with idiosyncratic hepatotoxicity and the “multiple determinant hypothesis” for the manifestation of idiosyncratic drug toxicity. Chem Biol Interact. 2002 Nov 10;142(1–2):7–23.