
1. Introduction
Diabetes is a chronic endocrine disorder and complex metabolic disease characterized by dysregulation of glucose homeostasis and hyperglycemia affecting the body’s metabolism. Long-term hyperglycemia leads to multiple structural and functional abnormalities and causes damages to many organs including eyes, feet, kidneys, nerves, heart, and blood vessels with diabetes complications such as retinopathy, nephropathy, neuropathy, cardiovascular disease, and impaired wound healing [Citation1].
The loss of functional pancreatic β-cells, mainly by increased β-cell apoptosis is a central feature of both type 1 diabetes (T1D) and type 2 diabetes (T2D) [Citation2–Citation5]. Other possible causes of this reduced β-cell mass in diabetes include β-cell dedifferentiation [Citation6,Citation7] and failure of adaptive expansion due to impaired β-cell proliferation [Citation8]. In T1D, selective β-cell destruction occurs through auto-immune-mediated processes; infiltration of activated CD4+/CD8+ T cells, B cells, and macrophages in the pancreatic islets. Interaction of antigen presenting cells as well as β-cells directly with T cells leads to high local concentrations of pro-inflammatory cytokines, chemokines, ROS, and other apoptotic triggers like the perforin/granzyme B and Fas/FasL systems [Citation9]. High expression of innate immune receptors by injured β-cells further amplify their own destruction through lethal cross-communication between infiltrating immune cells and the targeted β-cells [Citation10]. Ultimately, the auto-immune reaction against β-cell antigens results in β-cell apoptosis, insulin deficiency, and hyperglycemia.
In T2D, there are multiple stimuli including glucotoxicity, lipotoxicity, islet amyloid polypeptide, and inflammation, which can trigger endoplasmic reticulum stress and/or oxidative stress leading to loss of β-cell function and mass [Citation11]. Islet β-cell failure is progressive in T2D; it occurs already in patients with impaired fasting glucose levels [Citation12] and potentiates when hyperglycemia is established and β-cells are unable to sustain compensation for the increased insulin demand. Also, β-cell apoptosis is evident in human islets from both lean and obese T2D patients as determined by multiple complementary approaches like in situ Terminal deoxynucleotidyl transferase dUTP nick end labeling and Capase-3 stainings as well as electron microscopy-based determination of apoptotic cells [Citation4,Citation13,Citation14]. Therefore, targeting β-cell apoptosis and dysfunction represents an attractive therapeutic approach for the treatment of diabetes by rescuing the functional β-cell mass.
2. β-cell’s Hippo signaling
Because of the fundamental importance of pancreatic β-cell death in the pathogenesis of both T1D and T2D, therapeutical strategies for apoptosis blockade require the mechanistic dissection of complex regulatory signaling governing the apoptosis of pancreatic β-cells. Identification of these intracellular signaling pathways is crucial for the better understanding of the molecular changes underlying β-cell pathogenesis and would offer novel targets for therapeutic diabetes intervention. First discovered using genetic screens in Drosophila and subsequently established in the mammalian system, the Hippo pathway has emerged as a complex signaling network that regulates organ size by controlling critical cellular states, such as proliferation, apoptosis and differentiation [Citation15]. Hippo pathway components are highly conserved in both Drosophila and mammals [Citation16]; they include Hippo (Hpo/MST1/2 in mammals), Salvador (Sav, Sav1/WW45 in mammals), Warts (Wts/LATS1/2 in mammals), and Yorkie (Yki/YAP in mammals). The regulation of the Hippo pathway is complex and may interconnect and cooperate with other signaling pathways involved in cell survival and proliferation regulation. In mammals, when the Hippo pathway is on, MST1/2 kinase complexes with a scaffold protein, Sav1, phosphorylates and activates the LATS1/2 kinase. Activated LAST1/2 together with its associated proteins MOB1A/B subsequently phosphorylate and inactivate transcriptional co-activators YAP and TAZ, which leads to YAP and TAZ cytoplasmic retention and inactivation by their interaction with 14–3–3 proteins. Upon multiple additional phosphorylations, YAP and TAZ are marked for degradation by the ubiquitin–proteasome system. When the Hippo pathway is off, the kinases MST1/2 and LATS1/2 are inactive and consequently, YAP/TAZ will be translocated into the nucleus and interact with TEAD1-4 and other transcription factors leading to gene transcription, cell proliferation, and growth [Citation17].
As an emerging significant player in many diseases, the Hippo pathway has attracted increasing attention also by diabetes research. Recently, the Hippo kinase MST1 has been identified as a key mediator of pancreatic β-cell apoptotic signaling and β-cell dysfunction in human and rodent β-cells in vitro and in vivo [Citation18,Citation19]. MST1 induces β-cell death and impaired insulin secretion through the mitochondrial-dependent pathway of apoptosis and direct phosphorylation of the β-cell transcription factor PDX1 promoting its degradation [Citation18]. Such direct action of MST1 supports the emerging role of the Hippo pathway in the regulation of β-cell survival and function. Analysis of clinical samples indicates that Hippo signaling operates in islets from T2D patients [Citation18]. Of interest, transcriptional co-activator YAP, a down-stream effector of the Hippo pathway, which controls pancreatic progenitors expansion in the embryonic phase of pancreas development, is not expressed in terminally differentiated mature human and mouse β-cells [Citation20,Citation21]. Intriguingly, re-expression of a constitutively active form YAP promotes human β-cell proliferation without affecting its differentiation status or secretory function. Furthermore, YAP protects β-cells from apoptosis triggered by multiple diabetic conditions [Citation22,Citation23] so that one can assume a complex network of ‘β-cell Hippo signaling’.
3. Switching Hippo ‘ON’ to Hippo ‘OFF’ by Merlin
Neurofibromatosis 2 (NF2/Merlin) is a tumor suppressor protein, which belongs to the ezrin–radixin–moesin family of actin-binding proteins and regulates the Hippo signaling pathway in both Drosophila and mammals [Citation24,Citation25]. Merlin is highly conserved in mammals and plays a key role in organ size control and development through the regulation of cell proliferation and apoptosis [Citation15]. Merlin regulates the Hippo signaling pathway by controlling the Hippo kinases cassettes MST1/2 and LATS1/2 [Citation25,Citation26]. Merlin initiates Hippo signaling by two different mechanisms: (1) direct activation of the MST–Sav complex and subsequent MST1/2-induced LATS1/2 phosphorylation and activation [Citation25] or (2) direct recruitment of LATS1/2 to the membrane [Citation27]. In the latter alternative model, Merlin functions as scaffold for MST-induced LATS phosphorylation without altering MST1/2 auto-phosphorylation [Citation27].
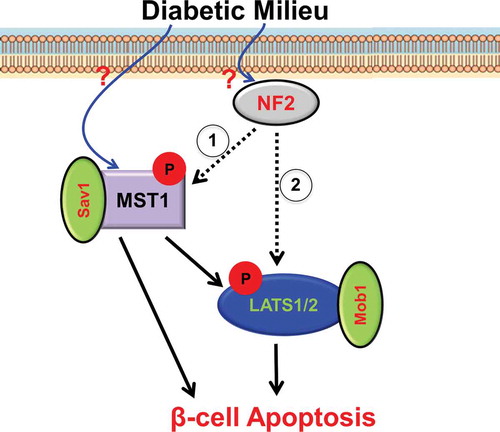
The Hippo pathway has emerged as a critical regulator of β-cell survival and proliferation, yet mechanisms that underlie signaling initiation remain elusive. Merlin is expressed in β-cell lines as well as in primary human and mouse islets. Its knockdown rescues β-cells from apoptosis under diabetogenic, that is, glucotoxic, glucolipotoxic, and pro-inflammatory conditions without compromising β-cell function or β-cell functional identity genes [Citation28]. Loss of Merlin reduces pro-diabetic milieu-induced LATS1/2 but not MST1/2 phosphorylation in pancreatic β-cells. In turn, LATS2 reconstitution abrogates the therapeutic benefit of Merlin depletion on β-cell survival under diabetic conditions. This suggests that Merlin might directly bind and regulate LATS1/2 phosphorylation, in accordance with the second alternative model of Hippo activation by Merlin [Citation27] (). These findings not only identify Merlin as Hippo initiator regulating apoptosis in β-cells but also highlight early upstream signal transductions activated by diabetic conditions. These seem to be critical gaps in β-cell death signaling pathways. Therapeutically, silencing of such early and upstream apoptogenic signals at very early stages of β-cell demise would be highly efficient and most suitable for a timely diabetes therapy. Consistently, Merlin is a principal inducer of apoptosis in the heart by activating MST1/Hippo signaling and its knockdown or genetic deletion blocks cardiac apoptosis and, in turn, provides protection against ischemic-reperfusion injury [Citation29].
Figure 1. Our view how Merlin/NF2 regulates β-cell apoptosis.
Through still unknown up-stream mechanism(s), a diabetogenic milieu activates the Hippo pathway in β-cells. Then, Merlin/NF2 initiates Hippo signaling by either direct activation of the MST1–Sav1 complex and subsequent MST1-induced LATS1/2 phosphorylation and activation (1; classical model) or direct activation of LATS1/2 (2; alternative model). This results in collective transcriptional and post-transcriptional events leading to β-cell apoptosis and impaired function.
Merlin may have additional functions out of context of the Hippo cassette and may regulate the Hippo pathway through cross-communication with other signaling pathways. Activation of Merlin induces cell apoptosis and blocks tumor initiation through the inhibition of mechanistic target of rapamycin complex 1 (mTORC1) signaling [Citation30,Citation31]. Conversely, loss of Merlin promotes mTORC1 signaling in malignant mesothelioma and is sensitive to rapamycin [Citation30]. Tumor samples isolated from patients with Neurofibromatosis type II (NF2; caused by Merlin gene mutations) as well as from Merlin-deficient mouse embryonic fibroblasts exhibit constitutively elevated mTORC1. This shows a clinically established negative regulation of mTORC1 by Merlin [Citation31]. Of note, mTORC1 signaling is also elevated in both Merlin-deficient β-cells and human islets, confirming the existence of such Merlin’s inhibitory action on mTORC1 also in pancreatic β-cells [Citation28]. As mTORC1 is a mitogenic signal in β-cells whose transient upregulation is pro-proliferative, the modulation of Merlin might be useful for β-cells that normally do not undergo proliferation and for enhancing the regenerative potential of damaged β-cells in the context of diabetes.
4. Concluding remarks
Multiple lines of evidence uncover the importance of Hippo signaling in pancreas development as well as in the regulation of β-cell survival, proliferation, and regeneration. These studies have greatly advanced our understanding of the physiological regulation of the β-cell and disease mechanisms at the cellular and molecular level. Next, it is essential to identify the primary signals, which initiate the Hippo pathway and to understand how these signals functionally control Hippo downstream cascades at basal and disease state. The establishment of Merlin as a previously uncharacterized and disease-relevant regulator of Hippo kinases in β-cells provides an insight into the complicated network of ‘β-cell Hippo signaling’ as well as a potential therapeutic opportunity to block β-cell failure in diabetes. Because of the powerful impact of Hippo signaling manipulations such as Merlin loss on β-cell survival/apoptosis, such strategies require tight control of the oncogenic potential in long-term studies. Having established the antiapoptotic effect of Merlin deficiency in β-cells and isolated human islets in vitro, the question remains what the impact of Merlin ablation would be in pancreatic β-cells during metabolic adaptations under conditions of increased β-cell stress and demand in vivo. Also, which stimuli could block such activated Merlin in diabetic β-cells in vivo, and could such strategy be therapeutically used for β-cell-directed therapy in diabetes? Generation of β-cell-specific Merlin knockout mice as well as further comprehensive, in-depth and long-term analyses of the complex Hippo signaling would provide further insights into the physiological role of Merlin in β-cells in vivo.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Additional information
Funding
References
- Calcutt NA, Cooper ME, Kern TS, et al. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov. 2009;8(5):417–429.
- Kurrer MO, Pakala SV, Hanson HL, et al. Beta cell apoptosis in T cell-mediated autoimmune diabetes. Proc Natl Acad Sci U S A. 1997;94(1):213–218.
- Mathis D, Vence L, Benoist C. Beta-cell death during progression to diabetes. Nature. 2001;414(6865):792–798.
- Butler AE, Janson J, Bonner-Weir S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110.
- Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death? Science. 2005;307(5708):380–384.
- Cinti F, Bouchi R, Kim-Muller JY, et al. Evidence of beta-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab. 2016;101(3):1044–1054.
- Jeffery N, Harries LW. Beta-cell differentiation status in type 2 diabetes. Diabetes Obes Metab. 2016;18:1167–1175.
- Tiwari S, Roel C, Tanwir M, et al. Definition of a Skp2-c-Myc pathway to expand human beta-cells. Sci Rep. 2016;6:28461.
- Thomas HE, Kay TW. Intracellular pathways of pancreatic beta-cell apoptosis in type 1 diabetes. Diabetes Metab Res Rev. 2011;27(8):790–796.
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5(4):219–226.
- Johnson JD, Luciani DS. Mechanisms of pancreatic beta-cell apoptosis in diabetes and its therapies. Adv Exp Med Biol. 2010;654:447–462.
- Ritzel RA, Butler AE, Rizza RA, et al. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006;29(3):717–718.
- Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia. 2009;52(6):1083–1086.
- Tomita T. Immunocytochemical localisation of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology. 2010;42(5):432–437.
- Saucedo LJ, Edgar BA. Filling out the Hippo pathway. Nat Rev Mol Cell Biol. 2007;8(8):613–621.
- Dong J, Feldmann G, Huang J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133.
- Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13(4):246–257.
- Ardestani A, Paroni F, Azizi Z, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. 2014;20(4):385–397.
- Ardestani A, Maedler K. MST1: a promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia. 2016;59(9):1843–1849.
- Gao T, Zhou D, Yang C, et al. Hippo signaling regulates differentiation and maintenance in the exocrine pancreas. Gastroenterology. 2013;144(7):1543-1553, 1553.e1.
- George NM, Day CE, Boerner BP, et al. Hippo signaling regulates pancreas development through inactivation of Yap. Mol Cell Biol. 2012;32(24):5116–5128.
- Yuan T, Rafizadeh S, Azizi Z, et al. Proproliferative and antiapoptotic action of exogenously introduced YAP in pancreatic beta cells. JCI Insight. 2016;1(18):e86326.
- George NM, Boerner BP, Mir SU, et al. Exploiting expression of Hippo effector, Yap, for expansion of functional islet mass. Mol Endocrinol. 2015;29(11):1594–1607.
- Hamaratoglu F, Willecke M, Kango-Singh M, et al. The tumour-suppressor genes NF2/Merlin and expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8(1):27–36.
- Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19(1):27–38.
- Yu J, Zheng Y, Dong J, et al. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and expanded. Dev Cell. 2010;18(2):288–299.
- Yin F, Yu J, Zheng Y, et al. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154(6):1342–1355.
- Yuan T, Gorrepati KD, Maedler K, et al. Loss of Merlin/NF2 protects pancreatic beta-cells from apoptosis by inhibiting LATS2. Cell Death Dis. 2016;7:e2107.
- Matsuda T, Zhai P, Sciarretta S, et al. NF2 activates Hippo signaling and promotes ischemia/reperfusion injury in the heart. Circ Res. 2016;119(5):596–606.
- Lopez-Lago MA, Okada T, Murillo MM, et al. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol. 2009;29(15):4235–4249.
- James MF, Han S, Polizzano C, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29(15):4250–4261.