
ABSTRACT
Introduction
The process of immunoglobulin class switch recombination (CSR) occurs in secondary lymphoid organs. This highly regulated process is essential for the development of different antibody isotype maturation and long-life memory/plasma cell generation. Patients with impaired CSR present heterogeneous noninfectious complications.
Areas covered
We provide an overview of recent advancements in the tight regulation of B cells before and during the CSR at different levels of cytokine stimulations, intracellular signaling, transcription-factor activation, gene transcription, and epigenetic controls.
Expert opinion
Besides recurrent infections which result from the lack of production of class-switched immunoglobulins, intrinsic B cell signaling pathways and regulatory component defects have distinct roles in other immune-related clinical manifestations including autoimmunity, atopy, lymphoproliferation, and cancer.
1. Introduction
Over the course of evolution, vertebrates have developed a highly efficient system to battle pathogens, with the adaptive humoral immunity arm providing the antibodies mediating a variety of effector functions of the immune system [Citation1]. After the completion of differentiation in the bone marrow, immature B cells enter the bloodstream and tissues to comprise the mature B cell pool [Citation2]. In secondary lymphoid organs, B cells form germinal centers (GCs) where the highly regulated processes of immunoglobulin (Ig) class switch recombination (CSR) and antibody maturation occur [Citation3]. CSR is a fundamental B cell process that harbors the full potential of the immune system’s components to fight off infection and to keep baseline immune regulation [Citation4]. Although most of the B cell development schemes are modeled with knowledge generated from mouse studies [Citation5,Citation6], a plethora of human in vitro studies and clinical findings from patients with inborn errors of immunity have identified most of the key components and mechanisms of CSR [Citation7–9].
CSR physiologically occurs in the light zone (LZ), although it has been suggested that it also happens extrafollicularly [Citation10]. The process consists of a specialized intrachromosomal deletional recombination occurring in the switch regions (S-regions) that are located upstream of every heavy chain constant (C-region) region, except Cδ (). The CSR process is initiated by activation-induced cytidine deaminase (AID, encoded by the AICDA gene), where conversion of dC to dU inside the switch (S)-regions leads to a series of well-described events [Citation11], ultimately mediating the change of the Ig isotype. A correct CSR process needs to be tightly regulated before and during the AID activity using different levels of cytokine stimulations, intracellular signaling, transcription factor (TF) activation, gene transcriptions, and epigenetic controls. Defects in these regulatory elements can both lead to infections (mainly due to lack of antibody production and class-switched isotypes) and also some noninfectious complications (e.g. autoimmunity, atopy, lymphoproliferation, and cancer) [Citation12–14]. Here, we review the main known regulatory processes and drivers of CSR with a focus on their role in noninfectious complications of human inborn errors of immunity.
Figure 1. Main cytokine/antigen stimulations, T cell cross-linkages, intracellular signaling pathways, activated transcription factors, gene transcriptions, and epigenetic regulators during the process of class switch recombination of B cells. Monogenic diseases in humans leading to only infectious complications are blue, molecules involved only in autoimmunity are shown in red, molecules involved only in lymphoproliferation/malignancy are shown in yellow, molecules involved both in autoimmunity and lymphoproliferation/malignancy are shown in orange, and the rest of the molecules with noninfectious complications are shown in pink (for details, please see ).
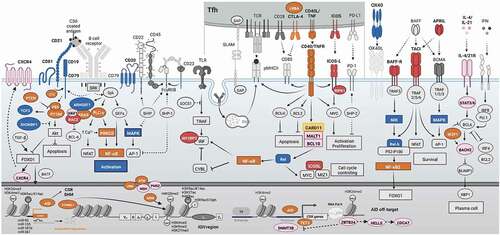
Table 1. Known human inborn errors of immunity with defects in CSR, cytokine, and transcription factors associated with noninfectious complications.
2. Microenvironmental cues on TFs to promote CSR
Germinal centers are the microanatomical structures that regulate B cell functions and provide the essential cues to promote B cell survival, proliferation, differentiation, and Ig production [Citation3,Citation15]. Physiologically, a complex network of extracellular stimuli is physically restricted inside the GC to induce a plethora of signaling pathways and promote essential (biological) processes, including TF-mediated induction of CSR. The four main driving forces for the CSR during GC B cells differentiation are as follows: external signals of B cell receptors (BCR, by antigen engagement), T cell co-stimulators (mainly tumor necrosis factor [TNF] superfamily receptors CD40L/CD40 and ICOS/ICOSL), T cell cytokine/interleukin receptors (mainly IL-21R and IL-4R), and pattern recognition receptors (mainly Toll-like receptors, TLRs) [Citation9,Citation15–17]. Other B cell cross-activation via survival signals (e.g. B-cell activating factor [BAFF] and A proliferation-inducing ligand [APRIL] stimulating TNF receptors superfamily 13B/C) and Ig receptors (e.g. fragment constant region [Fc] receptors) seems to be dispensable for the CSR process [Citation18].
At the time of pathogen encounter, stimulation via cytokines with low levels of BCR and T-cell co-stimulatory signals may lead to impaired GC B cell generation. Although cytokines can activate Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway and interferon [IFN] regulatory factors (IRFs, mainly IRF8), this can lead to B cell lymphoma 6 (BCL-6) expression and GC development but repression of BCL-2 expression (). Therefore, these GC B cells lack sufficient survival signals and undergo apoptosis fate without CSR initiation [Citation16,Citation19].
If the cognate selected BCR improves the affinity to antigen (original naive B cells or due to early somatic hypermutation), it can lead to only BCR signaling but low T cell co-stimulations. Improved BCR engagement can activate downstream tyrosine kinases (e.g. SYN/LYN) and the induction of KRAS/ERK signaling which accelerate repression of BCL-6 and improve BCL-2 survival signals. However, due to the absence of sufficient T cell stimulation and non-canonical nuclear factor-κB (NF-κB) support, BTB domain and CNC homology 2 (BACH2) protein expression will be at a high level which suppresses the B lymphocyte-induced maturation protein 1 (BLIMP1) and plasma cell developmental program. Therefore, this B cell will continue to generate pre-memory B cells in the LZ with increased expression of CD27, CD21, CCR2, TLR, and Fc receptors [Citation20–22].
Upon receiving stronger BCR signaling, intermediate T cell help (via T follicular helper cells or Tfh) can be achieved due to better affinity maturation. Stronger BCR activity of a GC B cell stimulates B cell linker protein (BLNK) and suppresses phosphatase and tensin homolog (PTEN) that can intensify the phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) signaling pathways. mTOR can subsequently activate forkhead box O1 (FOXO1) that increases CXCR4 expression mediating migration toward the dark zone of the GC. Improved induction of T cell stimulation can also activate the non-canonical NF-κB pathway molecules that are important for controlling of the cell cycle and epigenetic regulators (mainly with the expression of MYC). However, since this signal is not optimal, it cannot reverse the negative impact of PI3K/AKT/mTOR on BCL-6 expression and BACH2 that again fails to produce plasma cells. In fact, these cells can migrate to the dark zone for another round of somatic hypermutation since they have a high level of FOXO1 and NF-κB signals that activate AID targeting the VDJ regions during rapid B cell proliferation. After generation of a better-fitted antibody with improved avidity, they can return to the LZ and continue the process of CSR via the residual AID expression targeting the S-regions [Citation19,Citation23,Citation24].
Optimally, the reentry of GC B cells to the LZ with a high-affinity BCR leads to expanded T cell help, favoring both PI3K/AKT and NF-κB signaling pathways. Strong NF-κB signaling represses Casitas B-lineage lymphoma (CBL) expression and allows for upregulation of IRF4. IRF4 strongly represses BCL-6 and BACH2 expression and leads to the expression of BLIMP1 and X-box-binding protein 1 (XBP-1) required for plasma cell differentiation and expression of CD138, CD78, and a high level of CD27 [Citation25–27].
3. CSR regulation via specific GC cytokines
High BCR signaling and intermediate T cell stimulation could recruit the selected GC B cells for reentry to the DZ, AID activation, and subsequently CSR in the LZ. Although a plethora of cytokines have been reported to induce CSR in B cells (including IL-2, IL-12, IL-13, IL-15, TNF, and IFNs reviewed elsewhere [Citation28]), IL-21 and IL-4 are worth mentioning as two of the central cytokines involved in B cell CSR. It has been demonstrated that IL-21 alone can mediate switching to IgG1 and IgG3 on stimulated human naive B cells, and in combination with IL-4, it can potentiate switching to IgG1, IgG3, IgA, and IgE isotypes [Citation29–42] . When naive B cells from different human tissues (umbilical cord blood, spleen, tonsils, and adult peripheral blood) were stimulated with IL-21 in combination with CD40L, class-switching to IgG1, IgG3, IgA1, and IgE was observed [Citation29,Citation31,Citation32,Citation34–36]. Finally, IL21R-deficient naive B cells cannot class-switch upon IL-21 stimulation [Citation43], further highlighting the central role of this cytokine in the process of CSR.
Physiologically, IL-21 is highly produced by Tfh cells [Citation44–46] and can act directly on human naive B cells to induce AID expression, the master regulator of CSR initiation. The importance of IL-21 on GC B cells is also highlighted by the high expression of IL-21 R on GC B cells [Citation31]. Importantly, IL-21 signals through the JAK/STAT pathway (Jak1 and Jak3, and Stat1, Stat3, Stat5), and STAT3 mutations have been associated with elevated IgE serum levels, with the other isotypes remaining unaffected [Citation47–50]. Although IL-2 unresponsiveness of naive B cells through an IL-21-induction manner has been reported in STAT3-deficient patients, potentially amplifying their humoral immunodeficiency, it has not been investigated mechanistically how STAT3 mutations impact only IgE class-switching [Citation51], or how STAT3 can promote CSR in general. It is, however, apparent that the IL21R/STAT3 signaling axis is not the only one involved in the hyper IgE syndrome, as patients with polarized Th2 signaling including cases with IL6/R deficiencies may preset a similar phenotype [Citation52].
However, the functions of IL-6, IL-10, and TGF-β on B cells’ fate are also remarkable. More specifically, human B cell stimulation through the CD40 or BCR pathways, with IL-10 addition can increase the IgM, IgA, and IgG1 levels [Citation53], while alone can potentiate CSR to IgG1 and IgG3 in human naive B cells [Citation54], while it also increases AICDA expression. Furthermore, IL-10 promotes the generation of plasma cells that secrete IgM, IgG, and IgA [Citation55], and co-stimulation with TGFβ increases switching to IgA [Citation56]. Finally, IL-6 can increase B cell survival and differentiation [Citation57], enhances the production of IgE of human B cells stimulated with IL-4 [Citation58,Citation59], and augments IgM secretion when combined with IL-12 [Citation57].
4. CSR regulation via AID transcriptional controlling
Current knowledge, mostly from mouse models, suggests that both canonical and non-canonical NF-κB pathways are induced upon BCR cross-linking and T cell co-stimulation and TLR- ligation, with a synergistic effect to promote AID expression and mediation of CSR. AID as a CSR central enzyme is regulated on multiple levels: transcriptionally, post-transcriptionally, translationally, and post-translationally; here, we review the current literature on the regulation of AID by a range of TFs at all levels.
TLR agonists along with BCR signaling can potentiate CSR in human naive B cells, suggesting that TLR ligation could be a third signal along with T cell help, in the form of co-stimulatory molecules and cytokines [Citation60]. In line with that, TLR9 engagement on human B cells initiates germline transcription of Cγ1, Cγ2, and Cγ3, a process associated with increased AID transcription, through the non-canonical NF-κB pathway, and cooperates with IL-10 through STATs and IRFs to promote class-switching to IgG1, IgG2, and IgG3, respectively [Citation61]. On the same study, BCR engagement, along with IL-10, CD40L, and BAFF stimulation, was able to enhance IgG class-switching. Subsequent NF-κB activation leads to Hoxc4 expression, which binds to the region I of the Aicda promoter to enhance AID expression; an effect furtherly enhanced by estrogen-mediated expression of Hoxc4 [Citation62–64]. In line with that, Hoxc4 expression is upregulated upon CD40 ligation or lipopolysaccharide (LPS, activator of TLR4) plus IL-4 stimulation of human B cells, with Hoxc4-deficient Ramos cells displaying severely reduced CSR; an effect, however, rescued by forced AID expression [Citation65]. HOXC4 upregulation has also been reported in human autoimmunity (in systemic lupus erythematosus patients’ B cells), with a several-fold correlational increase in HOXC4 and AID, compared to healthy controls [Citation66]. As autoimmune patients have high-affinity class-switched auto-Abs, and a Hoxc4-deficient SLE-prone mouse model (Hoxc4−/− MRL/Fas lpr/lpr) displays decreased CSR and SHM, HOXC4 may have a similar role in human CSR; however, HOXC4-deficient patients with immunodeficiency characteristics have not been reported to date. Aberrant expression of AID and NF-κB signaling has also been reported in cases of B cell lymphoma and gastric cancer [Citation67–69], highlighting both the importance of NF-κB signaling and the severity of aberrant AID expression and its off-target effects.
Elegant luciferase experiments on human B cell lines have demonstrated several TFs (OCT1, OCT2, OCAB, HOXC4, PAZ5, SP1, SP3, and NFKB2) binding to the promoter region I of AICDA, with most of them displaying enhanced binding activity upon CD40L and IL-4 stimulation. The TFs PAX5, E2A, and BATF have also been reported to bind to the regions II/III of AICDA, suggesting potential cooperative effects between them to promote CSR [Citation70,Citation71]. As mentioned above, FOXO1 is also very crucial for the induction of GC and it induces BATF expression in the LZ GC B cells [Citation72]. Although this is not a direct induction, BATF can be upregulated by FOXO1 due to higher expression of CD40 and BCR signaling components that subsequently promote BATF expression (). A mouse study has demonstrated the need for (the TF) Ascl2 in the development of the GC reaction and transcriptional activation of AID. Ascl2 positively regulates GC B cell-related gene signatures, including that of the Aicda, and negatively regulates the expression of genes involved in plasma and memory B cell development [Citation73].
5. Epigenetic regulation of CSR
An open chromatin state of the Ig encoding region is essential for the mediation of CSR as evidenced by germline transcripts of Ig heavy chain gene (IGH), and histone modification signatures. IgM+ B cells are constitutively marked with the open chromatin-associated histone modification H3K27me3 in the Sμ region [Citation74–81], and downregulation of histone methyltransferase expression experiments leading to absent H3K27m3 signatures on a mouse cell line revealed that germline transcription is important but not the only factor involved in CSR [Citation81]. Furthermore, H3K9me3 signatures, normally associated with closed chromatin, found exclusively in Sμ donor regions are associated with CSR, as recruitment of other proteins, HP1, KAP1 and AID is important for CSR itself, with Kap1-deficient mouse B cells displaying reduced CSR [Citation82]. In line with that, IgA-switching can be impaired upon Suv39 methyltransferase deletion [Citation83].
Naive B cells express low levels of AID, while activated B cells undergoing CSR increase their AID transcription. This effect is, at least in part, mediated by hypermethylation of the Aicda promoter region in naive B cells, while activation of B cells results in H3K4me3 and H3K9ac/K14ac signatures associated with euchromatinic state and de-repression of the Aicda gene [Citation84]. Activation signatures H3K9ac and H3K4me3 are enriched in the S-regions compared to the upstream and downstream IGH regions [Citation74,Citation75] and are increased upon naive B cell stimulation [Citation76,Citation79,Citation80,Citation82]. In line with that, Set1 methyltransferase knockdown experiments potentiate a reduction in H3K4me3 signatures and a reduction of class-switched B cells [Citation81]. Moreover, Spt6 depletion experiments (impaired H3K4me3 signatures) cause a reduction in CSR to IgA [Citation85].
H3K4me3, H3K36me3, H2BK5ac, H3K9ac/K14ac, H3K27ac, and H4K8ac open-state chromatin signatures are constitutively present in the Sμ donor region of naive B cells [Citation86]. 14-3-3 adaptor proteins interact with AID and H3K9acS10ph modifications in the 5′-AGCT-3′-rich motifs of the S regions to mediate CSR in activated B cells [Citation87]. It has been suggested that 14-3-3 adaptor proteins act as readers of the histone code and recruit other effector molecules involved in the process. Other proteins recruited to the S-regions have been proposed; Rev1 [Citation88], RPA [Citation77], and 53BP1, potentially functioning as a scaffold for CSR mediation. Moreover, H4K20me2 has been proposed to recruit 53BP1 for CSR mediation [Citation89]. As we now begin to understand the specialized ‘histone code’ written in the S-regions that are to undergo CSR, it becomes more and more apparent that chromatin structure is important. However, caution is needed to recap the phenotypes in primary B cells, and future studies are needed to evaluate the impact of specific interleukins/cytokines or T helper polarization within GC on an accessible frame of S-regions for specific isotype switching to IgG, IgA, and IgE. Moreover, very little knowledge is available regarding the epigenetic regulation of other CSR-related receptors, TFs and modifiers; however, some recently discovered genetic defects in DNMT3B, ZBTB24, CDCA7, HELLS, and even TET2 with prominent B cell impairment highlighted the importance of DNA methylation and histone modification on the expression profiles of these CSR elements (). In more detail, TET-2 deficiency has been associated with reduced AID expression and IgG1 B cell formation in vitro [Citation90], while in vivo, it is involved in lymphangiogenesis through DNA hypermethylation in genes associated with GC B cell exit [Citation91]. Interestingly, it has been suggested that BATF facilitates the recruitment of TET to the Aicda enhancer [Citation90].
As cytokine stimulation induces a plethora of TFs that are critically involved in CSR (IL2/IL4/IL5 for IgM, IL2/IL4/IFNγ for IgG, IL5/IL10/TGFβ for IgA, and IL4/IL9/IL13/IFNγ for IgE), and the epigenetic landscape is evidently important for CSR, it is reasonable to suggest an interplay between cytokines, TFs, and the epigenetic landscape of the IGH locus. It has been reported that NF-κB blockade can reduce H3K4me3 and H3K9acS10ph signatures in the Sμ, Sγ1, and Sγ3 regions upon cytokine stimulation, and BCL-6 to interact with HDAC1 [Citation92], HDAC4, HDAC5, and HDAC7 [Citation93] to exert its full transcriptional repressing potential, possibly contributing to isotype-specific CSR.
6. CSR regulation via post-transcriptional and post-translational control of AID
Four miRNAs are known to date to regulate AICDA mRNA levels: miR-155, miR-361, miR-181b, and miR-93 [Citation94–96]. Interestingly, the master TF for GC formation BCL-6 is repressing the expression of miR-155 and miR-361, creating a double-negative gate for the upregulation of AID. Furthermore, miR-155 deficiency is reported in Burkitt’s lymphoma cases [Citation97], suggesting that aberrant AID overexpression increases AID off-target effects and may contribute to the characteristic translocations reported in the disease. More than 20 factors have been reported to interact with AID to influence its activity, stability, nuclear/cytoplasmic localization, and targeting to specific DNA sites. Remarkably, the TF YY1 interacts with AID to maintain its stability and increase its nuclear levels; loss of YY1 reduces AID nuclear levels [Citation98]. Recently some signaling proteins including CTNNBL1 have been identified for post-translational regulation of AID-dependent antibody diversification in humans (). It has been suggested that CTNNBL1 can guide AID for translocation into the nucleus and spliceosome-complex localization [Citation99].
It has been well described that BCR engagement alone cannot induce CSR on B cells, while co-stimulation with cytokines can [Citation100,Citation101]. Furthermore, it has been reported that TRAF3 deficiency can circumvent this BCR unresponsiveness, successfully inducing AID and Cγ1-germline transcripts (GLTs), potentiating CSR in naive B cells activated with anti-IgM/IL-4, and the expression of IgG1 and IgE on B cells [Citation102]. Thus, it has been shown that TRAF3 is a checkpoint molecule directly suppressing BCR signaling, restricting its signaling strength and increasing its signaling threshold, and preventing AID expression and CSR. Thus, TRAF3 serves as a negative BCR-signaling and NF-κB2 suppressor, licensing the process of CSR. Finally, TRAF3 is degraded upon CD40- and BAFFR-ligation, derepressing the NF-κB2 and inducing AID expression.
7. Expert opinion on noninfectious clinical manifestations of CSR defect including TF deficiencies
CSR defects can be caused by intrinsic B cell defects, a T cell–B cell cooperation defect, or an unknown DNA repair deficiency [Citation1,Citation8,Citation12]. Infections due to the absence of class-switched Igs are the most common presentations in patients with CSR defects; however, due to the intrinsic or extrinsic roles of defective regulatory molecules in the immune and nonimmune systems, other manifestations can be observed [Citation103–105]. summarizes the main noninfectious complications reported in CSR-deficient patients due to B cell receptor, Tfh co-stimulation, cytokine/interleukin receptor interaction, NF-κB pathway, PI3K pathway, and DNA repair and epigenetic defects. On the other hand, defects with specific BCR co-stimulatory (CD19 complex defects) and selected NF-κB-associated (REL complex defects) elements usually present with an infection-only phenotype and have less predisposition to noninfectious complications [Citation52].
The majority of these monogenic CSR defects are associated with autoimmune manifestations, except for a minority of cases with BCR, NF-κB, DNA repair pathway and epigenetic defects () [Citation106]. The CSR defects can be associated with disturbed tolerance to self-antigens through both central and peripheral tolerance [Citation52,Citation107,Citation108]. The main mechanisms of central tolerance which cooperate in the induction of tolerance in B and T cells in bone marrow and thymus, respectively, include the deletion of high-affinity autoreactive lymphocytes which can be diminished by decreased functional elements of lymphocyte intracellular signaling both via BCR/self-antigen recognition or other survival stimulation that may lead to a release of self-reactive lymphocyte in the periphery [Citation13,Citation109]. Moreover, abrogation in peripheral tolerance or regulatory signaling (e.g. apoptosis/anergy of cells with self-reactive antigen receptor or dysfunctional regulatory T cells) is more prone to be observed in CSR defect cases. This phenomenon is mainly due to the increased rate of SHM in Ig variable regions without proper productive switching in constant regions (possible errors in clonal somatic mutations making new self-reactive receptors) and is accompanied by T cell developmental defects [Citation110,Citation111]. Several studies provided conclusive evidence that SHM generates spontaneous autoreactive B cells that participate in systemic autoimmunity and the first autoantibodies detected in the serum of these cases are high-avidity IgGs and not IgM [Citation112,Citation113]. Other proposed mechanisms of autoimmunity in CSR-deficient cases besides defective elimination of self-reactive T and B cells have been reported particularly with defects in the generation of regulatory B cells (Bregs), increased proportion of CD21low B cells, persistent inflammation due to pathogens or immune dysregulation, increased apoptosis and production of immune complexes, epigenetic abnormalities (DNA methylation, small non-coding RNA transcripts, and histone modifications), and the role of the intestinal microbiome in affecting the polarization of helper T cells [Citation108,Citation114–117].
The second most frequent noninfectious complication among these monogenic CSR deficiencies is lymphoproliferation. These complications can mainly be presented in patients with TNF/TNF receptor defects (due to the importance of their function in Epstein–Barr-virus-specific CD8 memory T cell generation), PI3K defects (due to overactivation of proliferation and defective apoptosis of non-selected GC B cells), and DNA repair defects (due to higher level of double-stranded DNA lesions and lymphomagenesis of non-repaired lesions after AID activity) [Citation13,Citation118–124]. Similarly, malignancy (mainly B cell lymphoma) can be observed in these patients with a late-onset presentation and continuation of lymphoproliferation [Citation13,Citation14,Citation125–127]. However, other DNA-repair genes that have not been linked with lymphoproliferation but still are crucial for several different pathways including ATM, MRE11, and NBS1 deficiencies can be associated with other hematological malignancies (including leukemia) and solid tumors [Citation126,Citation128]. Interestingly, this subset of patients as well as epigenetic modifiers of DNA repair and CSR genes also have other noninfectious neurological complications due to the impact of these defective proteins in the progressive neurodegeneration and oxidative stress defects [Citation126,Citation129]. These noninfectious manifestations can lead to developmental/cognitive delay due to structural damage or progressive neuroinflammation. DNA repair proteins are directly involved in the response to endogenous damages in the nervous system required for neuronal differentiation and function. These damages initiate neuronal apoptosis to eliminate the abnormal neurons; therefore, monogenic defects of these elements can lead to progressive cerebellar ataxia, basal ganglia dysfunction, and even microcephaly due to massive apoptosis. Similarly, it has been suggested that epigenetic regulation plays a critical function in neural adaptive gene expression and neurogenesis. Subsequent to the mutations in these epigenetic factors, facial abnormality and developmental delay can present progressively due to abrogated structural and anatomic development as well as damaged neuro-homeostasis [Citation130–132].
Other clinical phenotypes that have been demonstrated by patients with CSR defects are noninfectious, non-autoimmune enteropathies that are most likely due to the local function of these proteins in the mucosal layers of the gastrointestinal tract. Gene defects with these presentations constitute Tfh co-stimulation defects (ICOS/ICOSL and CTLA4/LRBA/DEF and RIPK1 deficiencies), NF-κB pathway defects (NF-κB1/2 and IKBKG deficiencies), and cytokine stimulations (IL21/R and BACH2 deficiencies) [Citation52,Citation133–137]. Many unsolved CSR-defect patients presenting with common variable immunodeficiency with normal peripheral B cells but decreased levels of IgG, IgA, and/or IgM also display this phenotype that can be a consequence of lack of IgA in the mucosal layer, microbiota dysbiosis, initiation of systemic inflammatory responses, specific human leukocyte antigen (HLA) genotypes, and also specific innate immune cell interactions [Citation138–140]. Atopy including allergic manifestations, hypersensitivity reactions, and asthma can be also detected in a minority of patients mainly with cytokine stimulation (IL-6 and STAT3 deficiencies) and PI3K signaling (mainly PIK3CG and PLCG2 deficiencies) defects [Citation52,Citation137,Citation141]. Finally, other functional defects can be observed in connective tissues, cardiovascular and dermatologic systems with CSR-defect patients that have been listed in [Citation52,Citation141].
Noninfectious complications in CSR defects have emerged as a major clinical challenge in a substantial proportion of patients with a significant impact on increased morbidity and mortality. The targeted treatment for these complications needs to be designed based on the current molecular defects discovered and their pathological impacts on both immune and nonimmune cells.
Article highlights
The majority of patients with CSR defects manifest with complications except for a selected group of patients with B cell receptor co-stimulation (CD19 complex) and selected NF-κB-associated elements (REL complex) defects.
The majority of CSR defects can present autoimmunity; however, lymphoproliferation, malignancy, neurological defect, enteropathy, and atopy are more pathway-specific with distinct monogenic immunodeficiencies.
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Durandy A, Kracker S. Immunoglobulin class-switch recombination deficiencies. Arthritis Res Ther. 2012;14(4):218.
- Vale AM, Kearney JF, Nobrega A, et al. Development and function of B cell subsets. Molecular Biology of B Cells: Elsevier. 2015: 99–119
- Victora GD, Nussenzweig MC. Germinal Centers. Annu Rev Immunol. 2022;40(1):413–442.
- Martin A, Chahwan R, Parsa JY, et al. Somatic hypermutation: the molecular mechanisms underlying the production of effective high-affinity antibodies. Molecular Biology of B Cells: Elsevier. 2015: 363–388
- Palanichamy A, Bauer JW, Yalavarthi S, et al. Neutrophil-mediated IFN activation in the bone marrow alters B cell development in human and murine systemic lupus erythematosus. J Immunol. 2014;192(3):906–918.
- Sanz I, Wei C, Jenks SA, et al. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front Immunol. 2019;10:2458.
- Pan-Hammarstrom Q, Zhao Y, Hammarstrom L. Class switch recombination: a comparison between mouse and human. Adv Immunol. 2007;93:1–61.
- Yazdani R, Fekrvand S, Shahkarami S, et al. The hyper IgM syndromes: epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol. 2019;198:19–30.
- Yazdani R, Abolhassani H, Kiaee F, et al. Comparison of common monogenic defects in a large predominantly antibody deficiency cohort. J Allergy Clin Immunol Pract. 2019;7:864–78 e9.
- Roco JA, Mesin L, Binder SC, et al., Class-switch recombination occurs infrequently in germinal centers. Immunity. 2019;51(2):337–50 e7.
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26(1):261–292.
- Amirifar P, Yazdani R, Azizi G, et al. Known and potential molecules associated with altered B cell development leading to predominantly antibody deficiencies. Pediatr Allergy Immunol. 2021;32(8):1601–1615.
- Abolhassani H, Hammarstrom L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood. 2020;135(9):656–667.
- Abolhassani H, Aghamohammadi A, Fang M, et al. Clinical implications of systematic phenotyping and exome sequencing in patients with primary antibody deficiency. Genet Med. 2019;21(1):243–251.
- Abolhassani H, Parvaneh N, Rezaei N, et al. Genetic defects in B-cell development and their clinical consequences. J Investig Allergol Clin Immunol. 2014;24(1):6–22.
- Mayer CT, Gazumyan A, Kara EE, et al. The microanatomic segregation of selection by apoptosis in the germinal center. Science. 2017;358(6360):eaao2602.
- Stewart I, Radtke D, Phillips B, et al. Germinal center B cells replace their antigen receptors in dark zones and fail light zone entry when immunoglobulin gene mutations are damaging. Immunity. 2018;49(3):477–89 e7.
- Willis SN, Tellier J, Liao Y, et al. Environmental sensing by mature B cells is controlled by the transcription factors PU.1 and SpiB. Nat Commun. 2017;8(1):1426.
- Ersching J, Efeyan A, Mesin L, et al. Germinal center selection and affinity maturation require dynamic regulation of mTORC1 kinase. Immunity. 2017;46(6):1045–58 e6.
- Kometani K, Nakagawa R, Shinnakasu R, et al. Repression of the transcription factor Bach2 contributes to predisposition of IgG1 memory B cells toward plasma cell differentiation. Immunity. 2013;39(1):136–147.
- Tamahara T, Ochiai K, Muto A, et al. The mTOR-Bach2 cascade controls cell cycle and class switch recombination during B cell differentiation. Mol Cell Biol. 2017;37(24):e00418–17.
- Fischer SF, Bouillet P, O’Donnell K, et al. Proapoptotic BH3-only protein Bim is essential for developmentally programmed death of germinal center-derived memory B cells and antibody-forming cells. Blood. 2007;110(12):3978–3984.
- Luo W, Weisel F, Shlomchik MJ. B Cell Receptor and CD40 signaling are rewired for synergistic induction of the c-Myc transcription factor in germinal center B cells. Immunity. 2018;48(2):313–26 e5.
- Zhu Z, Shukla A, Ramezani-Rad P, et al. The AKT isoforms 1 and 2 drive B cell fate decisions during the germinal center response. Life Sci Alliance. 2019;2(6):e201900506.
- Zhang TT, Gonzalez DG, Cote CM, et al. Germinal center B cell development has distinctly regulated stages completed by disengagement from T cell help. Elife. 2017;6:e19552.
- Ise W, Fujii K, Shiroguchi K, et al. T follicular helper cell-germinal center B cell interaction strength regulates entry into plasma cell or recycling germinal center cell fate. Immunity. 2018;48(4):702–15 e4.
- Saito M, Gao J, Basso K, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. 2007;12(3):280–292.
- Moens L, Tangye SG. Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. Front Immunol. 2014;5:65.
- Bryant VL, Ma CS, Avery DT, et al. Cytokine-mediated regulation of human b cell differentiation into Ig-secreting cells: predominant role of IL-21 Produced by CXCR5 + T follicular helper cells. J Immunol. 2007;179(12):8180–8190.
- Parrish-Novak J, Dillon SR, Nelson A, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408(6808):57–63.
- Good KL, Bryant VL, Tangye SG. Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol. 2006;177(8):5236–5247.
- Avery DT, Deenick EK, Ma CS, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. 2010;207(1):155–171.
- Diehl SA, Schmidlin H, Nagasawa M, et al. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J Immunol. 2008;180(7):4805–4815.
- Pene J, Gauchat JF, Lecart S, et al. Cutting edge: IL-21 is a switch factor for the production ofIgG 1 and IgG 3 by human B cells. J Immunol. 2004;172(9):5154–5157.
- Avery DT, Bryant VL, Ma CS, et al. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol. 2008;181(3):1767–1779.
- Ettinger R, Sims GP, Fairhurst AM, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. 2005;175(12):7867–7879.
- Avery DT, Ma CS, Bryant VL, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood. 2008;112(5):1784–1793.
- Pene J, Guglielmi L, Gauchat JF, et al. IFN-γ-Mediated inhibition of human IgE synthesis by IL-21 Is associated with a polymorphism in the IL-21R Gene. J Immunol. 2006;177(8):5006–5013.
- Brenne AT, Ro TB, Waage A, et al. Interleukin-21 is a growth and survival factor for human myeloma cells. Blood. 2002;99(10):3756–3762.
- Kuchen S, Robbins R, Sims GP, et al. Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007;179(9):5886–5896.
- Deenick EK, Avery DT, Chan A, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med. 2013;210(12):2739–2753.
- Recher M, Berglund LJ, Avery DT, et al. IL-21 is the primary common gamma chain-binding cytokine required for human B-cell differentiation in vivo. Blood. 2011;118(26):6824–6835.
- Kotlarz D, Zietara N, Uzel G, et al. Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. J Exp Med. 2013;210(3):433–443.
- Crotty S. Follicular Helper CD4 T Cells (TFH). Annu Rev Immunol. 2011;29(1):621–663.
- Ma CS, Deenick EK, Batten M, et al. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209(7):1241–1253.
- Tangye SG, Ma CS, Brink R, et al. The good, the bad and the ugly - TFH cells in human health and disease. Nat Rev Immunol. 2013;13(6):412–426.
- Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper-Ige syndrome. N Engl J Med. 2007;357(16):1608–1619.
- Minegishi Y, Saito M, Tsuchiya S, et al., Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–1062.
- Tavassoli M, Abolhassani H, Yazdani R, et al. The first cohort of Iranian patients with hyper immunoglobulin E syndrome: a long-term follow-up and genetic analysis. Pediatr Allergy Immunol. 2019;30(4):469–478.
- Aghamohammadi A, Moghaddam ZG, Abolhassani H, et al. Investigation of underlying primary immunodeficiencies in patients with severe atopic dermatitis. Allergol Immunopathol (Madr). 2014;42(4):336–341.
- Berglund LJ, Avery DT, Ma CS, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood. 2013;122(24):3940–3950.
- Tangye SG, Al-Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2020;40(1):24–64.
- Rousset F, Garcia E, Defrance T, et al. Interleukin 10 is a potent growth and differentiation factor for activated human B lymphocytes. Proc Natl Acad Sci U S A. 1992;89(5):1890–1893.
- Briere F, Servet-Delprat C, Bridon JM, et al. Human interleukin 10 induces naive surface immunoglobulin D+ (sIgD+) B cells to secrete IgG1 and IgG3. J Exp Med. 1994;179(2):757–762.
- Arpin C, Dechanet J, Van Kooten C, et al. Generation of memory B cells and plasma cells in vitro. Science. 1995;268(5211):720–722.
- Defrance T, Vanbervliet B, Briere F, et al. Interleukin 10 and transforming growth factor beta cooperate to induce anti-CD40-activated naive human B cells to secrete immunoglobulin A. J Exp Med. 1992;175(3):671–682.
- Dubois B, Massacrier C, Vanbervliet B, et al. Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J Immunol. 1998;161(5):2223–2231.
- Gauchat JF, Aversa G, Gascan H, et al. Modulation of IL-4 induced germline epsilon RNA synthesis in human B cells by tumor necrosis factor-alpha, anti-CD40 monoclonal antibodies or transforming growth factor-beta correlates with levels of IgE production. Int Immunol. 1992;4(3):397–406.
- Itoh K, Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J Immunol. 1995;154(9):4341–4350.
- Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36(4):810–816.
- He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173(7):4479–4491.
- Heldring N, Pike A, Andersson S, et al. Estrogen receptors: how do they signal and what are their targets. Physiol Rev. 2007;87(3):905–931.
- Pauklin S, Sernandez IV, Bachmann G, et al. Estrogen directly activates AID transcription and function. J Exp Med. 2009;206(1):99–111.
- Mai T, Zan H, Zhang J, et al. Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. J Biol Chem. 2010;285(48):37797–37810.
- Park SR, Zan H, Pal Z, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol. 2009;10(5):540–550.
- White CA, Seth Hawkins J, Pone EJ, et al. AID dysregulation in lupus-prone MRL/ Faslpr/ lpr mice increases class switch DNA recombination and promotes interchromosomal c-Myc/IgH loci translocations: modulation by HoxC4. Autoimmunity. 2011;44(8):585–598.
- Perez-Duran P, de Yebenes VG, Ramiro AR. Oncogenic events triggered by AID, the adverse effect of antibody diversification. Carcinogenesis. 2007;28(12):2427–2433.
- Casellas R, Basu U, Yewdell WT, et al., Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16(3):164–176.
- Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Annu Rev Pathol. 2013;8(1):79–103.
- Sayegh CE, Quong MW, Agata Y, et al. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–593.
- Ise W, Kohyama M, Schraml BU, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol. 2011;12(6):536–543.
- Inoue T, Shinnakasu R, Ise W, et al. The transcription factor Foxo1 controls germinal center B cell proliferation in response to T cell help. J Exp Med. 2017;214(4):1181–1198.
- Sun L, Zhao X, Liu X, et al. Transcription factor Ascl2 promotes germinal center B cell responses by directly regulating AID transcription. Cell Rep. 2021;35(9):109188.
- Daniel JA, Santos MA, Wang Z, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329(5994):917–923.
- Pavri R, Gazumyan A, Jankovic M, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143(1):122–133.
- Wang L, Wuerffel R, Feldman S, et al. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J Exp Med. 2009;206(8):1817–1830.
- Yamane A, Resch W, Kuo N, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12(1):62–69.
- Wang L, Whang N, Wuerffel R, et al. AID-dependent histone acetylation is detected in immunoglobulin S regions. J Exp Med. 2006;203(1):215–226.
- Chowdhury M, Forouhi O, Dayal S, et al. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc Natl Acad Sci U S A. 2008;105(41):15872–15877.
- Kuang FL, Luo Z, Scharff MD. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc Natl Acad Sci U S A. 2009;106(13):5288–5293.
- Stanlie A, Aida M, Muramatsu M, et al. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc Natl Acad Sci U S A. 2010;107(51):22190–22195.
- Jeevan-Raj BP, Robert I, Heyer V, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208(8):1649–1660.
- Bradley SP, Kaminski DA, Peters AH, et al. The histone methyltransferase Suv39h1 increases class switch recombination specifically to IgA. J Immunol. 2006;177(2):1179–1188.
- Crouch EE, Li Z, Takizawa M, et al. Regulation of AID expression in the immune response. J Exp Med. 2007;204(5):1145–1156.
- Begum NA, Stanlie A, Nakata M, et al. The histone chaperone Spt6 is required for activation-induced cytidine deaminase target determination through H3K4me3 regulation. J Biol Chem. 2012;287(39):32415–32429.
- Li G, White CA, Lam T, et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep. 2013;5(3):702–714.
- Xu Z, Fulop Z, Wu G, et al., 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol. 2010;17(9):1124–1135.
- Zan H, White CA, Thomas LM, et al. Rev1 recruits ung to switch regions and enhances du glycosylation for immunoglobulin class switch DNA recombination. Cell Rep. 2012;2(5):1220–1232.
- Pei H, Wu X, Liu T, et al. The histone methyltransferase MMSET regulates class switch recombination. J Immunol. 2013;190(2):756–763.
- Lio CJ, Shukla V, Samaniego-Castruita D, et al. TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci Immunol. 2019;4(34):eaau7523.
- Rosikiewicz W, Chen X, Dominguez PM, et al. TET2 deficiency reprograms the germinal center B cell epigenome and silences genes linked to lymphomagenesis. Sci Adv. 2020;6(25):eaay5872.
- Dhordain P, Lin RJ, Quief S, et al. The LAZ3(BCL-6) oncoprotein recruits a SMRT/mSIN3A/histone deacetylase containing complex to mediate transcriptional repression. Nucleic Acids Res. 1998;26(20):4645–4651.
- Lemercier C, Brocard MP, Puvion-Dutilleul F, et al. Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J Biol Chem. 2002;277(24):22045–22052.
- de Yebenes VG, Belver L, Pisano DG, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. J Exp Med. 2008;205(10):2199–2206.
- Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer. 2011;11(1):347.
- Basso K, Schneider C, Shen Q, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med. 2012;209(13):2455–2465.
- Kluiver J, van den Berg A, de Jong D, et al. Regulation of pri-microRNA BIC transcription and processing in Burkitt lymphoma. Oncogene. 2007;26(26):3769–3776.
- Zaprazna K, Atchison ML. YY1 controls immunoglobulin class switch recombination and nuclear activation-induced deaminase levels. Mol Cell Biol. 2012;32(8):1542–1554.
- Kuhny M, Forbes LR, Cakan E, et al. Disease-associated CTNNBL1 mutation impairs somatic hypermutation by decreasing nuclear AID. J Clin Invest. 2020;130(8):4411–4422.
- Heltemes-Harris LM, Gearhart PJ, Ghosh P, et al. Activation-induced deaminase-mediated class switch recombination is blocked by anti-IgM signaling in a phosphatidylinositol 3-kinase-dependent fashion. Mol Immunol. 2008;45(6):1799–1806.
- Pone EJ, Zhang J, Mai T, et al. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nat Commun. 2012;3(1):767.
- Chen Z, Krinsky A, Woolaver RA, et al. TRAF3 acts as a checkpoint of B cell receptor signaling to control antibody class switch recombination and anergy. J Immunol. 2020;205(3):830–841.
- Azizi G, Bagheri Y, Tavakol M, et al. The clinical and immunological features of patients with primary antibody deficiencies. Endocr Metab Immune Disord Drug Targets. 2018;18(5):537–545.
- Bazregari S, Azizi G, Tavakol M, et al. Evaluation of infectious and non-infectious complications in patients with primary immunodeficiency. Cent Eur J Immunol. 2017;42(4):336–341.
- Aghamohammadi A, Abolhassani H, Latif A, et al. Long-term evaluation of a historical cohort of Iranian common variable immunodeficiency patients. Expert Rev Clin Immunol. 2014;10(10):1405–1417.
- Rizvi FS, Zainaldain H, Rafiemanesh H, et al. Autoimmunity in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol. 2020;16(12):1227–1235.
- Asgardoon MH, Azizi G, Yazdani R, et al. Monogenic primary immunodeficiency disorder associated with common variable immunodeficiency and autoimmunity. Int Arch Allergy Immunol. 2020;181(9):706–714.
- Cunningham-Rundles C. Autoimmunity in primary immune deficiency: taking lessons from our patients. Clin Exp Immunol. 2011;164(Suppl 2):6–11.
- Abolhassani H, Amirkashani D, Parvaneh N, et al. Autoimmune phenotype in patients with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2013;23(5):323–329.
- Azizi G, Kiaee F, Hedayat E, et al. Rheumatologic complications in a cohort of 227 patients with common variable immunodeficiency. Scand J Immunol. 2018;87(5):e12663.
- Azizi G, Abolhassani H, Kiaee F, et al. Autoimmunity and its association with regulatory T cells and B cell subsets in patients with common variable immunodeficiency. Allergol Immunopathol (Madr). 2018;46(2):127–135.
- Yu L, Robles DT, Abiru N, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A. 2000;97(4):1701–1706.
- Guo W, Smith D, Aviszus K, et al. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J Exp Med. 2010;207(10):2225–2237.
- Azizi G, Ahmadi M, Abolhassani H, et al. Autoimmunity in primary antibody deficiencies. Int Arch Allergy Immunol. 2016;171(3–4):180–193.
- Fairweather D, Kaya Z, Shellam GR, et al. From infection to autoimmunity. J Autoimmun. 2001;16(3):175–186.
- Azizi G, Abolhassani H, Asgardoon MH, et al. Autoimmunity in common variable immunodeficiency: epidemiology, pathophysiology and management. Expert Rev Clin Immunol. 2017;13(2):101–115.
- Gupta S, Demirdag Y, Gupta AA. Members of the regulatory lymphocyte club in common variable immunodeficiency. Front Immunol. 2022;13:864307.
- Abolhassani H. Specific immune response and cytokine production in CD70 deficiency. Front Pediatr. 2021;9:615724.
- Ghosh S, Kostel Bal S, Edwards ESJ, et al. Extended clinical and immunological phenotype and transplant outcome in CD27 and CD70 deficiency. Blood. 2020;136(23):2638–2655.
- Abolhassani H, Edwards ES, Ikinciogullari A, et al. Combined immunodeficiency and Epstein-Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. J Exp Med. 2017;214(1):91–106.
- Alkhairy OK, Perez-Becker R, Driessen GJ, et al. Novel mutations in TNFRSF7/CD27: clinical, immunologic, and genetic characterization of human CD27 deficiency. J Allergy Clin Immunol. 2015;136(3):703–12 e10.
- Dimitrova D, Nademi Z, Maccari ME, et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J Allergy Clin Immunol. 2022;149(1):410–21 e7.
- Jamee M, Moniri S, Zaki-Dizaji M, et al. Clinical, immunological, and genetic features in patients with activated PI3Kdelta syndrome (APDS): a systematic review. Clin Rev Allergy Immunol. 2020;59(3):323–333.
- Maccari ME, Abolhassani H, Aghamohammadi A, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase delta syndrome: the European Society for Immunodeficiencies-activated Phosphoinositide 3-kinase Delta Syndrome Registry. Front Immunol. 2018;9:543.
- Ye X, Maglione PJ, Wehr C, et al. Genomic characterization of lymphomas in patients with inborn errors of immunity. Blood Adv. 2022. DOI:10.1182/bloodadvances.2021006654.
- Abolhassani H, Wang Y, Hammarstrom L, et al. Hallmarks of cancers: primary antibody deficiency versus other inborn errors of immunity. Front Immunol. 2021;12:720025.
- Kiaee F, Azizi G, Rafiemanesh H, et al. Malignancy in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol. 2019;15(10):1105–1113.
- Tak Manesh A, Azizi G, Heydari A, et al. Epidemiology and pathophysiology of malignancy in common variable immunodeficiency? Allergol Immunopathol (Madr). 2017;45(6):602–615.
- Kiaee F, Zaki-Dizaji M, Hafezi N, et al. Clinical, immunologic and molecular spectrum of patients with immunodeficiency, centromeric Instability, and Facial Anomalies (ICF) syndrome: a systematic review. Endocr Metab Immune Disord Drug Targets. 2021;21(4):664–672.
- Dehkordy SF, Aghamohammadi A, Ochs HD, et al. Primary immunodeficiency diseases associated with neurologic manifestations. J Clin Immunol. 2012;32(1):1–24.
- Chavoshzadeh Z, Hashemitari A, Darougar S. Neurological manifestations of primary immunodeficiencies. Iran J Child Neurol. 2018;12(3):7–23.
- Yildirim M, Ayvaz DC, Konuskan B, et al. Neurologic involvement in primary immunodeficiency disorders. J Child Neurol. 2018;33(5):320–328.
- Abolhassani H, El-Sherbiny YM, Arumugakani G, et al. Expanding clinical phenotype and novel insights into the pathogenesis of ICOS deficiency. J Clin Immunol. 2020;40(2):277–288.
- Alkhairy OK, Abolhassani H, Rezaei N, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. 2016;36(1):33–45.
- Tesch VK, Abolhassani H, Shadur B, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. J Allergy Clin Immunol. 2020;145(5):1452–1463.
- Lorenzini T, Fliegauf M, Klammer N, et al. Characterization of the clinical and immunologic phenotype and management of 157 individuals with 56 distinct heterozygous NFKB1 mutations. J Allergy Clin Immunol. 2020;146(4):901–911.
- Tsilifis C, Freeman AF, Gennery AR. STAT3 Hyper-IgE syndrome-an update and unanswered questions. J Clin Immunol. 2021;41(5):864–880.
- Andersen IM, Jorgensen SF. Gut inflammation in CVID: causes and consequences. Expert Rev Clin Immunol. 2022;18(1):31–45.
- Yazdani R, Azizi G, Abolhassani H, et al. Selective IgA deficiency: epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Scand J Immunol. 2017;85(1):3–12.
- Al-Nesf MA, Morgan D, Mohamed-Ali V. Primary immunodeficiency and the microbiome. Curr Opin Pediatr. 2021;33(6):633–638.
- Tangye SG, Al-Herz W, Bousfiha A, et al. The ever-increasing array of novel inborn errors of immunity: an interim update by the IUIS committee. J Clin Immunol. 2021;41(3):666–679.