
ABSTRACT
Introduction
Type 1 diabetes (T1D) is a prevalent, and yet uncurable, autoimmune disease targeting insulin-producing pancreatic β-cells. Despite a known genetic component in T1D onset, genetics alone cannot explain the alarming worldwide rise in T1D incidence, which is attributed to a growing impact of environmental factors, including perturbations of the gut microbiome.
Areas covered
Intestinal commensal bacteria plays a crucial role in host physiology in health and disease by regulating endocrine and immune functions. An aberrant gut microbiome structure and metabolic function have been documented prior and during T1D onset. In this review, we summarize and discuss the current studies depicting the taxonomic profile and role of the gut microbial communities in murine models of T1D, diabetic patients and human interventional trials.
Expert opinion
Compelling evidence have shown that the intestinal microbiota is instrumental in driving differentiation and functions of immune cells. Therefore, any alterations in the intestinal microbiome composition or microbial metabolite production, particularly early in life, may impact disease susceptibility and amplify inflammatory responses and hence accelerate the course of T1D pathogenesis.
1. Introduction
Type 1 diabetes (T1D) is an organ-specific autoimmune disease caused by autoreactive T lymphocytes that attack insulin-producing pancreatic β-cells leading to progressive loss of β-cells and deficiency on insulin production.
The incidence of T1D is rapidly increasing worldwide at a rate of 3–5% per year [Citation1]. Notably, the highest incidence rates are reported in western countries in Europe and North America [Citation2,Citation3], hinting to a contribution of environmental factor, e.g. diet and lifestyle, in T1D onset. Indeed, despite a strong genetic component, genetics alone cannot explain the alarming rise in T1D incidence. Genetic susceptibility is accounted in approximately 70% of T1D cases by carrying human leukocytes antigens (HLA) high-risk alleles [Citation4,Citation5]. Nonetheless, less than 10% of individuals genetically susceptible develop T1D [Citation6]. Environmental factors are therefore pivotal modulators of T1D pathogenesis. For instance, viral infections, particularly by enterovirus and Coxsackie B, may accelerate T1D onset possibly via molecular mimicry of islet autoantigens or through upregulation of interferon and downstream HLA molecules, thereby increasing autoantigen presentation [Citation7,Citation8]. Other important environmental factors comprise exposure to dietary antigens (e.g. cow’s milk proteins), vitamin D deficiency, antibiotic usage and alterations of the gut microbiota and intestinal barrier [Citation9–13].
In regard to the latter, compelling evidence of the past decades highlight the importance of the gut microbiome as modulator of T1D susceptibility. In this review, we will summarize and discuss the current knowledge and significant findings on the role of the gut microbiome in T1D progression and development.
1.1. Immune mediators in T1D pathogenesis
The progressive destruction of β-cells, which starts years prior to diagnosis, is largely driven by autoreactive CD4 + and CD8 + T effector cells. Islet antigen–specific CD4 + T cells support the activation of B and T cells, and secrete pro-inflammatory cytokines that activate macrophages and islet-specific CD8 + T cells. Pathogenic T cells are known to react to a set of β-cell-derived autoantigens, such as insulin, preproinsulin (PPI), insulinoma-associated protein 2 (IA-2), islet-specific glucose 6 phosphatase catalytic subunit-related protein (IGRP), zinc transporter 8 (ZnT8), glutamic acid decarboxylase 65 (GAD65), and neoantigens derived by cross-linking of proinsulin peptides to other peptides in the secretory granules hybrid insulin) or posttranslational modifications of β-cell proteins [Citation14–17]. These autoantigens are also targeted by autoantibodies, which are detectable in the circulation prior to the clinical manifestation of T1D (referred as seropositivity); the number of autoantibodies can be used as prognostic marker as it associates with an increased risk of eventually developing T1D [Citation18–22]. Nonetheless, the role of B cells in T1D is still controversial with murine studies suggesting that B cells are necessary in the disease pathogenesis, independently of their ability to secrete autoantibodies, and mainly through B cell ability to act as antigen-presenting and cytokine-producing cells [Citation23–26].
Recently, Gearty et al. identified, in a murine model of T1D, a stem-like autoimmune progenitor TCF1hi CD8 + T cell population present in pancreatic lymph nodes that self-renew and migrate to the pancreas, where they differentiate and constantly sustain β-cell destruction [Citation27].
Both costimulatory (CD28, CD2, ICOS, CD40-ligand) and co-inhibitory (CTLA-4, PD-1, Lag-3, OX40) molecules regulate T cell activation and can be successfully targeted for autoimmune diseases [Citation28,Citation29]. For instance, abatacept, a soluble form of CTLA-4 that blocks CD28-mediated costimulatory signaling in T cells, have been shown to decelerate the rate of β-cell loss in T1D patients [Citation30]. Moreover, follicular helper T(fh) cells, which provide help to B cell differentiation into antibody-producing plasma and memory B cells, have been found to predict the response to abatacept, with higher proportion of ICOS + Tfh cells linked to poorer clinical response [Citation31].
Defects in function or reduction in frequencies of immunoregulatory lymphocytes such as regulatory B and T cells (e.g. Tr1 [type 1 regulatory] and FOXP3 + Treg) have been implicated in T1D pathogenesis [Citation32–34]. Particularly, Treg are major checkpoints of autoimmunity and suppressor of effector T cells and are characterized by the expression of the master transcriptional regulator FOXP3. The studies of Lindley et al. and Long et al. have reported defective features of Treg cells in T1D as Tregs from T1D patients displayed a defective ability to suppress T cell proliferation ex vivo and an impaired IL-2 receptor signaling, resulting in diminished maintenance of FOXP3 expression [Citation35,Citation36]. The importance of Treg in limiting T1D pathogenesis has been shown in diabetic mice where depletion of Treg accelerates T1D development while adoptive transfer of Treg prevents diabetes development [Citation37,Citation38]. In line with this, therapy with autologous Treg in T1D patients has been proven to prolong the survival of β-cells [Citation39]. Nonetheless, autologous Treg therapy has its challenges as ex vivo Treg expansion increases their number but reduces their suppressive capacity [Citation40]. Interestingly, the frequencies of autoreactive CD8 + T with an exhausted phenotype have been associated with better preservation of β-cell function in human T1D [Citation41,Citation42], suggesting that therapies inducing T cell functional exhaustion may be beneficial in autoimmune diabetes.
T1D pathogenesis is not solely accounted by immune cells since the response NFкBNFкBof β-cells to inflammatory and metabolic stress can induce β-cell death and accelerate T1D [Citation43]. Indeed, a recent study, employing imaging mass cytometry to simultaneously measure 35 insulitis biomarkers, unveiled that β-cell destruction is preceded by loss of functional β-cell markers [Citation44]. Moreover, HLA hyperexpression in β-cells is an early event in insulitis [Citation43,Citation45,Citation46].
1.2. Microbiota and immunity
The gut microbiome harbors circa 500–1000 distinct bacterial species [Citation47]. Quantity and diversity of microbial species increase longitudinally from the stomach to the colon, the latter containing the most dense and metabolically active commensal community (estimated 1014 bacteria) [Citation48,Citation49]. Five bacteria phyla dominate the gastrointestinal tract comprising Firmicutes, Bacteroides, Proteobacteria, and Actinobacteria, and Verrucomicrobia with Firmicutes and Bacteroides being the most abundant in the adult’s intestines [Citation47,Citation48].
Early-life colonization of mucosal surfaces is critical for the maturation of the host immune system [Citation50]. Indeed, studies in germ-free (GF) mice revealed deficits in the development of the gut-associated lymphoid tissue (GALT), with fewer and smaller mesenteric lymph nodes, lower proportion of FOXP3+ Treg cells and IgA-secreting plasma cells, as well as suppression of myeloid cell differentiation [Citation51–56]. Moreover, GF mice exhibit a distorted morphology of the intestinal mucosa with an altered mucosal layer, production of antimicrobial peptides (AMP), and lower expression of innate pattern recognition receptors, which, by sensing bacterial products, drive inflammatory signaling and thus production of mucus and AMP [Citation57–59]. Importantly, these defects in GF mice are reversed within a few weeks after gut colonization with commensal bacteria [Citation51–53,Citation55,Citation56,Citation60].
Thus, commensal gut bacteria are pivotal for immune cell development and gut homeostasis, and they also constantly shape immune cell differentiation. For instance, Bacteroides fragilis exerts systemic anti-inflammatory activities by promoting FOXP3 + Treg expansion and function with higher secretion of the immunosuppressive interleukin (IL)-10 [Citation53,Citation61,Citation62]. Other commensals have been identified for their ability to drive Treg differentiation and expansion, including Clostridia, Escherichia, Akkermansia, Bacteroides, Lactobacillus, and Streptococcus strains [Citation63,Citation64]. Instead, the pioneer study of Ivanov et al. demonstrated that intestinal colonization with the commensal bacteria Savagella, a segmented filamentous bacterium, is sufficient to drive Th17 cell differentiation in the intestinal lamina propria [Citation65]. Similarly, colonization by other mucosal-associated bacteria, including Escherichia coli, Bifidobacterium adolescentis, and Staphylococcus aureus, also induce Th17 cells responses [Citation63].
According to the ‘balanced signal hypothesis,’ specific commensals provide anti-inflammatory/tolerogenic signals while other microbes induce inflammatory responses [Citation66]. Therefore, perturbations in the microbiome structure, as documented in many inflammatory diseases, alter the homeostatic balance and may thus increase the susceptibility to disease or aggravate its clinical course.
Commensal gut bacteria produce a myriad of metabolites from de novo synthesis and metabolism of dietary or host-derived compounds. The repertoire of microbial metabolites can directly interact with host intestinal cells or be absorbed in the circulation and influence systemic immune responses [Citation67]. Bacterial fermentation of indigestible complex carbohydrates results in the production of short-chain fatty acids (SCFAs), predominantly butyrate, acetate, and propionate, which are largely absorbed by colonocytes through diffusion or co-transport [Citation68]. SCFAs have been intensively studied owing to their benefits for host metabolism and inflammation. SCFAs act as histone deacetylase (HDAC) inhibitors and ligands for G-protein coupled receptors (GPCR). Via GPCR signaling or epigenetic modifications, butyrate has been shown to harness the NFкB-elicited production of pro-inflammatory cytokines in macrophages, dendritic cells (DC), and neutrophils and concomitantly to promote the function and expansion of Treg cells [Citation63,Citation69–76]. Furthermore, butyrate can fortify mucus production and tight-junction expression and hence maintain the gut barrier impermeability [Citation69].
Another mechanism by which intestinal bacteria may control the trajectory of T1D is by expressing peptides with a certain homology to the host β-cell-derived autoantigens, thereby triggering a immune attack by bacteria-specific T cells [Citation77]. Tai and colleagues discovered that a microbial mimic peptide derived from a Mgt protein of intestinal Leptotrichia goodfellowii, a member of the Fusobacteria phylum, can activate IGRP206-214-specific NY8.3 CD8 + T cells in vitro and accelerate T1D onset in NY8.3 non-obese diabetic (NOD) mice [Citation78]. In another study, the commensal Parabacteroides distasonis was found to express a mimotope for the insulin B-chain 9–23, which in vitro and in NOD mice induce cross-reactivity with insulinB9-23-specific T cells. Moreover, colonization of NOD mice with P.distasonis stimulates insulin autoantibody seroconversion and accelerates T1D [Citation79]. Surprisingly, gut microbial mimicry can also be beneficial in T1D; indeed, Nanjundappa et al. disclosed that Bacteroides-derived integrase encodes a low-avidity mimotope of IGRP206-214. Monocolonization of GF NOD mice with integrase-competent/deficient/transgenic Bacteroides proved that the mimotope recruits diabetogenic CD8 + T cells to the gut and suppress dextran sulfate sodium (DSS)-induced colitis by killing autoantigen-loaded DC [Citation80].
2. The diabetic microbiome
Taxonomic alterations of the intestinal microbiota have been unveiled by cross-sectional case-control studies as well as longitudinal studies and include a reduction in the proportion of Lactobacillus and Bifidobacterium, Prevotella, Akkermansia, and Faecalibacterium prausnitzii and increase in abundance of Bacteroides dorei and B. vulgatus [Citation81–86]. Common traits of the human gut microbiome in presence of diabetes are lower Firmicutes to Bacteroidetes ratio, as well as decreased diversity and richness [Citation86–89].
Till now, the largest longitudinal study to investigate the microbiome functional and taxonomic profile in association with islet autoimmunity is The Environmental Determinants of Diabetes in the Young (TEDDY) study, which includes six clinical research centers across the the United States and Europe, and has recruited several thousands of newborns with HLA-conferred predisposition to T1D or first-degree relatives with T1D with biospecimens collection from 3 months of age. Metagenomic analysis of a total of 10,913 samples from 783 children reveals that, although a big variation in the microbiota structures among children is present, a common trait of the microbiomes of diabetic children is the reduced expression of genes involved in fermentation and synthesis of SCFA. However, no specific taxa were consistently found associated with the overall reduction in SCFA biosynthesis among the infants that developed T1D [Citation90]. An earlier study supports the findings of a functionally aberrant microbiota in T1D, as metagenomic data analysis shows that several pathways important for bacteria metabolism and function, including carbohydrate metabolism, adhesions, motility, phages, prophages, sulfur metabolism, and stress responses, are altered in the microbiome of T1D patients [Citation84].
In support of protective effects of SCFA in human T1D, de Groot et al. observed a decrease in butyrate-producing species and butyryl-CoA transferase genes in the fecal microbiota of 53 adult T1D patients as compared to matched healthy controls. Accordingly, the plasma levels of the SCFA acetate and propionate were also found reduced in the T1D group [Citation91].
Interestingly, the study of van Heck et al. shows that not only the microbiome structure of long-standing T1D patients differs from the one of healthy controls but clinical characteristics associated to diabetes, such as HbA1c, diabetes duration, micro- and macro-vascular complications, explain significant part of the variance in microbial profiles. Moreover, diabetic complications (nephropathy, retinopathy, and neuropathy) are specifically associated with distinct specie abundancy, with the strongest associations identified between diabetic nephropathy and the levels of Clostridium species, genus Bacteroides, and family Bacteroidaceae [Citation92]. It is not surprising that diabetic kidney diseases strongly associate with microbiome composition. Indeed, during chronic kidney dysfunction, the colon becomes the main route of excretion of nitrogen and uremic solutes, resulting in an aberrant microbiome. The uremic state, dietary limitations, and reduced intestinal transit exert a selective pressure on the microbial communities in the gut leading to expansion of proteolytic bacteria, which fuel the formation of hepatic uremic toxins (p-cresyl sulfate, indoxyl sulfate, trimethylamine N-oxide) from microbial precursors. As these microbiota-derived uremic toxins further harm kidney parenchymal cells and renal function, a vicious cycle is created between gut dysbiosis and kidney dysfunction [Citation93].
Notably, the existence of a “diabetogenic microbiota” appears to precede T1D development [Citation87,Citation90]. Indeed, the fecal microbiota of children that progressed to T1D was characterized by higher levels of Bifidobacterium pseudocatenulatum, Roseburia hominis, and Alistipes shahii species, whereas non-progressors displayed a higher abundance of Streptococcus thermophilus and Lactococcus lactis species. Moreover, the distinction between healthy and autoimmune states at the microbiome levels was seen in higher levels of Lactobacillus rhamnosus and Bifidobacterium dentium in healthy controls, and higher proportion of Streptococcus group mitis/oralis/pneumoniae species in the children with autoimmunity [Citation90]. A nested case-control analysis within the TEDDY cohort reveals subtle composition differences between healthy children and children that developed islet autoimmunity (sub-cohort of 632 children) or T1D (sub-cohort of 196 children). When comparing the 50 most abundant genera present in the microbiota of seropositive and matched controls, only delicate differences emerged with higher relative abundance of an unclassified Erysipelotrichaceae in autoimmunity cases. Instead, comparison of diabetic children versus controls identified five bacterial genera associated with T1D onset, with higher abundance of Streptococcus sp. and Lactococcus sp., and Parabacteroides being the most dominant in the T1D group. Several bacteria genera were found significantly less abundant in the T1D cases, including four unclassified Ruminococcaceae, Lactococcus, Streptococcus, and Akkermansia [Citation94].
To unravel the changes in the intestinal microbiota that accompanies or may cause T1D progression, Alkanani et al. analyzed the taxonomic profile of stool samples from 35 new-onset T1D patients, 21 seropositive subjects, 32 seronegative first-degree relatives of seropositive individuals, and 23 unrelated healthy controls [Citation95]. Although canonical discriminant analysis showed that the microbiomes of related seronegative and seropositive are not totally distinct, they separate from the ones of unrelated healthy controls and new-onset T1D individuals. Moreover, the taxonomic profile of seropositive and seronegative subjects was different, with a higher abundance in the Firmicutes genera Catenibacterium, the Bacteroidetes family and genus Prevotellaceae and RC9 gut group, and a concomitant constriction in taxa belonging to Bacteroides in the seropositive group. In addition, the T1D and seropositive groups showed reduced abundance of Lactobacillus and Staphylococcus than the microbiome of healthy controls. The latter harbored higher levels of the Firmicutes genera Lactobacillus and Staphylococcus and lower levels of the Bacteroidetes family and genus Prevotellaceae and RC9 gut group than in autoantibodies-positive subjects. Interestingly, a subanalysis of differences in microbiome structure between high-risk and low-risk for T1D groups (defined by presence of two-to-four autoantibodies versus one autoantibody) revealed an increase in the levels of the genera Bacteroides and Akkermansia but a decrease in the abundance of Prevotella, Butyricimonas, Coprococcus, and Butyrivibrio in high-risk subjects. These findings suggest that seropositivity and increased risk of T1D development are associated with changes in the gut microbiota taxonomy, especially in the abundance of bacteria belonging to the phyla Bacteroidetes and Firmicutes [Citation95].
2.1. Protective bacteria strains in autoimmune diabetes
Although differences in the taxonomic profile of the colonic microbiota have been described between healthy individuals and T1D patients, it remains challenging to identify specific beneficial strains or consortium of bacteria that hamper or decelerate the course of T1D development. NOD mice have been largely used to study the role of the gut microbiome as a regulator of T1D progression, since the NOD strain is a polygenic model for spontaneous autoimmune diabetes, in which T1D incidence strongly depends on environmental/microbial exposure [Citation96–99].
To understand the potential contribution of the gut microbiota to disease onset, Hanninen and colleagues investigated the differences in gut microbiome composition between the low-incidence colony NOD/MrkTac and the high-incidence colony NOD/Jax, and discovered that the bacteria Akkermansia muciniphila was abundant in the first but absent in the latter. Notably, transfer of Akkermansia muciniphila to the high-incidence colony improved intestinal homeostasis and gut barrier function by promoting mucus production, secretion of the antimicriobial Reg3γ protein, and reducing serum endotoxin. Moreover, intestinal A. muciniphila shaped the pancreatic immune landscape as shown by a reduction in immune cell infiltration and islet expression of Toll-like receptor (TLR)2 and TLR4, and an increase in regulatory FOXP3 + Treg and anti-inflammatory cytokines IL-10 and TGF-β, with an overall delay in diabetes development [Citation100]. Importantly, in humans, A. muciniphila levels were reported to be associated with diminished risk of developing T1D-associated autoantibodies in children at risk [Citation84,Citation101].
In another report, Kriegel et al. studied the variation in intestinal colonization by segmented filamentous bacteria (SFB) in the NOD strain, discovering a connection between SFB colonization and disease penetrance in female NOD mice. In fact, the authors report a clear-cut segregation of high incidence with SFB negativity as 91% of female NOD mice lacking intestinal SFB develop T1D by 30 weeks of age, whereas solely 16% of SFB colonized mice display diabetes within 30 weeks. SFB-positivity was associated with a Th17 signature and expansion of Th17 CD4 T cells in the small intestinal lamina propria, but did not affect other T cell subtypes [Citation102]. As the pathogenic role of Th17 in T1D is controversial, an attractive explanation of the protective function of intestinal Th17 cell is that the marked increase in the Th17 compartment can lead to the inhibition of islet-reactive Th1 cells. This hypothesis is supported by the reported Th17-mediated suppression of Th1 cells in a colitis murine model [Citation103].
In the BioBreeding diabetes prone (BB-DP) model of T1D, analysis of fecal bacteria communities between diabetes-resistant and diabetes-prone rats revealed a higher abundance of Lactobacillus and Bifidobacterium in the microbiome of the BB diabetes-resistant animals [Citation104]. Particularly, two Lactobacillus strains with ferulic acid esterase activity, L. johnsonii N6.2 and L. reuteri, were dominant in the diabetes-resistant group and are thought to be beneficial by releasing ferulic acid, which can improve insulin secretion and hyperglycemia [Citation105]. To unveil whether these two strains may antagonize T1D onset, Valladares et al. administered L. johnsonii or L. reuteri to BB-DP rats to find that L. johnsonii, but not L. reuteri, delayed T1D onset when administered post-weaning. In response to L. johnsonii intake, the ileal mucosa displayed a blunted oxidative stress rate and lower levels of pro-inflammatory cytokines tumor necrosis factor (TNF)-α and interferon (IFN)-γ [Citation106].
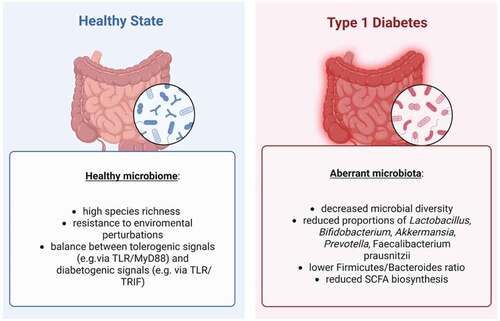
shows the main differences between the gut microbiota in healthy and diabetic states.
Figure 1. Overview of the main differences between healthy microbiota and aberrant microbiome in T1D.
3. Microbiota-host interactions as determinants of T1D progression
Innate immune cells are shaped by the environment, including commensal microbes, through interactions with Toll-like receptors and nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs) [Citation107]. In NOD mice, innate immune cells, including DC, macrophages, and neutrophils, are the first to infiltrate the pancreas as early as 3 weeks of age. Once in the pancreas, innate immune cells attract T lymphocytes from 4 to 6 weeks of age [Citation107,Citation108], as proven by the fact that depletion of DC or monocytes halts lymphocyte infiltration of pancreas [Citation109,Citation110].
In a pioneer study, Wen et al. disclosed that TLR signaling participates in T1D onset as NOD mice lacking the adaptor signaling protein MyD88, common adaptor molecule to multiple TLRs, were protected against the development of T1D when housed under normal specific-pathogen free (SPF) conditions. Importantly, the authors found that MyD88-deficiency resulted in local tolerance to diabetogenic peptides, specifically in T cells found in pancreatic lymph nodes, which drain both the pancreas and intestine. The protective effect of MyD88-deficiency was mediated by the microbiota; indeed, germ-free MyD88-deficient NOD mice robustly developed diabetes, while colonization with a consortium of human gut microbes restored the protection. Thus, the interaction of host innate immunity and commensal gut microbes shapes the predisposition to T1D [Citation96]. In a later study, Burrows et al. investigated the contribution of different innate immune components in T1D development by deleting crucial adaptor molecules of TLR, NOD1/2, and inflammasome signaling. The investigators found that lack of the TRIF adaptor, linked to the upstream TLR4 and TLR3 receptors, in MyD88-/- NOD mice reversed the protection given by MyD88-deficiency, whereas depletion of the inflammasome mediator caspase 1 or of the NOD1/2 signaling adaptor RIP2 kinase did not modify the outcome of disease. These data indicate that commensal microbes instigate both protective and diabetogenic signals through engagement of different innate immune pathways and TRIF-dependent signaling mediates microbiota-induced tolerance. Particularly, TLR4 signaling was found to be protective against T1D incidence, as lack of TLR4 in SPF NOD mice accelerates T1D while TLR4-/- GF mice showed a similar incidence to wild-type mice. This indicates that the TLR4-TRIF pathway is anti-diabetogenic in a microbiota-dependent manner [Citation98].
In line with the hypothesis that the gut microbiota generates different ‘signals,’ forced perturbations of the microbiome by consumption of acidified water or transfer of segmented filamentous bacteria changed T1D incidence and insulitis severity in NOD mice, accelerating the diabetogenic process in the first case and delaying it in the second [Citation111].
Interestingly, in a recent study, Tchitchek et al. disclosed that cytokine administration can impact the taxonomic profile and metabolic functionality of the gut microbiota, and through these changes, it can halt autoimmunity in NOD mice. Indeed, the authors supplemented mice with low-dose IL-2 to expand the Treg population, resulting in protection from autoimmunity and changes in the microbiome composition. When they transfered the IL-2-modified microbiota to new NOD mice, the rate of T1D was delayed, indicating that low-dose IL-2 is protective through microbiota alterations. Notably, metagenomic analysis of fecal samples from IL-2-treated or untreated mice and from patients subjected to low-dose IL-2 treatment (using baseline and post-treatment samples) revealed that IL-2 induces functional alterations of gut microbes affecting mainly pathways involved in biosynthesis and energy metabolism, such as synthesis pathways for amino acid, SCFA, and L-arginine [Citation112].
3.1. Intestinal permeability to fuel insulitis
The effects of host-microbiota interactions may be exacerbated in state of compromised intestinal barrier functions. Indeed, with an increased intestinal permeability, intestinal toxins, dietary antigens, and bacterial components may translocate to the intestinal mucosa and, hence, to the circulation or be absorbed in the lymphatic system, thereby reaching the pancreatic lymph nodes to accelerate the diabetogenic process.
An aberrant intestinal barrier integrity has been documented in both human and animal studies in T1D [Citation12,Citation81]. Bosi et al. investigated intestinal permeability by a lactulose-mannitol test and found that it occurs already at the stage of seroconversion prior to clinical diagnosis [Citation113]. In line with this, gut permeability was reported in T1D patients and was found to be associated with changes in the microbiota taxonomic profile [Citation85,Citation114,Citation115].
The elegant study of Sorini et al. revealed that disruption of the intestinal barrier may lead to the activation of autoreactive T cell clone, resulting in destructive immune responses toward β-cells. Using BDC2.5XNOD mice, which do not spontaneously develop diabetes but carry a transgenic T cell receptor specific for β-cell autoantigens, the authors show that chemical destruction of intestinal barrier with low-dosage of DSS triggers the activation of islet-reactive T cells in the gut mucosa, enhances the migration of mucosal T cells to the pancreas, and induces insulitis, with 60% of DSS-treated mice developing diabetes versus 100% of untreated mice remaining diabetes-free [Citation116]. In support of a lymphatic route from the gut to the pancreatic lymph nodes, Poysti et al. used FITC-dextran injected in colon to prove an accumulation in colon-draining lymph nodes as well as pancreatic lymph nodes; moreover, using photoconvertible-reporter mice and GFP-labeled bacteria, the researchers could prove DC migration from colon to pancreas and bacterial translocation to the pancreas via lymph route and after uptake by gut-resident dendritic cells [Citation117]. In line with the findings of Sorini et al., a previous study displayed that streptozotocin-induced T1D is accompanied by changes in the gut microbiome, loss of intestinal barrier integrity, and escape of bacterial products, leading to the activation of NOD2 in macrophages and DC of pancreatic lymph nodes. In turn, the activated myeloid cells instruct T cells to differentiate into pathogenic Th1 and Th2 lymphocytes, thereby contributing to the onset of invasive insulitis. Accordingly, lack of NOD2 or of gut microbiota downgrades the inflammatory infiltrates in the pancreas and the loss in β-cell mass [Citation118]. In support of this communication loop between intestines and pancreas, Zhang et al. showed that gut bacteria-derived NOD1 ligands act as signaling modulators that are senses by pancreatic β-cells and modulate intracellular distribution of insulin vesicles. In a very elegant work, the authors elucidate how lysozyme 1 released by intestinal Paneth cells hydrolyze bacterial wall peptidoglycan liberating NOD1 or NOD2 ligands, which are detected in the serum and are sensed by β-cells. The latter express both the peptidoglycan receptor NOD1 and its adaptor molecule RIP2 upon ligand sensing. NOD1 and RIP2 localize to insulin vesicles, where they recruit Rab1a, which is a master regulator of intracellular vesicle trafficking, and hence modulates insulin trafficking. This elaborate regulation of insulin distribution by NOD1/RIP2 results in the control of insulin secretion upon glucose challenge [Citation119].
3.2. Role of microbial metabolites in T1D
Metabolites derived from bacterial metabolism of host or dietary products act at the interference between the gastrointestinal commensal microbiota and host immune cells. Microbiota-derived SCFA have been largely studies owing to their anti-inflammatory effects and metabolic benefits [Citation69]. The pioneer study of Mariño et al. shows that blood and fecal acetate and butyrate levels inversely correlate with disease progression in NOD mice, and administration of acetate- or butyrate-yielding diets, through acetylation or butyrylation of high-amylose starch (HAMSA/HAMSB diets), from 5 weeks of age protects NOD mice against T1D development and against destructive insulitis [Citation119]. Mechanistically, the authors found that dietary SCFA diminished the frequency of splenic CD8 T cells autoreactive to IGRP as well as the frequency of splenic antigen-presenting B cells, particularly marginal-zone B cells, but expanded the population of colonic and splenic FOXP3+ Tregs. Interestingly, specific effects of butyrate and acetate were found. Indeed, acetate delivery, but not butyrate, could downregulate the expression of major histocompatibility complex (MHC) class I and co-stimulatory molecule CD86 on splenic IgM + B220 + B cells and potently reduce the proportion of IGRP-reactive CD8 T cells in NOD.8.3. mice. The latter is an accelerated model of type 1 diabetes due to the expression of αβ T cell antigen receptors that recognize IGRP. Instead, the anti-diabetogenic effects of dietary butyrate were mainly attributable to its capacity of inducing de novo differentiation of Treg (iTreg). Indeed, transfer of highly-purified T cells from HAMSB-fed mice to NOD-SCID (severe combined immunodeficiency) mice, lacking B and T cells, resulted in protection from diabetes development for more than 20 weeks. In addition, using NOD.FOXP3-GFP mice expressing a GFP-tagged FOXP3, the authors could prove that butyrate stimulates the conversion of naïve FOXP3-negative T cells into iTreg, since transfer of FOXP3-negative T cells into NOD-SCID mice fed control or acetate/butyrate-rich diets resulted into enhanced Treg in spleen and pancreatic lymph nodes solely when butyrate was supplemented. Altogether, these underlying effects of acetate and butyrate explain the superior protective effects against diabetes onset of combining HAMSA and HAMSB diet [Citation120]. Based on the experimental evidence provided by Mariño etal., we can postulate the dietary interventions design to increase the delivery or microbiota-mediated production of SCFA could be studied in humans as an early intervention therapy, as data on diabetes reversal by HAMSA/HAMB diet are missing. In line with the findings of Mariño et al., administration of sodium butyrate via drinking water upon hyperglycemia in female NOD mice delays the progression of hyperglycemia, increases survival, and reduces insulitis and β-cell destruction. Notably, butyrate treatment also induced an expansion of FOXP3+ regulatory T cells in colon and GALT. Instead, depletion of Treg with anti-CD25 antibodies abolished the protective effects of butyrate against destructive insulitis. Moreover, 6-week butyrate treatment robustly enhanced the percentage of Tregs expressing gut homing receptors (CCR9, GPR15, and α4β7) in pancreata and pancreatic lymph nodes, while colonic Tregs displayed greater migration ex vivo [Citation121]. These findings indicate that butyrate-mediated protection against T1D requires Treg expansion in colon and GALT and butyrate also promotes their migration to the pancreas.
The study of Jia et al. highlights a novel role for butyrate in prenatal life. In fact, butyrate administration to pregnant dams, in which the microbiota has been disrupted by vancomycin treatment, could protect the offspring from the accelerated T1D development caused by maternal dysbiosis. Moreover, supplementation of butyrate to NOD mice during gestation and nursing blunted the recruitment of IFN-γ-producing T lymphocytes to the pancreas and the exacerbated secretion of IFN-γ and IL-1β [Citation122]. In line with this, in a model of streptozotocin-induced diabetes, intraperitoneal injections of sodium butyrate for 7 days following the streptozotocin insult to β-cells ameliorated glycemia rate, insulitis severity, and diabetes incidence. This was accompanied by a reduction in serum IL-1β, and pancreatic high-mobility group box 1 protein and NF-κB activation [Citation123].
The recent report of Okada et al. shows that while the initial activation of islet-autoreactive CD8 T cells occurs in pancreatic lymph nodes, the trafficking of islet-specific effector memory T cells to intestinal Peyer’s patches or mesenteric lymph nodes increases their cytotoxic activities in NOD mice through bystander microbiota effects, likely attributable to reduced production of beneficial metabolites. Indeed, administration through drinking water of butyrate decreases the cytotoxic potential of autoreactive CD8 T cells in multiple tissues [Citation124].
3.3. Early-life microbiota disturbances shaping diabetes susceptibility
The microbiome-host interactions become particularly relevant in early-life when the immune system is still developing [Citation94]. The early postnatal period is viewed as a critical time where exposure to environmental factors sculpt the immune system development as well as the host-microbiota interactions, thereby priming the immune system and influencing lifelong health and disease susceptibility [Citation125].
In the multicenter TEDDY study, the longitudinal analysis of the fecal microbiota of 903 children, between 3 and 46 months of age, revealed that the microbiome develops through three phases which occurs between 3 and 14 months of age, 15 and 30 months (transitional phase), and lastly 31 and 46 months (stable phase) [Citation94]. This development is affected by environmental factors and durably shapes the microbiome-host immunity crosstalk in a manner that influences disease pathophysiology later in life.
Breast milk is the most significant determinant of the taxonomic structure of the developing microbiome. In the TEDDY study, the microbiome in the first year of life was reported to be dominated by one of three Bifidobacterium species, B. bifidum, B. breve, and B. longum. Specifically, the microbiota of breast-fed infant contained strain-specific carriage of genes for the utilization of human milk oligosaccharide within a subset of Bifidobacterium longum [Citation94]. This is not surprising considering that human milk contains oligosaccharides for bacteria growth, IgA and antimicrobial peptides, which shape the microbiome structure [Citation126].
Another important player in the developing gut microbiota, particularly vaginal delivery, is associated with higher abundance of Bacteroides species, which in turn appear to drive species diversity and accelerate microbiome maturation [Citation94].
It is postulated that human milk can beneficially influence the infant immune system as it harbors bioactive products, such as hormones, cytokines, and antibodies. Additionally, prolonged breast-feeding will postpone the exposure to dietary antigens, such as cow’s milk and solid food, especially gluten-containing cereals [Citation127–129]. In regard to the latter, Marietta et al. have shown that raising NOD mice on gluten-free diets significantly reduced the incidence of diabetes. As expected, the gluten-containing and gluten-free diets impacted the microbiota composition with Bifidobacterium, Tannerella, and Barnesiella species being increased in presence of gluten and Akkermansia species being enriched in the gluten-free diet group. As proof-of-concept, supplementation of gluten to the gluten-free diet-fed group reversed the protective effects on diabetes onset as well as the changes in microbiota structure, resulting in lower abundancy of Akkermansia species and higher levels of Bifidobacterium, Tannerella, and Barnesiella [Citation130].
The long-term consequences of breastfeeding in the development of autoimmune diabetes have been a matter of debate and an unsettled issue with some studies reporting protective effects and others not [Citation127]. The hypothesis that breastfeeding may protect against type 1 diabetes originated in the 80s supported by a retrospective case-control study using epidemiological data from genetically similar Scandinavian populations. This study revealed that, on average, diabetic children were breast-fed for a shorter time or never breast-fed as compared to the control cases [Citation131]. Accordingly, analysis of dietary practice between 6 and 18 months of age in more than 150,000 children involved in two national population-based cohorts (one Danish and one Norwegian) revealed that children who were never breastfed had a twofold higher risk of T1D development than those who received breastfeeding; however, duration and frequency did not influence the hazard ratios [Citation132]. Similarly, a meta-analysis of 43 observational studies including 9874 patients with type 1 diabetes disclose that the overall risk of T1D is diminished after exclusive breast-feeding for more than 2 weeks [Citation133].
Notably, in NOD mice, administration of human milk oligosaccharides (HMO) in a specific early-life window (from 4 to 10 weeks of age) was found to reduce the incidence of diabetes and the severity of insulitis. Notably, HMO intake resulted in an overall increase in fecal SCFA and a rise in the relative abundance of Akkermansia, Ruminococcus, Oscillospira, Lachnospiraceae, and Coprococcus. Mechanistically, HMO, particularly in combination with acetate or butyrate, suppressed the surface expression of major histocompatibility complex II and of the co-stimulatory molecules CD80, CD86, and CD40 in LPS-activated DC. Instead, the surface expression of inhibitory molecules PD-1 and CD40-ligand was downregulated at steady-state, suggesting a lower ability of DC to stimulate, respectively, T or B cells upon HMO and SCFA exposure [Citation134].
Remarkably, harboring specific MHC alleles shapes the gut microbiome early in ontogeny. Silverman et al. unveiled that expression of the protective MHC-II Eα:Eβ complex, which protects NOD mice from spontaneous diabetes, vertically protects the NOD offspring from diabetes and insulitis by modifying the intestinal microbiota in early-life. In fact, antibiotic therapy with vancomycin in the last week of pregnancy, before or immediately after weaning induces insulitis in Eα16/NOD mice. Compared to NOD mice, the cecal microbiome of Eα16/NOD mice has greater species diversity and higher representation of Clostridiales and reduced proportion of the genus Blautia. The benefits of the MHC-II Eα molecule on the microbiota structure were confirmed by transferring the intestinal microbiota from Eα16/NOD mice to NOD recipients, which displayed significantly less sever insulitis following faecal microbiota transplatations (FMT) in early-life (2–5 weeks of age) [Citation135]. Thus, the protective effects of certain HLA alleles can be partially explained by shaping the microbiota during a delicate window of early microbiome and immune development.
The impact of MHC molecules and host immunity in general on the gut microbiome structure may explain why microbiota transfer cannot fully transfer the disease phenotype to recipient NOD mice. For instance, Mullaney et al. reported that cohousing NOD mice with disease-protected C57BL/6 mice did not affect the disease outcome and only cohousing from weaning resulted in a trend toward delayed diabetes incidence. Importantly, the microbiota of NOD mice, which express the unusual MHC II Ag7 complex, was resistant to changes both after short- and long-term cohousing, maintaining lower diversity and a different composition than C57BL/6 mice [Citation136]. Neuman et al., instead, used GF NOD mice and oral gavages to transfer the fecal microbiome from five adolescents with T1D, two of which were fast progressors (with >75% loss of β cell function within 1 year) and three slow progressors (<28% function loss over 1 year). They found that although the human microbiota modified the pace of diabetes progression, disease severity was not transferable to GF NOD mice [Citation137].
3.4. Impact of microbiota depletion in T1D onset
Disruption of gut microbial communities by antibiotic therapy has also been shown to modify T1D development. The study of Ruiz et al. showed that C57BL/6 pups treated with macrolide (the most frequent antibiotic subscribed to children within 2 years of age) at postnatal day 5 displayed and perturbed microbiota community, intestinal and splenic T cell populations with decreased Th17 and CD8 T lymphocytes and reduced the levels of secretory IgA. Transferring the perturbed microbiota of macrolide-treated pups proved that the microbiome alterations were sufficient and required to disturb the immune cell development [Citation138]. In line with this, early-life macrolide-based pulsed antibiotic treatment in NOD mice significantly increased the incidence of T1D and insulitis severity. Moreover, at pre-diabetic stage, the pulsed-antibiotic treatment reduced the proportion of Th17 and Treg cells in the intestinal lamina propria [Citation139].
A subsequent study supported the finding that early-life antibiotic exposure with pulsed macrolide treatment between 5 and 10 days of age is sufficient to accelerate diabetes onset in NOD mice and to persistently disturb the microbial communities, which show reduced species richness weeks after macrolide treatment. Furthermore, metagenomic analysis of fecal samples and whole transcriptome analysis of ileal samples revealed that one course of antibiotic can alter both the metabolic and biosynthesis capacity of commensal microbes (by altering genes involved in carbohydrate degradation, amino acid synthesis, and synthesis of bacterial components such as peptidoglycan) as well as the ileal transcriptomic profile with an overall decrease in genes encoding molecules involved in host antimicrobial defenses and microbe sensing by innate signaling pathways. As for the studies described above, a single macrolide treatment of 5 days affected the adaptive branch of the immune system by increasing the systemic and pancreatic proportions of activated memory CD62L + CD4 and CD8 T cells and B cells, which can explain the accelerated insulitis upon antibiotics [Citation140]. In a follow-up study from the same group, it emerges that transfer of maternal cecal microbiota to pups rescues the acceleration of T1D caused by the early-life exposure to macrolide. Indeed, cecal microbiota transplantation could restore the recipient microbiota toward baseline state both in terms of taxonomy and metabolic pathways. Moreover, maternal microbiota re-established, in part, the expression of innate immunity regulators in the ileum, including CD44, TLR2, and the antimicrobial Reg3γ [Citation141]. Overall, these studies support the concept of the neonatal window of opportunity, during which the microbiome-host interactions are crucial in imprinting the host health for life. Further research is however needed in order to advance targeted therapeutic approaches that may beneficially shape the microbiome structure and hence nurture immune tolerance and intestinal homeostasis.
Opposing the above-described studies, vancomycin treatment of NOD mice both in adult life (from 8 weeks of age until diabetes onset) and in neonatal life (from birth till 28 days) was shown to lower the percentage of diabetes by 30 weeks of age. Particularly, the treated adults displayed better glycemic levels and ameliorated insulitis score at diabetes onset. However, in agreement with others’ work, early life vancomycin treatment disrupted the intestinal immune balance by significantly increasing the frequency among CD4 T cells of IFN-γ- and TNF-α-producing Th1 cells, albeit without changing the proportion of IL-10-producing anti-inflammatory T cells. Nonetheless, when studying the vancomycin-induced changes in microbiota composition, Hänninen and colleagues found that vancomycin depleted many major genera of Gram-positive and Gram-negative microbes but propagated one single species: Akkermansia muciniphila, which became dominant. This mucin-degrading bacteria has been largely considered beneficial to human metabolic health and was shown to be protective in NOD mice against T1D development [Citation100]. Thus, the suppression of clinical diabetes onset upon vancomycin treatment can be attributed to the expansion of Akkermansia muciniphila as a result of loss of competing bacteria species [Citation142]. Interestingly, Hu et al. discovered that prenatal and neonatal antibiotic treatment protects the offspring from T1D. Indeed, administration of neomycin, polymyxin B, and streptomycin, with the scope of mainly targeting Gram-negative bacteria, to pregnant NOD mice or to dams after giving birth (antibiotic intake via maternal milk) markedly decreases the incidence of T1D in the offspring adult life as compared to untreated NOD mice or mice treated with antibiotics after weaning. Accordingly, introduction of gut microbiota from adult NOD mice reversed this protection, indicating that early changes in gut microbiome modulate disease progression. Moreover, these protective effects are long-lasting since adoptive transfer of splenocytes from the offspring of antibiotic-treated dams halt diabetes both in NOD-SCID mice (lacking mature B and T cells) as well as in irradiated NOD recipients. Mechanistically, the protection given by prenatal and neonatal antibiotic exposure can be explained by alterations in the gut microbiome and in the host immune system. Indeed, the protection against diabetes was accompanied by a significant reduction in Th1 responses in mesenteric and pancreatic lymph nodes and an overall increase in frequency of regulatory T cells in several lymphoid tissues. Underlying this effect on T lymphocytes was an induction of tolerogenic antigen-presenting cells in splenocytes as attested by the constriction of IFN-γ + CD11c +, IL-12 + CD11b +, and IL-17 + CD11b + DC and macrophages and expansion of IL-10-producing CD11b + cells. Furthermore, splenic antigen-presenting cells from mice born from antibiotic-treated dams had an impaired ability to stimulate autoreactive CD8 T cells ex vivo and delayed diabetes progression in NOD-SCID recipient mice after adoptive transfer [Citation143]. Hence, multiple mechanisms are at play during antibiotic administration:timing and bacteria targeted by antibiotics can shift the delicate balance between favorable and ‘diabetogenic’ strains.
Overall, the human observations and animal studies suggest that the microbiome impact on autoimmunity occurs before seroconversion or clinical development, in the earliest months of life in genetically predisposed individuals.
3.5. Host antimicrobial peptides as protective mediators in T1D
Several studies have now proven the existence of a communication loop between the pancreas and the intestine and the disruptions of this loop may lead to both gut and pancreatic disorders [Citation144].
Ahuja and colleagues demonstrate that deficiency of calcium channel Orai1 specifically in pancreatic acinar cells disrupts secretion of cathelicidine-related antimicrobial peptide (CRAMP), resulting in pancreatic injury and perturbations in commensal microbes and a bacterial outgrowth. Timed deletion of pancreatic Orai1 was sufficient to cause death in the majority of mice, associated with intestinal inflammation. However, these deleterious effects could be reversed by oral administration of pancreatic antimicrobials indicating that pancreatic antimicrobial peptides control intestinal homeostasis [Citation145]. Vice versa, studies have shown that gut microbes control the production of pancreatic antimicrobials. Indeed, in diabetic female NOD mice, the secretion of CRAMP by islets α-/β-cells was diminished as compared to the nonautoimmune BALB/c and C57BL/6 strains [Citation146]. Intraperitoneal administration of CRAMP reduced the frequency of IGRP-autoreactive IFN-γ + CD8 T cells and the incidence of autoimmune diabetes. Specifically, CRAMP administration shaped the phenotype of intra-islet macrophages with an increase in regulatory macrophages at the expenses of pro-inflammatory macrophages. This phenomenon was observed both in autoimmune NOD mice as well as in streptozotocin-injected BALB/c and C57BL/6 mice. Similarly, CRAMP injections prompted the accumulation of regulatory DC and IL-10-producing FOXP3+ Treg and Tr1 T cells. Importantly, the effects of CRAMP were possible through a microbiota-mediated mechanism by which the SCFA butyrate controls islet-derived CRAMP by signaling through G protein-coupled receptors GPR43 and GPR41 on β-cells. Indeed, butyrate injection failed to induce regulatory macrophages in islets of CRAMP-deficient C57BL/6 mice, showing the requirement of CRAMP for part of butyrate anti-diabetogenic effects. As diabetic NOD mice display lower levels of fecal SCFA than non-autoimmune strains, fecal microbiota transfer from the latter could restore CRAMP production, indicating that in NOD mice a faulty microbial SCFA production results in defective pancreatic CRAMP production and hence a shift toward inflammatory stimuli at the expenses of tolerance [Citation146]. In line with this, Pound et al. reported CRAMP reduction in islets of diabetes-prone BB-DP rats before insulitis onset and showed that CRAMP daily intraperitoneal injections stimulated β-cells neogenesis from the pancreas as it significantly enhanced the number of duct-associated extraislet insulin+ clusters in BB-DP rats, but not in control BB rats. Lastly, they disclosed that CRAMP treatment shifted the abundance of potentially beneficial bacteria (e.g. probiotic Lactobacillus species) toward levels found in the gut of control rats [Citation147].
By comparing newborn female NOD mice with nonautoimmune mice C57BL/6 and BALB/c, Liang et al. found that gut dysbiosis occurs as early as 2 weeks of age and it is accompanied by a defect in the production of the antimicrobial peptide CRAMP by colonic epithelial cells [Citation148]. In this elegant study, the authors revealed that this deficiency results in an aberrant, but transient, type I interferon signature in the colon of 3-weeks old NOD mice, which in turn imprints the phenotype of colonic classical DC (cDC) toward more inflammatory CD103 + CD11b + cDC2 at the expenses of having less tolerogenic CD103 + CD11b - cDC1. Lastly, with a series of experiments including treatment with CRAMP or blockage of type I IFN signaling at early stage (between 10 and 21 days of age), and administration of the probiotic CRAMP-expressing L. lactis to NOD dams 1 day before delivery, the authors reveal that lack of CRAMP early in life sustain dysbiosis thereby pathologically imprinting colonic DC, with a consequential reduction in the frequency of colonic and pancreatic Treg and an increase in incidence of T1D in adult life [Citation148]. Although this study is exclusively conducted in mice, it sheds light on the importance of early microbiome perturbations and downstream host transitional effects in T1D development; moreover, this work hints at the possibility of treating pregnant mothers, with diabetes or genetic susceptibility to T1D, with specific probiotics in order to prevent an aberrant pathogenic microbiota in newborns.
LL-37 is the only peptide in the Cathelicidin family present in humans and it exerts both immunomodulatory and antimicrobial activities [Citation149]. It remains to be investigated whether human intestinal epithelial cells lack the ability to produce LL-37 in autoimmune diabetes and whether oral administration may simulate the effects seen in NOD mice.
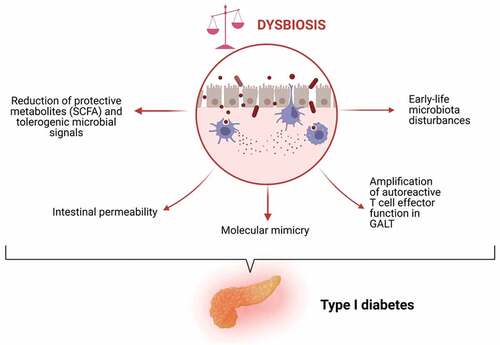
shows the potential mechanisms, by which an unbalanced microbiota can play a role in T1D onset.
Figure 2. Mechanisms by which dysbiosis may underpin T1D pathogenesis.
4. Microbiome-based human intervention trials
In light of the benefits observed in NOD mice fed a butyrate- or acetate-rich diet [Citation120], the group of Marino et al. performed a follow-up clinical trial to test the safety and therapeutic potential of 6-week dietary supplementation of butyrate and acetate in 20 subjects suffering from T1D [Citation150]. To deliver high yield of SCFA to the colon, the authors opted to administer 40 g/day of a high-maize starch modified by bonding acetate and butyrate (HAMSAB). Indeed, both butyrate and acetate were found increased in stool and plasma samples. However, the treatment did not improve fasting glycemia, fasting or postprandial C-peptide plasma levels. Of note, the mean diabetes duration of participant was 14 years; hence, it remains to be determined whether HAMSAB diet may be beneficial in protecting β-cell function in new-onset T1D patients. Nonetheless, when interrogating the relationship between SCFA and clinical parameters, plasma butyrate was found to negatively associate with HbA1c, daily basal insulin dosage, and time below target range (<3.9 mM glucose). The main effects of HAMSAB diet were observed in the immunophenotype profile after 6 weeks of dietary supplementation and particularly after a 6-week washout period with no HAMSAB intake. Indeed, 36 immune subpopulations out of 110 were affected by the HAMSAB diet in terms of proportions or phenotypic marker expression. For instance, the expression of the co-stimulatory molecule CD86 was downregulated on plasmacytoid DC and cDC, after HAMSAB treatment and reduced after the washout period on the marginal zone-like (CD27+IgD+IgMhigh) B cell subpopulation, which infiltrate pancreata and can present islet autoantigens to effector T cells [Citation150,Citation151]. Simultaneously, HAMSAB diet intake resulted in enhanced expression of the T cell inhibitory molecules CTLA4 and TIGIT. The levels of CTLA4 were significantly increased on effector CD4 T cells and CD8 T cells following the washout period; whereas, TIGIT expression was enhanced on conventional CD4 T cells and FOXP3+ regulatory T cells. Hence, the loss of CD86 on DC and marginal zone-like B cells together with increased expression of immune checkpoint inhibitors on T cells may imply that SCFA hamper antigen presentation and activation of pathogenic T cells. Moreover, the circulating levels of several pro-inflammatory cytokines were diminished by SCFA supplementation and even after the washout time [Citation150]. Overall, it is intriguing that these favorable immunological changes persist for 6 weeks after the last HAMSAB intake and raises the question of whether intermitted short (e.g. 6 weeks) dietary interventions can hamper T1D progression by promoting a systemic immunotolerogenic profile.
In contrast to the benefits on immunity achieved by HAMSAB diet, daily oral administration of 4 grams of sodium butyrate did not result in alterations in innate or adaptive immune responses in long-standing type 1 diabetic patients. De Groot et al. performed a controlled, double-blind cross-over study in 30 T1D patients with placebo/butyrate treatments and washout periods lasting one month each [Citation152]. In this study, both T cell proliferation to β-cell-derived autoantigens and number of autoreactive CD8 T cells were not altered by butyrate treatment. Similarly, phenotypic analysis of monocyte subsets did not show significant changes, except for expression intensity of CD11b after butyrate. In line with this, ex vivo monocyte stimulation with TLR2 and TLR4 ligands resulted in alike cytokine secretion after placebo or butyrate. At the fecal microbiota levels, moderate changes were induced by butyrate intake, with Lachnospira pectinoschiza, Ruminococcaceae spp., Marvinbryantia spp., Erysipelotrichaceae spp., Bifidobacterium adolescentis/faecale/stercoris, Dorea formicigenerans, and several Lachnospiraceae spp. being the most discriminative taxa between placebo and butyrate treatments. The discordancy between the findings of de Groot et al. and Bell et al. can be explained by the use of targeted antibody panels in the first study instead of the mass cytometry approach of the second study, which enabled the identification of 110 immunophenotypes; moreover, the delivery of butyrate via a modified maize starch appears a better form of delivering SCFA to the colon as de Groot et al. did not observe an increase in fecal SCFA in contrast to the report by Bell et al. Lastly, oral butyrate supplementation increased, albeit not significantly, the secretion of C-peptide during mix meal test. This may have been expected given that the duration of diabetes of the participants spanned from 4 to 16 years (mean of 8 years) arguing that the window of intervention is too late or the treatment period is too short to revive the proportion of still intact islets [Citation152].
Recently, our group has conducted the first clinical trial employing faecal microbiota transplantations in 20 new-onset type 1 diabetic individuals to interrogate the magnitude of microbiota-mediated effects in T1D progression [Citation153]. The participants were diagnosed with T1D for a maximum period of 6 weeks, presented residual β-cell function, and were randomized in two groups receiving three consecutive autologous or allogeneic (from healthy donors) FMTs over a period of 4 months and followed for up to 12 months. The most striking impact of colonic microbiota transplantations was the long-term preservation of β-cell function. Indeed, at 12-month follow-up (8 months after the last FMT) both the levels of C-peptide at fasting state and during the postprandial response were maintained similarly to baseline rates in the autologous FMT group but gradually reduced over the 12-month period in the allogeneic FMT group. Notably, the proportion of circulating effector CD4 + CXCR3 + and CD8 + CXCXR3 + T cells was differentially changed at 12 months compared to baseline in the two treatment groups, being decreased in the responder group. The chemokine receptor CXCR3 is pivotal for activated CD4 and CD8 T cell trafficking to inflamed organs, such the pancreas in T1D where β-cells secrete the ligands CXCL9/10/11 attracting activated T cells [Citation154–156]. Interestingly, when looking at the changes in small intestinal and colonic microbiota, the sulfate-reducing strain fecal Desulfovibrio piger and duodenal Prevotella spp. were identified as best predictors of FMT treatment groups and, respectively, positively and negatively associated with beneficial changes in C-peptide concentrations. Remarkably, predictive modeling revealed that baseline microbiota characteristics, including taxonomy and metabolic capacity, as well as baseline duodenal expression of genes involved in cytokine signaling and maintenance of epithelial barrier could accurately predict the response to FMTs at 12 months [Citation153]. This suggests that the intestinal characteristics of the host largely influence strain engraftment after colonic microbiota transplantation, as well as the response to incoming microbes by parenchymal and immune cells of the intestinal mucosa, which in turn can imprint systemic and organ-specific immune reactions.
Previous studies have investigated the impact of prebiotics in diabetes progression. Prebiotics are substrates utilized by commensal microbes that provide benefits to the host [Citation157]. For instance, prebiotics have been shown to ameliorate glycemia (HbA1c levels), postprandial glycemic responses, and circulating inflammatory markers (plasma CRP, TNF-α, LPS) in patients with type 2 diabetes [Citation158,Citation159]. A 2019 study conducted by Ho et al. disclosed a positive effect of prebiotic on β-cell function in children/adolescents, who had been diagnosed with T1D for at least 1 year [Citation160]. The 43 participants were randomized into two groups receiving daily oral prebiotics (oligofructose-enriched inulin) or placebo (maltodextrin) for 3 months. Interestingly, the group receiving prebiotics showed a robust improvement in C-peptide levels after 3 months, while the placebo group displayed a decline in C-peptide rates compared to baseline [Citation160]. Prebiotic treatment also improved the intestinal permeability, evaluated with the lactulose and mannitol intake test, and expanded the abundance of Bifidobacterium, which produce the beneficial SCFA [Citation157].
In view of the benefits provided by Lactobacillus johnsonii N6.2 in diabetes-prone rates, there is an ongoing trial set to investigate the potential therapeutic benefits against islet autoimmunity in 57 children and adolescents (8–17 years old) through daily intake of capsule harboring either placebo or Lactobacillus johnsonii N6.2 for 24 weeks (https://clinicaltrials.gov/ct2/show/NCT03961854). L. johnsonii N6.2 has already been shown to be safe and well-tolerated in 42 healthy individuals participating in a cross-over randomized trial where the participants received daily capsules containing L. johnsonii N6.2 or placebo for 8 weeks. The strain intake resulted in changes in the host metabolome with an increase in tryptophan-derived metabolites. In addition, L. johnsonii also led to enhanced frequency of effector Th1 cells (CD45RO+CD183+CD196−) and cytotoxic CD8+ T cells in the bloodstream [Citation161].
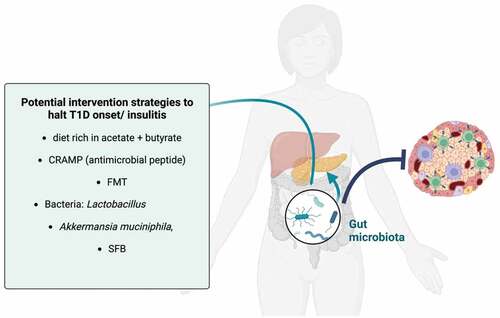
shows the putative intervention strategies targeting the gut microbiota that could hamper insulitis and diabetogenesis, based on the research discoveries described in this review.
Figure 3. Therapeutic strategies targeting the gut microbiota to harness insulitis/T1D progression.
5. Expert opinion
T1D is still an uncurable disease and despite the advances of glucose monitoring approaches and insulin replacement therapies, many patients experience short- and long-term complications, such as vascular and neuronal complications resulting in cardiovascular disease, nephropathy, neuropathy, or retinopathy. It is therefore important to intervene in the prevention and pathophysiology of disease. The gut microbiota constitutes a novel target of intervention, particularly in early-life or early-disease stage. The intestinal microbial communities can be viewed as modifiable components of immunity as they can influence the function and differentiation of innate and adaptive immune cells in a co-evolving microbiota-immune cell interplay [Citation50,Citation51].
Although a common taxonomic signature of diabetic microbiota has not been identified, common traits to the microbiome of T1D patients from different geographical regions are the loss in bacterial species diversity and ability to produce the protective microbial SCFA [Citation87–90]. Strategies aiming to re-establish a healthy microbiome or focusing on improving its composition should be studied in murine models and subsequently in human clinical trials. In this regard, the work of de Groot et al. [Citation153] is particularly important as it discloses that autologous FMTs in T1D patients are safe and halt the loss of pancreatic β-cell mass/functionality. Although the mechanism of this phenomenon was not unraveled, possible explanations may be that the entry of millions of indigenous bacteria in the small intestine recruits to the intestinal mucosa pathogenic T lymphocytes, which cross-react with both bacterial mimotopes and islet-derived autoantigens, and/or endogenous bacteria induce long-term immune tolerance as compared to engraftment with exogenous bacteria (from healthy donors), to which the host immune system has not developed an immunotolerant and symbiotic relationship. Moreover, the use of autologous microbiota for transplantation has the advantage of avoiding the challenges of selecting appropriate healthy donors based on time-consuming in-depth screening for illnesses, dietary habits, drug consumption, and detection of pathogens in fecal stool samples. Moreover, it can ensure a better engraftment than in the case of transplanting exogenous bacteria.
As more and more microbial strains have been identified for their beneficial impact on the immune system, particularly for dampening inflammation and expanding tolerogenic immune populations, autologous FMTs can be accompanied by the inoculum of beneficial strains, such as Akkermansia muciniphila, segmented filamentous bacteria, Lactobacillus johnsonii, or Bacteroides fragilis [Citation62,Citation100,Citation102,Citation106].
This review highlights the importance of microbial metabolites, namely SCFAs, in the maintenance of gut and immune homeostasis and in providing some degree of protection against T1D as shown by translational and clinical studies. Microbiota-derived small-molecule metabolite can be detected in the circulation and is increasingly being recognized as pivotal modulators of host physiology and immune cell functions. With the advances in multi-omics approaches and computational frameworks, we envision that more metabolites of microbial origin will be identified as predictors of T1D outcome and/or progression pace, and will subsequently be studied for their function not solely on immune cells but also on endocrine cells, including pancreatic β-cells. Moreover, the progress in the microbiome field has also brought attention to choices in analytical methods and technical cofounders in microbiota analysis [Citation162–164].
The studies of de Groot et al. [Citation153] and Bell et al. [Citation150] underline the importance of carefully designing the delivery strategy of microbial metabolites and take into account their primary site of absorption/action when planning metabolite-based human intervention studies. Similarly, both for metabolite- or bacteria-based therapeutic approaches, the window of intervention may be critical for achieving robust clinical benefits. In light of the translational studies showing the importance of early-life microbiome-immunity cross-talks [Citation138–141] and of the success and safety of maternal FMTs in cesarean section-born infants [Citation165], the long-term impacts of improving the microbiota with prebiotic/probiotic/metabolite supplementations or of re-shaping the microbiome structure and, consequently, the gut-immunity loop with autologous or maternal FMTs should be investigated in children genetically predisposed to T1D.
Finally, defining beneficial commensal strains and novel immunomodulatory microbial metabolites as well as uncovering how they influence host immunity and pancreatic functions will considerably expand the possibility of microbiome-based therapeutics against T1D and improve the design of intervention modality.
Article highlights
In the past decades, the incidence of T1D has drastically increased worldwide.
Both genetic and environmental factors govern T1D onset and perturbations of the gut microbiota have been linked to T1D development.
The gut microbiome and the host immune system coevolve after birth and reciprocally control each other’s.
Particular commensal species provide tolerogenic signals to immune cells, whereas others can induce or amplify inflammation.
Disturbances in the gut microbiome structure linked to T1D modify the trajectory of T1D development, affecting seroconversion and insulitis severity.
Declaration of interest
M Nieuwdorp is founder and member of the Scientific Advisory Boards of Caelus Health, The Netherlands. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Writing of this review was supported by the Health-Holland TKI-PPP and Dutch Kidney Foundation Innovation grants awarded to E Rampanelli and by a ZonMw-NW VICI grant 2020 (09150182010020) awarded to M Nieuwdorp.
Additional information
Funding
References
- Wang Z, Xie Z, Lu Q, et al. Beyond genetics: what causes type 1 diabetes. Clin Rev Allerg Immunol. 2016;52920:273–584.
- International Diabetes Federation, IDF diabetes atlas, 10th edition 2021. https://diabetesatlas.org
- Fenneman AC, Rampanelli E, Yin YS, et al. Gut microbiota and metabolites in the pathogenesis of endocrine disease. Biochem Soc TArans. 2020;48:915–931.
- Roep BO, Tree TI. Immune modulation in humans: implications for type 1 diabetes mellitus. Nat Rev Endocrinol. 2014;10:229–242.
- Hummel M, Bonifacio E, Schmid S, et al. Brief communication: early appearance of islet autoantibodies predicts childhood type 1 diabetes in offspring of diabetic patients. Ann Intern Med. 2004;140:882–886.
- Achenbach P, Bonifacio E, Koczwara K, et al. Natural history of type 1 diabetes. Diabetes. 2005;54(suppl.2):S25–31.
- Kaufman DL, Erland MG, Clare-Salzler M, et al. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. JClin Invest. 1992;89:283–292.
- Yoon JW, Onodera T, Notkins AL. Virus-induced diabetes mellitus. XV. Beta cell damage and insulin-dependent hyperglycemia in mice infected with coxsackie virus B4. J Exp Med. 1978;148:1068–1080.
- Luopajarvi K, Savilahti E, Virtanen SM, et al. Enhanced levels of cow’s milk antibodies in infancy in children who develop type 1 diabetes later in childhood. Pediatr Diabetes. 2008;9:434‐41.
- Du T, Zhou Z, You S, et al. Regulation by 1, 25‐dihydroxy‐vitamin D3 on altered TLRs expression and response to ligands of monocyte from autoimmune diabetes. Clin Chim Acta. 2009;402(1–2):133‐8.
- Ghazarian L, Diana J, Simoni Y, et al. Prevention or acceleration of type 1 diabetes by viruses. Cell Mol Life Sci. 2013;70(2):239‐55.
- Li X, Atkinson MA. The role for gut permeability in the pathogenesis of type 1 diabetes—a solid or leaky concept? Pediatr Diabetes. 2015;16(7):485‐92.
- Wernroth ML, Fall K, Svennblad B, et al. Early childhood antibiotic treatment for otitis media and other respiratory tract infections is associated with risk of type 1 diabetes: a nationwide register-based study with sibling analysis. Diabetes Care. 2020;43(5):991–999.
- James EA, Pietropaolo M, Mamula MJ. Immune recognition of β-cells: neoepitopes as key players in the loss of tolerance. Diabetes. 2018;67(6):1035–1042.
- Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. 2017;127(8):2881–2891.
- Rodriguez-Calvo T, Krogvold L, Amirian N, et al. One in Ten CD8+ cells in the pancreas of living individuals with recent-onset type 1 diabetes recognizes the preproinsulin epitope PPI15-24. Diabetes. 2021;70:752–758.
- Burrack AL, MartinovA T, Fife BT. T cell-mediated beta cell destruction: autoimmunity and alloimmunity in the context of type 1 diabetes. Front Endocrinol. 2017;8:343.
- Krischer JP, Lynch KF, Schatz DA, et al. The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: the TEDDY study. Diabetologia. 2015;58(5):980–987.
- Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA. 2007;104(43):17040–17045.
- Jacobsen LM, Bocchino L, Evans-Molina C, et al. The risk of progression to type 1 diabetes is highly variable in individuals with multiple autoantibodies following screening. Diabetologia. 2020;63(3):588–596.
- Vehik K, Bonifacio E, Lernmark Å, et al. Hierarchical order of distinct autoantibody spreading and progression to type 1 diabetes in the TEDDY study. Diabetes Care. 2020;43(9):2066–2073.
- Orban T, Sosenko JM, Cuthbertson D, et al. Pancreatic islet autoantibodies as predictors of type 1 diabetes in the Diabetes Prevention Trial-Type 1. Diabetes Care. 2009;32(12):2269–2274.
- Akashi T, Nagafuchi S, Anzai K, et al. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol. 1997;9(8):1159–1164.
- Fiorina P, Vergani A, Dada S, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes. 2008;57(11):3013–3024.
- Wong FS, Wen L, Tang M, et al. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes. 2004;53(10):2581–2587.
- Smith MJ, Cambier JC, Gottlieb PA. Endotypes in T1D: b lymphocytes and early onset. Curr Opin Endocrinol Diabetes Obes. 2020;27(4):225–230.
- Gearty SV, Dündar F, Zumbo P, et al. An autoimmune stem-like CD8 T cell population drives type 1 diabetes. Nature. 2022;602(7895):156–161.
- ElTanbouly MA, Noelle RJ. Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat Rev Immunol. 2021;21(4):257–267.
- Edner NM, Carlesso G, Rush JS, et al. Targeting co-stimulatory molecules in autoimmune disease. Nat Rev Drug Discov. 2020;19(12):860–883.
- Orban T, Bundy B, Becker DJ, et al. Costimulation modulation with Abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care. 2014;37:1069–1075.
- Edner NM, Heuts F, Thomas N, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol. 2020;21:1244–1255.
- Boldison J, Da Rosa LC, Davies J, et al. Dendritic cells license regulatory B cells to produce IL-10 and mediate suppression of antigen-specific CD8 T cells. Cell Mol Immunol. 2020;17:843–855.
- Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia. 2017;60:1839–1850.
- Raffin C, Vo LT, Bluestone JA. Treg cell-based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20:158–172.
- Lindley S, Dayan CM, Bishop A, et al. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–99.
- Long SA, Cerosaletti K, Bollyky PL, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes. 2010;59:407–415.
- Ellis JS, Wan X, Braley-Mullen H. Transient depletion of CD4+ CD25+ regulatory T cells results in multiple autoimmune diseases in wild-type and B-cell-deficient NOD mice. Immunology. 2013;139:179–186.
- Tang Q, Henriksen KJ, Bi M, et al. In Vitro–expanded antigen-specific regulatory t cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465.
- Marek-trzonkowskaan N, Myśliwiec M, Dobyszuk A, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — results of one year follow-up. Clinic Immunol. 2014;153:23–30.
- Bluestone JA, Buckner JH, Fitch M, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med. 2015;7:15ra189.
- Wiedeman AE, Muir VS, Rosasco MG, et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J Clin Invest. 2020;130:480–490.
- Diggins KE, Serti E, Muir V, et al. Exhausted-like CD8+ T cell phenotypes linked to C-peptide preservation in alefacept-treated T1D subjects. JCI Insight. 2021;6:e142680.
- Roep BO, Thomaidou S, van Tienhoven R, et al. Type 1 diabetes mellitus as a disease of the β-cell (do not blame the immune system? Nat Rev Endoc. 2021;17:150–161.
- Damond N, Engler S, Zanotelli VRT, et al. A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab. 2019;29:755–768.
- Foulis AK, Jackson R, Farquharson MA. The pancreas in idiopathic Addison’s disease–a search for a prediabetic pancreas. Histopathology. 1988;12:481–490.
- Coppieters KT, Dotta F, Amirian N, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med. 2012;209:51–60.
- Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118.
- Donaldson GP, Lee M, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microb. 2016;14:20–32.
- Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164:337–340.
- Gensollen T, Iyer SS, Kasper DL, et al. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–544.
- Round JL, Mazmanian SK. The gut microbiome shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2000;9:313–323.
- Hapfelmeier S, Lawson MA, Slack E, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328:1705–1709.
- Mazmanian SK, LiuC H, Tzianabos AO, et al. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122(1):107e18.
- Gordon HA. Morphological and physiological characterization of germfree life. Ann N Y Acad Sci. 1959;78:208e20.
- Chung H, Pamp SJ, Hill JA, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578e93.
- Khosravi A, Yanez A, Price JG, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe. 2014;15:374e81.
- Cash HL, Whitham CV, Behrendt CL, et al. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126e30.
- Pickard JM, Zeng MY, Caruso R, et al. Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. 2017;279:70–89.
- Thoo L, Noti M, Krebs P. Keep calm: the intestinal barrier at the interface of peace and war. Cell Death Dis. 2019;10:849.
- Johansson MEV, Jakobsson HE, Holmén-Larsson J, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. 2015;18:582–592.
- Wang Y, Telesford KM, Ochoa-Repáraz J, et al. Intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun. 2014;5:4432.
- Telesford KM, Yan W, Ochoa-Reparaz J, et al. A commensal symbiotic factor derived from Bacteroides fragilis promotes human CD39(+)Foxp3(+) T cells and treg function. Gut Microbes. 2015;6:234–242.
- Brown EM, Kenny DJ, Xavier RJ. Gut microbiota regulation of T cells during inflammation and autoimmunity. Annu Rev Immunol. 2019;37:599–624.
- Atarashi K, Tanoue T, Oshima K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236.
- Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498.
- Chervonsky AV. Microbiota and autoimmunity. Cold Spring Harb Perspect Biol. 2013;5:a007294.
- Skelly AN, Sato Y, Kearney S, et al. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat Rev Immunol. 2019;19:305–323.
- den Besten G, van Eunen K, Groen AK, et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54:2325–2340.
- Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16:341–352.
- Quivy V, Van Lint C. Regulation at multiple levels of NF-kappaB-mediated transactivation by protein acetylation. Biochem Pharmacol. 2004;68:1221–1229.
- Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286.
- Vinolo MA, Rodrigues HG, Hatanaka E, et al. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011;22:849e55.
- Millard AL, Mertes PM, Ittelet D, et al. Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin Exp Immunol. 2002;130:245e55.
- Chang PV, Hao L, Offermanns S, et al. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111:2247e52.
- Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455.
- Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450.
- Garabatos N, Santamaria P. Gut microbial antigenic mimicry in autoimmunity. Front Immunol. 2022;13:873607.
- Tai N, Peng J, Liu F, et al. Microbial antigen mimics activate diabetogenic CD8 T cells in NOD mice. J Exp Med. 2016;213:2129–2146.
- Girdhar K, Huang Q, Chow I, et al. A gut microbial peptide and molecular mimicry in the pathogenesis of type 1 diabetes. Proc Natl Acad Sci U S A. 2022;119:e2120028119.
- Nanjundappa RH, Ronchi F, Wang J, et al. A gut microbial mimic that hijacks diabetogenic autoreactivity to suppress colitis. Cell. 2017;171:655–667.
- de Goffau MC, Luopajarvi K, Knip M, et al. Fecal microbiota composition differs between children with beta‐cell autoimmunity and those without. Diabetes. 2013;62:1238‐44.
- Davis‐Richardson AG, Ardissone AN, Dias R, et al. Bacteroides dorei dominates gut microbiome prior to autoimmunity in Finnish children at high risk for type 1 diabetes. Front Microbiol. 2014;5:678.
- de Goffau MC, Fuentes S, van den Bogert B, et al. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. 2014;57:1569‐77.
- Brown CT, Davis‐Richardson AG, Giongo A, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS One. 2011;6:e25792.
- Maffeis C, Martina A, Corradi M, et al. Association between intestinal permeability and faecal microbiota composition in Italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab Res Rev. 2016;32:700‐9.
- Murri M, Leiva I, Gomez-Zumaquero JM, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med. 2013;11:46.
- Kostic AD, Gevers D, Siljander H, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17:260–273.
- Qi C, Zhang Q, Yu M, et al. Imbalance of fecal microbiota at newly diagnosed type 1 diabetes in Chinese children. Chin Med J. 2016;129:1298–1304.
- Giongo A, Gano KA, Crabb DB, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011;5:82–91.
- Vatanen T, Franzosa EA, Schwager R, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. 2018;562:589–594.
- de Groot PF, Belzer C, Ö A, et al. Distinct fecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS One. 2017;12:e0188475.
- van Heck JIP, Gacesa R, Stienstra R, et al. The Gut microbiome composition is altered in long-standing type 1 diabetes and associates with glycemic control and disease-related complications. Diabetes Care. 2022;45:2084–2094.
- Mosterd CM, Kanbay M, van den Born BJH, et al. Intestinal microbiota and diabetic kidney diseases: the role of microbiota and derived metabolites inmodulation of renal inflammation and disease progression. Best Pract Res Clin Endocrinol Metab. 2021;35:101484.
- Stewart CJ, Ajami NJ, O’Brien JL, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018;562:583–588.
- Alkanani AK, Hara N, Gottlieb PA, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes. 2015;64:3510–3520.
- Wen L, Ley RE, Volchkov PY, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113.
- Pozzilli P, Signore A, Williams AJK, et al. NOD mouse colonies around the world - recent facts and figures. Immunol Today. 1993;14:193–196.
- Burrows MP, Volchkov P, Kobayashi KS, et al. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc Natl Acad Sci USA. 2016;112:9973–9977.
- Gülden E, Ihira M, Ohashi A, et al. Toll-like receptor 4 deficiency accelerates the development of insulin-deficient diabetes in non-obese diabetic mice. PLoS ONE. 2013;8(9):e75385.
- Hänninen A, Toivonen R, Sakari P, et al. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut. 2018;67(8):1373–1374.
- Endesfelder D, Engel M, Davis-Richardson AG, et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome. 2016;4(1):17.
- Martin AK, Sefik E, Hill JA, et al. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108(28):11548–11553.
- O’Connor W Jr, Kamanaka M, Booth CJ, et al. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat Immunol. 2009;10(6):603–609.
- Roesch LFW, Lorca GL, Casella G, et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. SME J. 2009;3:536–548.
- Lai KK, Lorca GL, Gonzalez CF. Biochemical properties of two cinnamoyl esterases purified from a lactobacillus johnsonii strain isolated from stool samples of diabetes-resistant rats. Appl Environ Microbiol. 2009;75:5018–24.
- Valladares R, Sankar D, Li N, et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP Rats. PLoS One. 2010;5(5):e10507.
- Diana J, Gahzarian L, Simoni Y, et al. Innate immunity in type 1 diabetes. Discov Med. 2011;11(61):513–520.
- Pearson JA, Wong FS, Wena L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J Autoimmun. 2016;66:76–88.
- Lee KU, Amano K, Yoon JW. Evidence for initial involvement of macrophage in development of insulitis in NOD mice. Diabetes. 1988;37(7):989–991.
- Nikolic T, Geutskens SB, van Rooijen N, et al. Dendritic cells and macrophages are essential for the retention of lymphocytes in (peri)-insulitis of the nonobese diabetic mouse: a phagocyte depletion study. Lab Invest. 2005;85(4):487–501.
- Sofi MH, Gudi R, Karumuthil-Melethil S, et al. pH of drinking water influences the composition of gut microbiome and type 1 diabetes incidence. Diabetes. 2014;63(2):632–644.
- Tchitchek N, Tchoumba ON, Pires G, et al. Low-dose IL-2 shapes a tolerogenic gut microbiota that improves autoimmunity and gut inflammation. JCI Insight. 2022;7(17):e159406.
- Bosi E, Molteni L, Radaelli MG, et al. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia. 2006;49(12):2824–2827.
- Leiva-Gea I, Sánchez-Alcoholado L, Martín-Tejedor B, et al. Gut microbiota differs in composition and functionality between children with type 1 diabetes and MODY2 and healthy control subjects: a case-control study. Diabetes Care. 2018;41(11):2385–2395.
- Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55(5):1443–1449.
- Sorini C, Cosorich I, Lo Conte M, et al. Loss of gut barrier integrity triggers activation of islet-reactive T cells and autoimmune diabetes. Proc Natl Acad Sci USA. 2019;116(30):15140–15149.
- Pöysti S, Toivonen R, Takeda A, et al. Infection with the enteric pathogen C. rodentium promotes islet-specific autoimmunity by activating a lymphatic route from the gut to pancreatic lymph node. Mucosal Immunol. 2022;15(3):471–479.
- Costa FRC, Françozo MCS, de Oliveira GG, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med. 2016;213(7):1223–1239.
- Zhang Q, Pan Y, Zeng B, et al. Intestinal lysozyme liberates Nod1 ligands from microbes to direct insulin trafficking in pancreatic beta cells. Cell Res. 2019;29(7):516–532.
- Mariño E, Richards JL, McLeod KL, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol. 2017;18(5):552–562.
- Jacob N, Jaiswal S, Maheshwari D, et al. Butyrate induced Tregs are capable of migration from the GALT to the pancreas to restore immunological tolerance during type-1 diabetes. Sci Rep. 2020;10(1):19120.
- Jia L, Cao M, Chen H, et al. Butyrate ameliorates antibiotic-driven type 1 diabetes in the female offspring of nonobese diabetic mice. J Agric Food Chem. 2020;68(10):3112–3120.
- Guo Y, Xiao Z, Wang Y, et al. Sodium butyrate ameliorates streptozotocin-induced type 1 diabetes in mice by inhibiting the HMGB1 expression. Front Endocrinol. 2018;9:630.
- Okada M, Zhang V, Loaiza Naranjo JD, et al. Islet-specific CD8+ T cells gain effector function in the gut lymphoid tissues via bystander activation not molecular mimicry. Immunol Cell Biol. 2022. DOI:10.1111/imcb.12593.
- Renz H, Adkins BD, Bartfeld S, et al. The neonatal window of opportunity—early priming for life. J Allergy Clin Immunol. 2018;141(4):1212–1214.
- van den Elsen LWJ, Garssen J, Burcelin R, et al. Shaping the gut microbiota by breastfeeding: the gateway to allergy prevention? Front Pediatr. 2019;7:47.
- Nielsen DS, Krych L, Buschard K, et al. Beyond genetics. Influence of dietary factors and gut microbiota on type 1 diabetes. FEBS Lett. 2014;e588(22):4234–4243.
- Knip M, Virtanen SV, Akerblom HK, et al. Infant feeding and the risk of type 1 diabetes. Am J Clin Nutr. 2010;91(5):1506S–13S.
- Hummel S, Ziegler AG. Early determinants of type 1 diabetes: experience from the BABYDIAB and BABYDIET studies. Am J Clinic Nutrition. 2011;84(suppl_6):1821S–3S.
- Marietta EV, Gomez AM, Yeoman C, et al. Low incidence of spontaneous type 1 diabetes in non-obese diabetic mice raised on gluten-free diets is associated with changes in the intestinal microbiome. PLoS One. 2013;8(11):e78687.
- Borch-Johnsen K, Joner G, Mandrup-Poulsen T, et al. Relation between breast-feeding and incidence rates of insulin-dependent diabetes mellitus. A hypothesis. Lancet. 1984;2(8411):1083–1086.
- Lund-Blix NA, Sander SD, Størdal K, et al. Infant feeding and risk of type 1 diabetes in two large Scandinavian birth cohorts. Diabetes Care. 2017;40(7):920–927.
- Cardwell CR, Stene LC, Ludvigsson J, et al. Breast-feeding and childhood-onset type 1 diabetes: a pooled analysis of individual participant data from 43 observational studies. Diabetes Care. 2012;35(11):2215–2225.
- l X, Van’t Land B, Engen PA, et al. Human milk oligosaccharides protect against the development of autoimmune diabetes in NOD-mice. Sci Rep. 2018;8(1):3829.
- Silverman M, Kua L, Tanca A, et al. Protective major histocompatibility complex allele prevents type 1 diabetes by shaping the intestinal microbiota early in ontogeny. Proc Natl Acad Sci USA. 2017;114:9671–9676.
- Mullaney JA, Stephens JE, Geeling BE, et al. Early-life exposure to gut microbiota from disease-protected mice does not impact disease outcome in type 1 diabetes susceptible NOD mice. Imunol Cell Biol. 2019;97(1):97–103.
- Neuman V, Cinek O, Funda DP, et al. Human gut microbiota transferred to germ-free NOD mice modulate the progression towards type 1 diabetes regardless of the pace of beta cell function loss in the donor. Diabetologia. 2019;62(7):1291–1296.
- Ruiz VE, Kurtz, ZD, Battaglia T, et al. A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Comm. 2017;8(1):518.
- Livanos AE, Greiner TU, Vangay P, et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol. 2016;1(11):16140.
- Zhang X, Li J, Krautkramer KA, et al. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. eLife. 2018;7:e37816.
- Zhang X, Yin YS, Wang J, et al. Maternal cecal microbiota transfer rescues early-life antibiotic-induced enhancement of type 1 diabetes in mice. Cell Host Microbe. 2021;19:1259–1265.
- Hansen CHF, Krych L, Nielsen DS, et al. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia. 2012;55(8):2285–2294.
- Hu Y, Peng J, Tai N, et al. Maternal antibiotic treatment protects offspring from diabetes development in nonobese diabetic mice by generation of tolerogenic APCs. J Immunol. 2015;195(9):4176–4184.
- Tilg H, Adolph TE. Beyond digestion: the pancreas shapes intestinal microbiota and immunity. Cell Metab. 2017;25(3):495–496.
- Ahuja M, Schwartz DM, Tandon M, et al. Orai1-mediated antimicrobial secretion from pancreatic acini shapes the gut microbiome and regulates gut innate immunity. Cell Metab. 2017;25(3):635–646.
- Sun J, Furio L, Mecheri R, et al. Pancreatic β-cells limit autoimmune diabetes via an immunoregulatory antimicrobial peptide expressed under the influence of the gut microbiota. Immunity. 2015;43(2):304–317.
- Pound LD, Patrick C, Eberhard CE, et al. Cathelicidin antimicrobial peptide: a novel regulator of islet function, islet regeneration, and selected gut bacteria. Diabetes. 2015;64(12):4135–4147.
- Liang W, Enée E, Andre-Vallee C. Intestinal cathelicidin antimicrobial peptide shapes a protective neonatal gut microbiota against pancreatic autoimmunity. Gastroenterology. 2022;162(4):1288–302.e16.
- Pahar B, Madonna S, Das A, et al. Immunomodulatory role of the antimicrobial LL-37 peptide in autoimmune diseases and viral infections. Vaccines (Basel). 2020;8(3):517.
- Bell KJ, Saad S, Tillett BJ, et al. Metabolite-based dietary supplementation in human type 1 diabetes is associated with microbiota and immune modulation. Microbiome. 2022;10(1):9.
- Mariño E, Batten M, Groom J, et al. Marginal-zone B-cells of nonobese diabetic mice expand with diabetes onset, invade the pancreatic lymph nodes, and present autoantigen to diabetogenic T-cells. Diabetes. 2008;57(2):395–404.
- de Groot PF, Nikolic T, Imangaliyev S, et al. Oral butyrate does not affect innate immunity and islet autoimmunity in individuals with longstanding type 1 diabetes: a randomised controlled trial. Diabetologia. 2020;63(3):597–610.
- de Groot P, Nikolic T, Pellegrini S, et al. Faecal microbiota transplantation halts progression of human new-onset type 1 diabetes in a randomised controlled trial. Gut. 2021;70(1):92–105.
- Burke SJ, Karlstad MD, Eder AE, et al. Pancreatic β-cell production of CXCR3 ligands precedes diabetes onset. Biofactors. 2016;42(6):703–715.
- Frigerio S, Junt T, Lu B, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8(12):1414–1420.
- Roep BO, Kleijwegt FS, van Halteren AGS, et al. Islet inflammation and CXCL10 in recent-onset type 1 diabetes. Clin Exp Immunol. 2010;159(3):338–343.
- Gibson GR, Hutkins R, Sanders ME, et al. Expert consensus document: the international scientific association for probiotics and prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat Rev Gastroent Hepat. 2017;14(8):491–502.
- Kellow NJ, Coughlan MT, Reid CM, et al. Metabolic benefits of dietary prebiotics in human subjects: a systematic review of randomised controlled trials. Br J Nutr. 2014;111(7):1147–1161.
- Dehghan P, Gargari BP, Jafar-Abadi MA, et al. Inulin controls inflammation and metabolic endotoxemia in women with type 2 diabetes mellitus: a randomized-controlled clinical trial. Int J Food Sci Nutr. 2014;65(1):117–123.
- Ho J, Nicolucci AC, Virtanen H, et al. Effect of prebiotic on microbiota, intestinal permeability, and glycemic control in children with type 1 diabetes. J Clinc Endoc Metab. 2019;104(10):4427–4440.
- Marcial GE, Ford AL, Haller MJ, et al. Lactobacillus johnsonii N6.2 modulates the host immune responses: a double-blind, randomized trial in healthy adults. Front Immunol. 2017;8:655.
- Gambardella J, Castellanos V, Santulli G. Standardizing translational microbiome studies and metagenomic analyses. Cardiovasc Res. 2021;117(3):640–642.
- Allaband C, Lingaraju A, Ramos SF, et al. Time of sample collection critical for microbiome replicability. BioRxiv. 2022.
- Galloway-Peña J, Hanson B. Tools for analysis of the microbiome. Dig Dis Sci. 2021;65(3):674–685.
- Korpela K, Helve O, Kolho K, et al. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell. 2020;183(2):324–34.e5.