Abstract
Background Periprosthetic osteolysis precipitates aseptic loosening of components, increases the risk of periprosthetic fracture and, through massive bone loss, complicates revision surgery and ultimately is the primary cause for failure of joint arthroplasty. The anti-inflammatory properties of HMG-CoA reductase inhibitors belonging to the statin family are well recognized. We investigated a possible role for status in initiating the first stage of the osteolytic cycle, namely monocytic activation.
Methods We used an in vitro model of the human monocyte/macrophage inflammatory response to poly-methylmethacrylate (PMMA) particles after pretreat-ing cells with cerivastatin, a potent member of the statin family. Cell activation based upon production of TNF-α and MCP-1 cytokines was analyzed and the intracellular Raf-MEK-ERK signal transduction pathway was evaluated using western blot analysis, to identify its role in cell activation and in any cerivastatin effects observed.
Results We found that pretreatment with cerivastatin significantly abrogates the production of inflammatory cytokines TNF-α and MCP-1 by human monocytes in response to polymethylmethacrylate particle activation. This inflammatory activation and attenuation appear to be mediated through the intracellular Raf-MEK-ERK pathway.
Interpretation We propose that by intervening at the upstream activation stage, subsequent osteoclast activation and osteolysis can be suppressed. We believe that the anti-inflammatory properties of statins may potentially play a prophylactic role in the setting of aseptic loosening, and in so doing increase implant longevity.
Despite advances in operative technique, in component materials and their fixation, wear and its sequelae continue to be the main factors limiting the longevity—and ultimately the clinical success—of total joint arthroplasty. It is generally accepted that aseptic osteolyis is a manifestation of an adverse granulomatous cellular response to particle wear and corrosion debris (Glant et al. Citation1993, Shanbhag et al. Citation1995, Schmalzried et al. Citation1997).
The precise mechanism by which macrophages (the primary cellular initiators of this response) become activated remains unclear. Although mac-rophage phagocytosis of polyethylene, polymethylmethacrylate and titanium particles has been demonstrated, there is also much literature to support the concept of membrane receptor binding and subsequent cell activation (Horowitz et al. Citation1993, Catelas et al. Citation1998). Once activated, a cascade of proinflammatory events ensues, involving other cellular participants including osteoblasts, fibroblasts and osteoclasts, fueled by cytokines such as TNF-α, PGE2, IL-1 and IL-6, chemokines such as MCP-1, and metalloproteinases (Frokjaer et al. Citation1995, Algan et al. Citation1996, Takagi et al. Citation1998, Nakashima et al. Citation1999).
Our improved understanding of osteolysis has prompted much interest in the possible pharmacological manipulation and attenuation of the inflammatory response and subsequent osteoclast activation.
Statins are a family of inhibitors of the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase. They have revolutionized the treatment of hypercholesterolemia and atherosclerotic disease, and are amongst the most widely prescribed agents in cardiovascular practice (Vaughan et al. Citation2000, Vaughan Citation2003). The beneficial effects are usually attributed to reduction of endogenous cholesterol synthesis through competitive inhibition of the principal enzyme HMG-CoA reductase. Since mevanonate, the product of the HMG-CoA reduction reaction, is the precursor not only of cholesterol, but also of many other non-steroidal isoprenoidic compounds, inhibition of this key enzyme has pleotropic effects (Stancu and Sima Citation2001). In particular, the anti-inflammatory effects of the statin group are well recognized (Bellosta et al. Citation2000). We endeavored to examine the hypothesis that statins attenuate the inflammatory reaction incited by polymethylmethacrylate (PMMA) particles.
We used an in vitro model of the human mono-cyte/macrophage inflammatory response to PMMA particles after pretreating cells with cerivastatin, a potent member of the statin family. Cell activation based upon TNF-α and MCP-1 cytokine production was determined and the intracellular Raf-MEK-ERK signal transduction pathway was evaluated to identify its role in cell activation and in any cerivastatin effects observed.
Material and methods
Reagents
Sterile monomeric polymethylmetacrylate particles (Howmedica) were suspended in RPMI-1640 culture medium with 10% fetal bovine serum (FCS), penicillin (100 U/mL) and streptomycin sulphate (100 μg/mL) at a final concentration of 1,000 μg/mL (later referred to as complete RPMI-1640 medium). Cerivastatin sodium (MW 485.1, a gift from Bayer) was dissolved in complete RPMI-1640 medium to the desired concentrations. The MEK inhibitor UO126 (1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene; Promega) was dissolved in DMSO and diluted further in complete RPMI-640 medium to a working concentration of 20 μM.
Monocyte isolation
Peripheral whole blood was collected from healthy donors (n = 10) into heparinized bottles. The heparinized blood was diluted in equal volumes of complete RPMI-1640 medium and maintained at 37°C. The mononuclear cell population was isolated by density gradient centrifugation by layering on Ficoll-Paque (Amersham Biosciences) and centrifugation at 400 x g for 40 min. The buffy mononuclear layer was removed and washed with PBS three times, and the monocyte population was estimated using flow cytometry staining with a phycoerythrin (PE)-conjugated anti-CD 14 (Becton-Dickinson). Cells were plated in compete medium at 8 x 105 per mL in sterile 24-well plates. The monocyte population was allowed to adhere for 90 min at 37°C with an atmosphere of 5% CO2. Cells (lymphocytes) that had not adhered were then washed off with PBS and the remaining monocyte population was used. Untreated monocytes (n = 5 per group), made up to equal volumes with complete medium, acted as controls for baseline monocyte activity. A PMMA-stimulated group involved monocyte culture in PMMA at 1000 μg/mL for 23 h. The treatment groups involved pretreating monocytes with cerivastatin at concentrations of 150 and 300 μM for 1 h, followed by PMMA stimulation for 23 h. Similarily, a group was pretreated with the MEK pathway inhibitor UO126 prior to stimulation with PMMA. The culture medium, precooled to 4°C, was then aspirated and centrifuged for 15 min at 10,000 x g. The supernatants were removed and stored at −80°C for cytokine analysis.
Cell viability was determined using the Trypan blue exclusion technique at 3 time points during the 24-h incubation with PMMA at 1,000 μg/mL and cerivastatin at 300 μM.
Inflammatory cytokine production.
Supernatants were thawed once. The levels of TNFα and MCP-1 were measured using commercially available enzyme-linked immunosorbant assay kits (Quantikine; R & D Systems, Abingdon, UK).
Western blot
Monocytes (1 x 107) were challenged with PMMA for 15 and 45 min, with and without pretreatment with cerivastatin for 1 h. The cells were similarily challenged with PMMA for 45 min after 1 h of pretreatment with UO-126 (MEK inhibitor). They were then washed 3 times with cold PBS and scraped into 200 μL of cell lysis buffer (1% Triton X-100, 20 mM Tris, pH X, 137 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 2 mM Na3VO4, 10 μg/mL leupeptin, and 2 μg/mL aprotinin). Protein concentrations were determined using a Micro BCA protein assay kit (Pierce, Rockford, IL). 100 μL of each sample was mixed with 100 μL of the loading buffer (Laemmli's buffer: 2 mL 60 mM Tris-HCl, pH 6.8, 5 mL 10% SDS, 1 mL 2-mercaptoethanol, 2 mL glycerol, and 0.01% bromophemol blue). The protein samples were then denatured at 100°C for 10 min. Aliquots containing equal amounts of total proteins from each sample were separated in SDS-polyacrylamide gels and electrophoretically transferred onto a nitrocellulose membrane (Schleicher & Schuell). After blocking for 2 h with PBS containing 0.1% Tween 20 and 6% non-fat milk, the membrane was incubated with primary antibody ERK 1/2 (p 42/44 MARK 1:1000, Cell Signaling Technology, Inc., Beverly, MA) in the blocking solution overnight at 4°. The membrane was then washed and incubated with the secondary antibody (goat antirabbit IgG, Santa Cruz Biotechnology) for 1 h at temperature and finally developed using an enhanced chemiluminescence detector system (Santa Cruz Biotechnology) according to the manufacturer's instructions.
Statistics
Statistical analysis was performed using analysis of variance. P-values of less than 0.05 were considered significant.
Results
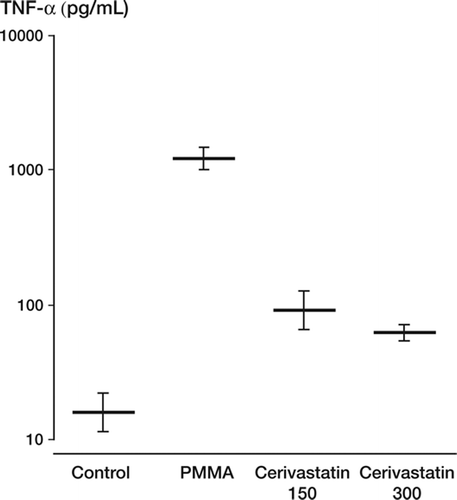
When monocytes were incubated with PMMA particles at 1,000 μg/mL, this resulted in upregulation of TNF-α expression relative to baseline (p = 0.002). Pretreatment with 150 μM cerivastatin markedly attenuated this response (p = 0.005) and a similar, but dose-dependent suppression was noted at the 300 μM statin concentration (p = 0.005) (). MCP-1 expression in response to PMMA activation also showed comparable results with a significant reduction on pretreatment with cerivastatin relative to PMMA-activated levels (p = 0.009) ().
Figure 1. TNF-α expression in pg/mL. The upregulation in expression (p = 0.002) on stimulation with PMMA particles is significantly attenuated on pretreatment with cerivastatin at 150 μM (p = 0.005).
The Raf-MEK-ERK transduction pathway was evaluated using the MEK inhibitor, UO-126. Inhalation of this pathway was found to suppress both PMMA-mediated TNF-α and MCP-1 (p = 0.003) expression, suggesting its integral role in PMMA activation and the possible site of statin activity ( and ).
Figure 2. TNF-α expression in pg/mL. Monocytes pre-treated with cerivastatin at 150 uM and the Raf-MEK-ERK inhihitor UO-126 showed significant (p = 0.003) reductions in TNF-α expression.
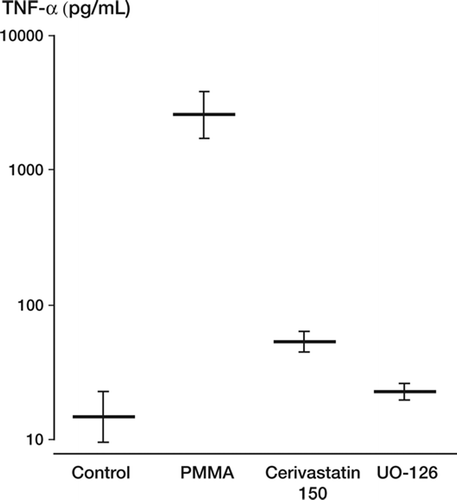
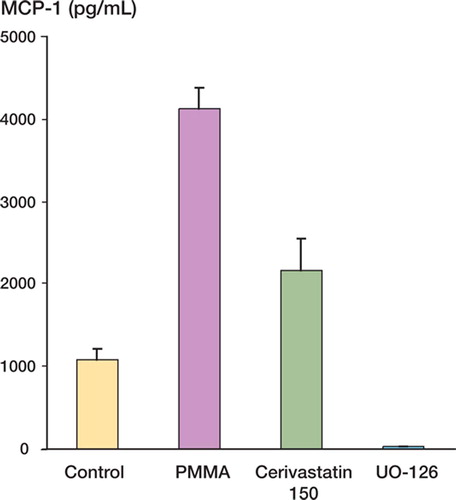
Figure 3. MCP-1 expression in pg/mL. Monocytes exposed to PMMA showed increased expression relative to the control. Pretreatment with cerivastatin at 150 μM and the Raf-MEK-ERK inhihitor UO-126 suppressed PMMA-activated MCP-1 production (p < 0.05).
Cell viability studies showed no significant difference in viability between PMMA- and cerivas-tatin-exposed cells compared to controls (p = 0.6).
Western blot analysis confirmed Raf-MEK-ERK activation in response to PMMA challenge with upregulation of the downstream ERK 1 and 2 kinases. Graduated increases in activity could be seen in response to particle challenge at 15 and 45 min (, lanes 2 and 4). Pretreatment with cerivastatin followed by similar particle challenge suppressed Raf-MEK-ERK pathway activity (lanes 3 and 5). Pretreatment with the selective Raf-MEK-ERK inhibitor UO-126 followed by 45 min of exposure to PMMA (, lane 6) mirrored the activity of the cerivastatin pretreatment group (, lane 5).
Figure 4. Western blot analysis illustrating the activity of the Raf-MEK-ERK intracellular pathway (ERK1 and 2) in response to PMMA particle challenge for 15 and 45 min (lanes 2 and 4). Similar challenges were repeated after 1 h of pretreatment with 150 μM cerivastatin (lanes 3 and 5). Monocytes were pretreated with UO-126 followed by PMMA challenge for 45 min (lane 6).

Discussion
Efforts to manipulate the inflammatory-osteolysis cascade have focused upon key steps in this process. Downstream interventions have attempted to modulate osteoclast differentiation and activation. Bisphosphonates, now widely prescribed in the treatment of osteoporosis, Paget's disease and hypercalcemia have stimulated much interest in them as a possible treatment or prophylaxis for periprosthetic osteolysis. Alendronate, a third-generation bisphosphonate, has been shown in animal models to be effective in attenuating particle-mediated osteolysis (Millett et al. Citation2002, Mulhall et al. Citation2002). However, results in long-term human trials are still awaited.
Increases in tissue levels of the osteoclast differentiation factors receptor activator of nuclear factor-κB (RANK) and its ligand (RANKL) have been found at sites of periprosthetic osteolysis (Mandelin et al. Citation2003). Osteoprotegerin (OPG), a soluble decoy receptor for RANKL, has been shown in vitro to inhibit osteoclastogenesis induced by joint fluid from failed total hip replacements (Kim et al. Citation2001). Recombinant adenovirus-medi-ated osteoprotegerin gene therapy has been shown to suppress titanium-induced osteolysis in mouse calvarial models (Ulrich-Vinther et al. Citation2002).
Horowitz identified TNF-α as one of the primary mediators released by particle-activated macrophages (Horowitz et al. Citation1994, Horowitz and Purdon Citation1995). It has been shown to control the release of other proinflammatory mediators, as well as accelerating osteoclast activation through RANKL-dependent and independent pathways (Shanbhag et al. Citation1997, Kobayashi et al. Citation2000). This has prompted the use of TNF-α antagonists such as etanercept and adenovirus-mediated gene delivery of TNF-α inhibitors, with promising anti-osteo-lytic results in mouse calvarial models (Childs et al. Citation2001a, Citation2001b, Schwarz et al. Citation2000).
In Citation1977, Willert and Semlitsch proposed that macrophages instigate the inflammatory response to wear debris, which initiates the cascade of events ultimately precipitating osteoclast activation and bone resorption. Macrophage activation, whether by phagocytosis or by particle-bound-ligand signaling, ultimately leads to the activation of cellular signal transduction pathways, culminating in NF- κB activation and binding to the promoter regions of TNF-α and other proinflammatory genes (Roebuck et al. Citation1999). We therefore targeted this, the most upstream mediator of the inflammatory cascade, with the assumption that by suppressing macrophage activation, subsequent amplifications would be thwarted.
Statins have been shown to reduce neointimal inflammation by reducing the recruitment of mac-rophages, the expression of MCP-1, and the activation of NF-κB in hypercholesterolemic animal models (Ortego et al Citation1999). Statins attenuate the expression of intracellular adhesion molecule-1 and IL-6 production by monocytes in response to endotoxin lipopolysaccharide (LPS) challenge (Bellosta et al. Citation2000) Similarily, inhibition of tran-sendothelial neutrophil migration and chemotaxis has been reported in rat models treated with flu-vastatin (Stancu and Sima Citation2001), while simvas-tatin has been shown to inhibit proinflammatory cytokine expression in monocytes of patients with hypercholesterolemia (Ferro et al. Citation2000).
Cerivastatin sodium is a hydrophobic, synthetic enantomer, and one of the most potent members of the HMG-CoA reductase inhibitors. Its anti-inflammatory effects are well recognized. It has been shown to suppress NF-κB signaling in response to LPS in a dose-dependent manner, and with greatest potency relative to other statins (Ando et al. Citation2000). It has also been shown to improve survival in vivo in murine sepsis models (Hilgendoroff et al. Citation2003).
We have examined the ability of cerivastatin to abrogate PMMA-mediated monocyte activation. The peripheral blood monocyte/macrophage model is a well-recognized in vitro model for particle stimulation (Shanbhag at al. Citation1995, Blain et al. Citation1997, Mulhall et al. 2002). This was felt to be the most appropriate model, as monocytes represent the circulatory precursors of tissue macro-phages, the established primary instigators of foreign-body inflammatory response and subsequent osteolysis.
The particulate cement material used to activate human monocytes was monomeric PMMA. Support for using the monomeric form comes from work comparing the release of TNF-α from different cement preparations. A consistent, equivalent dose-dependent response was noted in cells treated with prepolymerized polymethylmethacrylate particles of 1–12 μm size (phagocytosable) and the unpolymerized powder (Horowitz et al. Citation1993).
We measured production of the potent proinflammatory cytokine TNF-α and the chemokine monocyte chemotactic protein-1 (MCP- 1). MCP-1 functions mainly in mobilizing circulating monocytes, the precursors of macrophages and osteo-clasts, to the site of inflammation. Its upregulation has been demonstrated in response to polymethylmethacrylate stimulation (Nakashima et al.Citation1999). The Raf/MEK/ERK kinase transduction pathway is recognized as one of the prominent intracellular pathways responsible for activation of the transcription factor NF-κB and subsequent monocyte /macrophage activation (Roebuck et al. Citation1999, Abbas et al. Citation2003).
Pretreatment with cerivastatin significantly attenuated PMMA-mediated cell activation. We demonstrated that this attenuation is mirrored by the selective mitogen-activated protein kinase kinase (MAPKK or MEK) pathway inhibitor UO-126 (Favata et al. Citation1998) Western blot confirmed Raf/MEK/ERK activation by PMMA particles, and downregulation on pretreatment with statins or UO-126.
Periprosthetic osteolysis precipitates aseptic component loosening, increases the risk of periprosthetic fracture, and complicates surgical revision through massive bone loss. We have demonstrated in vitro that statins can abrogate particle-induced inflammatory responses in a dose-dependent manner, and that this is mediated intracellularily through its effect on the Raf/MEK/ERK transduction pathway. We propose that by attenuating this inflammatory response, the associated subsequent osteoclast activation and osteolysis is attenuated.
While polymethylmethacrylate particle debris is known to incite an inflammatory reaction, the most common particle in periprosthetic tissues is polyethylene—generated mainly from mode-1 bearing surface wear. Stainless steel, titanium, cobalt alloy, and ceramics all generate stimulatory particles. Thus, we have only focused on a single protagonist.
In light of the widespread prescription of statins in the control of hypercholesterolemia and prevention of coronary artery disease, we feel that in vivo osteolysis modeling experiments are necessary to evaluate their role in inhibition of osteolysis and promotion of implant longevity in greater depth.
No competing interests declared.
Contributions of authors
JHW and HPR supervised laboratory work. All other authors developed the hypothesis and wrote the paper.
- Abbas S, Clohisy JC, Abu-Amer Y. Mitogen-activated protein (MAP) kinases mediate PMMA-induction of osteo-clasts. J Orthop Res 2003; 21(6)1041–8
- Algan S M, Purdon M, Horowitz S M. Role of tumor necrosis factor alpha in particulate - induced bone resorption. J Orthop Res 1996; 14: 30–5
- Ando H, Takamura T, Ota T, Nagai Y, Kobayashi K. Ceriv-astatin improves survival of mice with lipopolysaccha-ride-induced sepsis. J Pharmacol Exp Ther 2000; 294(3)1043–6
- Bellosta S, Ferri N, Bernini F, Paoletti R, Corsini A. Non-lipid-related effects of statins. Ann Med 2000; 32(3)164–76
- Blain T A, Pollice P F, Rosier R N, Reynolds P R, Puzas J E, O'Keefe R J. Modulation of the production of cytokines in titanium- stimulated human peripheral blood monocytes by pharmacological agents. The role of cAMP-mediated signaling mechanisms. J Bone Joint Surg (Am) 1997; 79: 1519–28
- Catelas I, Huk O L, Petit A, Zukor D J, Marchand R, Yahia L. Flow cytometric analysis of macrophage response to polymethylmetacrylate particle debris: effect of size, morphology and surface area. J Biomed. Mater Res 1998; 41: 600–7
- Childs L M, Goater J J, O'Keefe R J, Schwartz E M. Effect of anti-tumor necrosis factor-a gene therapy on wear debris-induced osteolysis. J. Bone Joint Surg (Am) 2001a; 83: 1790–7
- Childs L M, Goater J J, O'Keefe R J, Schwartz E M. Efficacy of etanercept for wear debris-induced osteolysis. J. Bone Miner Res 2001b; 16: 338–47
- Favata M F, Horiuchi K Y, Manos E J, Daulerio A J, Strad-ley D A, Feeser W S, Van Dyk D E, Pitts W J, Earl R A, Hobbs F, Copeland R A, Magolda R L, Scherle P A, Trzaskos J M. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 1998; 273(29)18623–32
- Ferro D, Parrotto S, Basili S, Alessandri C, Violi F. Simv-astatin inhibits the monocyte expression of proinflamma-tory cytokines in patients with hypercholesterolemia. J Am Coll Cardiol 2000; 36(2)427–31
- Frokjaer J, Deleuran B, Lind M, Overgaard S, Soballe K, Bunger C. Polyethylene particles stimulate monocyte chemotactic and activating factor production in synovial mononuclear cells in vivo. An immunohistochemical study in rabbits. Acta Orthop Scand 1995; 66: 303–7
- Glant T T, Jacobs J J, Molnar G, Shanbhag A S, Valyon M, Galante J O. Bone resorption activity of particulate-stim-ulated macrophages. J Bone Min Res 1993; 8: 1071–9
- Hilgendorff A, Muth H, Parviz B, Staubitz A, Haberbosch W, Tillmanns H, Holschermann H. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int J Clin Pharmacol Ther 2003; 41(9)397–401
- Horowitz S M, Purdon M A. Mechanisms of cellular recruitment in aseptic loosening of prosthetic joint implants. Calcif Tissue Int 1995; 57: 301–5
- Horowitz S M, Doty S B, Lane J M, Burstein A H. Studies of the mechanism by which the mechanical failure of pol-methylmethacrylate leads to bone resorption. J Bone Joint Surg(Am) 1993; 75: 802–13
- Horowitz S M, Doty S B, Rapuano B P, Lane J M, Burstein A H. Role of the macrophage and the osteoblast in aseptic loosening. Calcific Tissue Int 1994; 54: 320–4
- Kim K J, Kotake S, Udagawa N, Ida H, Ishii M, Takei L, Kubo T, Takagi M. Osteoprotegerin inhibits invitro mouse osteoclast formation induced by joint fluid from failed total hip arthroplasty. J Biomed Mater Res 2001; 58(4)393–400
- Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Morinaga T, Higashio K, Martin T J, Suda T. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med 2000; 191: 275–86
- Mandelin J, Li T F, Lihjestrom M, Kroom M E, Hanemaaijer R, Santavirta S, Konttinen Y T. Imbalance of RANKL/ RANK/OPG system in interface tissue in loosening of total hip replacement. J Bone Joint Surg (Br) 2003; 85(8)1196–201
- Millett P J, Allen M J, Bostrom M P. Effects of alendronate on particle induced osteolysis in a rat model. J Bone Joint Surg (Am) 2002; 84(2)236–49
- Mulhall K J, Curtin W A, Given F H. Inhibition of polymeth-ylmethacrylate particle induced monocyte activation and IL-1β and TNFα expression by the antioxidant agent N-acetlycysteine. Acta Orthop Scand 2002; 73(2)206–12
- Nakashima Y, Sun D-H, Trindade M C D, Chun L E, Song Y, Goodman S B, Schurman D J, Maloney W J, Smith R L. Induction of macrophage C-C chemokine expression by titanium alloy and bone cement particles. J Bone Joint Surg (Br) 1999; 81: 155–62
- Ortego M, Bustos C, Hernandez-Presa M A, Tunon J, Diaz C, Hernandez G, Egido J. Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis 1999; 147(2)253–61
- Roebuck K A, Jacobs J J, Glant T T. New Horizons in Orthopaedic Research: cellular signal transduction pathways. J Bone Joint Surg (Am) 1999; 81: 599–602
- Schmalzried T P, Akizuki K H, Fedenko A N, Mirra J. The role of access of joint fluid to bone in periarticular oste-olysis. A report of four cases. J Bone Joint Surg (Am) 1997; 79: 447–52
- Schwarz E M, Looney R J, O'Keefe R J. Anti-TNF-alpha therapy as a clinical intervention for periprosthetic oste-olysis. Arthritis Res 2000; 2(3)165–8
- Shanbhag A S, Jacobs J J, Black J, Galante J O, Glant T T. Human monocyte response to particulate biomaterials generated in vivo and in vitro. J Orthop Res 1995; 13: 792–801
- Shanbhag A S, Hasselman C T, Rubash H E. The John Charnley Award. Inhibition of wear debris mediated osteolysis in a canine total hip arthroplasty model. Clin Orthop 1997, 344: 33–43
- Stancu C, Sima A. Statins: Mechanism of action and effects. J Cell Mol Med 2001; 5(4)378–87
- Takagi M, Santavirta S, Ida H, Ishii M, Mandelin J, Kont-tinen Y T. Matrix metalloproteinases and tissue inhibitors of metalloproteinases in loose artificial hip joints. Clin Orthop 1998, 352: 35–45
- Ulrich-Vinther M, Carmody E E, Goater J J, Søballe K, O'Keefe R J, Schwarz E M. Recombinant adeno- associated virus-mediated osteoprotegerin gene therapy inhibits wear debris-induced osteolysis. J Bone Joint Surg (Am) 2002; 84(8)1405–12
- Vaughan C J. Prevention of stroke and dementia with statins: Effects beyond lipid lowering. Am J Cardiol 2003; 91(4A)23B–29B
- Vaughan C J, Gotto A M, Jr, Basson C T. The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol 2000; 35(1)1–10
- Willert H-G, Semlitsch M. Reactions of the articular capsule to wear products of artificial joint prostheses. J Biomed Mater Res 1977; 11: 157–64