
ABSTRACT
Introduction: Multiple myeloma (MM) remains an incurable disease with a median overall survival of approximately 5 years. Gain or amplification of 1q21 (1q21+) occurs in around 40% of patients with MM and generally portends a poor prognosis. Patients with MM who harbor 1q21+ are at increased risk of drug resistance, disease progression, and death. New pharmacotherapies with novel modes of action are required to overcome the negative prognostic impact of 1q21+. Areas covered: This review discusses the detection, biology, prognosis, and therapeutic targeting of 1q21+ in newly diagnosed and relapsed MM. Patients with MM and 1q21+ tend to present with higher tumor burden, greater end-organ damage, and more co-occurring high-risk cytogenetic abnormalities than patients without 1q21+. The chromosomal rearrangements associated with 1q21+ result in dysregulation of genes involved in oncogenesis. Identification and characterization of the 1q21+ molecular targets are needed to inform on prognosis and treatment strategy. Clinical trial data are emerging that addition of isatuximab to combination therapies may improve outcomes in patients with 1q21+ MM. Expert opinion: In the next 5 years, the results of ongoing research and trials are likely to focus on the therapeutic impact and treatment decisions associated with 1q21+ in MM.
1. Introduction
The occurrence and accumulation of different combinations of chromosomal and molecular abnormalities increase the risk of a poor outcome in multiple myeloma (MM) [Citation1,Citation2]. The International Myeloma Working Group (IMWG) has defined the presence of t(4;14), del(17p), t(14;16), t(14;20), hypodiploidy, and gain or amplification of 1q21 (1q21+) as conferring a high risk for a worsened outcome [Citation3].
Copy number gain of 1q21 is among the most common chromosomal aberrations in MM, observed in approximately 28−44% of patients at diagnosis [Citation4–9]. It manifests in a tumor subclone as a secondary genomic event, becoming more frequent, and amplified as the tumor progresses [Citation10]. In MM cells harboring 1q21+, oncogenesis and drug resistance are driven, at least in part, by upregulation of genes located in the 1q21 amplicon. Several genes on chromosome 1q are strongly associated with aggressive MM cell phenotypes, including CKS1B, PSMD4, ANP32E, and MCL-1. 1q21+ is divided into either 1q21 gain (3 copies) or 1q21 amplification (≥4 copies), with worse prognosis for patients with amplification than those with gain, though this finding has not been universal [Citation8,Citation10,Citation11]. Co-existence of other chromosomal abnormalities such as t(4;14), t(14;16), del(1p), and del(17p) are common and further worsen the prognosis for patients with 1q21+ [Citation10].
The availability of new treatments over the last 20 years have improved outcomes substantially in MM but 1q21+ remains an area of unmet need. In various therapy settings, presence of 1q21+ predisposes to reduced progression-free survival (PFS) and overall survival (OS) [Citation5,Citation12]. The adverse impact of 1q21+ in MM has continued into the era of treatment with immunomodulatory drugs (IMiDs) and proteasome inhibitors (PIs) as evidenced by meta-analysis of the National Cancer Research Institute Myeloma XI and Medical Research Council Myeloma IX trials showing that presence of 1q21+ was associated with a 68% greater risk for death [Citation13].
This review summarizes the prognostic significance of 1q21+, highlighting the inability of various antimyeloma drug combinations in overcoming the adverse prognostic impact of 1q21+. It also provides clues for some new therapeutic combinations, which can ameliorate the adverse prognostic impact of 1q21+. Understanding the biology of 1q21+ will advance our knowledge to better risk-stratify MM patients, target 1q21+ candidate gene(s) and facilitate more appropriate tailored treatment strategies.
1.1. Detection and mechanism of 1q gain/amplification in MM
The 1q21 copy number is detected in CD138+ plasma cells by interphase fluorescent in situ hybridization (iFISH) using a specific probe covering the 1q21.3 chromosomal region, and more specifically, 350 kb around the CKS1B gene [Citation7,Citation8]. Hanamura and colleagues report that cutoffs used for determining a detrimental effect of 1q21+ by iFISH vary between studies from 5% to 30% of plasma cells [Citation10]. However, most studies report that 20% of plasma cells is the best cutoff for reporting poor outcomes [Citation7,Citation8,Citation10,Citation12,Citation14]. Receiver-operating characteristics analysis of 333 patients with newly diagnosed MM (NDMM) and 92 patients with relapsed refractory MM (RRMM) with 1q21+ who received either thalidomide- or bortezomib-based treatments in a randomized clinical trial suggested that a cutoff of 20% was the optimal value for predicting poor outcomes [Citation14]. More expedient assays for the detection of 1q21+ are in development to better capture the genomic diversity of MM. These include next-generation sequencing, comparative genomic hybridization, and multiplex ligation-dependent probe amplification [Citation15–17].
Gain 1q21+ is detected in approximately 17−23% of patients with monoclonal gammopathy of undetermined significance (MGUS), 28−49% of patients with smoldering MM and/or NDMM, and up to 42–80% of patients with RRMM [Citation4–8,Citation14,Citation18–21]. 1q21+ is associated with adverse prognosis in both NDMM and RRMM [Citation5,Citation7,Citation22–24], with amplification (≥4 copies) of 1q associated with the worst outcome, suggesting a dosage effect of genes located on chromosome 1q [Citation24–26]. Over-expression of the CKS1B gene was initially defined as a driver event [Citation7,Citation22,Citation27,Citation28] but later many other genes located on 1q (MUC1, MCL1, ANP32E, BCL9, PSMD4, PDZK1, NEK2, ARNT, ILF2, and ADAR1) have been linked to disease biology, myeloma cell growth, poor prognosis, poor survival, and drug resistance [Citation29–38].
It is likely that the combined effects of multiple genes contribute to the poor prognosis of patients with 1q21+ [Citation30,Citation39]. Dysregulated transcriptional program associated with 1q21+ have also been linked to the MM biology [Citation40]. Gene expression profiling (GEP) of high-risk patients shows that genes mapping at 1q21, 1q22, and 1q43–q44 are overexpressed in high-risk disease: University of Arkansas for Medical Science (UAMS70) gene signature shows that genes residing on 1q21 contribute critically to the high-risk designation in the GEP70 gene signature [Citation41,Citation42].
The number of cells with 1q gain and total copy number of the 1q chromosome increase with disease progression [Citation8]. The incidence of 1q21 amplification in MM cell lines is 91% [Citation8]. The progressive increase in 1q copy number and cells with 1q gain from diagnosis to relapse suggest a clonal evolution and therapeutic selection of MM cells with 1q21+ [Citation8] (). Based on GEP, MM has been classified into seven subgroups [Citation42]. Of these, the proliferation subgroup (PR) dominated at relapse and was characterized by increases in 1q copy number and cells with 1q gain, indicating a role for genes located on 1q in driving cell proliferation [Citation42].
Figure 1. Accumulation of 1q21+ clones.
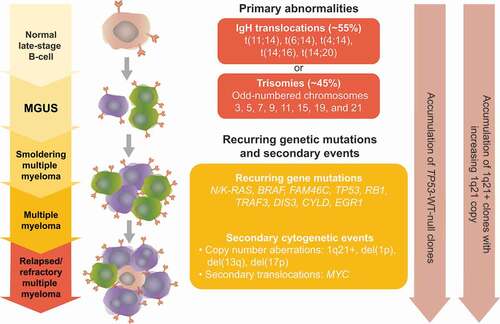
The exact mechanism underlying the accumulation of copy number alterations in 1q21+ MM and their role in driving aggressive disease are starting to be elucidated [Citation9]. In the late 1990s, Sawyer and colleagues elegantly showed that 1q21+ is brought about by duplications of all or part of chromosome 1q, whole-arm jumping translocations of 1q (JT1q), and trisomy of chromosome 1 [Citation43]. Recurring JT1q were detected on chromosomes 5, 8, 12, 14, 16, 17, 19, 21, and 22 [Citation43]. The primary mechanism of 1q21 amplification is JT1q12 [Citation44,Citation45]. The event initiating instability of chromosome 1 is postulated to be de-condensation of the peri-centromeric heterochromatin, which facilitates recombination and formation of unstable 1q translocations [Citation43,Citation46]. In particular, pericentromeric weakness of the 1q12 region, which lies adjacent to 1q21, results in JT1q and segmental duplications of 1q21 [Citation31,Citation47]. Segmental duplications can occur when 1q jumps to a nonhomologous chromosome followed by, either, 1q12–1q21 segment duplication or duplication of the proximal adjacent nonhomologous chromosome segment prior to 1q jumping or inserting itself into a new location [Citation31].
Locus specific FISH and spectral karyotyping of patients with ≥4 copies of 1q21 revealed four types of chromosomal anomalies that distinguish this phenotype, namely, JT1q12 gains, deletion of the receptor chromosome including 17p, insertions into the receptor chromosome, and breakage-fusion-bridge cycle amplifications [Citation2]. Such aberrant chromosomes undergo multiple rounds of breakage and fusion cycles and mitotic segregation errors, which finally result into either whole-chromosome arm or segmental duplications [Citation2,Citation31,Citation44]. The epigenetic factors responsible for driving the pericentromeric heterochromatin de-condensation in MM are still unknown. In solid tumors, aberrant expression of histone lysine demethylase 4A (KDM4A) and hypoxic stress induce aberrant amplification of 1q21 [Citation48,Citation49].
Whether 1q21+ is a cause or a consequence of disease in MM still remains an unanswered question [Citation9]. For instance, JT1q12 also provides a mechanism for the deletion of 17p [Citation44], recognized as another cytogenetic abnormality that predisposes to a poor prognosis [Citation3]. Certainly, 1q21+ reflects a genome instability phenotype, which results in transcription remodeling and drives an aggressive disease, however, the driver event which leads to 1q21+ in MM remains elusive.
2. Co-occurrence of 1q21+ with other cytogenetic abnormalities
The presence of other cytogenetic abnormalities along with adverse clinical disease markers have called into question the power of 1q21+ as an independent prognostic variable [Citation7,Citation9,Citation50–52]. 1q21+ may require associations with other high-risk factors to maintain an adverse effect on survival and the impact of 1q21+ requires assessment in conjunction with accompanying high-risk factors and the different treatment regimens available [Citation51,Citation52]. 1q21+ often coexists with other cytogenetic abnormalities such as t(4;14), t(14;16), del(1p), and del(17p) that are known to confer an additional adverse impact on survival () [Citation7,Citation52–54]. In addition, patients with NDMM with 1q21+ tend to be older and present with greater end-organ damage and higher tumor burden, anemia, thrombocytopenia, hypercalcemia, elevated lactate dehydrogenase, higher proportion of bone marrow plasma cells, higher International Staging System (ISS) stage, and more high-risk cytogenetic abnormalities than patients with normal 1q21 [Citation4,Citation12,Citation50,Citation55]. In several studies, three or four or more copies of 1q21 was associated with worse PFS and OS, and a shorter time to next treatment, relative to fewer 1q21 copies [Citation11,Citation55,Citation56].
Figure 2. Co-occurrence of high-risk cytogenetic abnormalities and genes associated with 1q21 abnormalities.
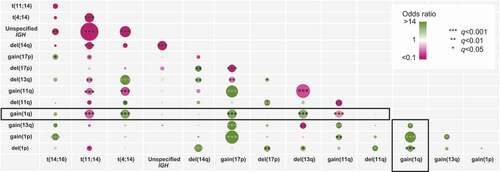
The number and type of high-risk cytogenetic abnormalities co-occurring with 1q21+ has a profound influence on survival. Generally, patients with one or two high-risk abnormalities [featuring t(4;14), t(14;16), t(14;20) del(17p), 1q21+, del(1p), or TP53 mutations] have lower OS than patients with a standard risk profile [Citation57]. A retrospective analysis of 201 NDMM patients treated with bortezomib, lenalidomide, and dexamethasone induction indicated that 1q21+, detected in 47% of cases, was significantly associated with t(4;14) and t(14;16) [Citation55]. Patients with 1q21+ co-occurring with t(4;14), t(14;16), or del(17p) had worse PFS than patients with no other high-risk cytogenetic abnormalities [Citation55]. Thus, co-occurrence of 1q21+ with t(4;14), t(14;16), or del(17p) should be considered in the ‘double hit’ category, and affected patients are at high risk for early disease progression [Citation55]. Similarly, among 104 NDMM patients treated with various induction regimens, presence of del(13q14) or del(17p13) in addition to 1q21+ foreshortened PFS and OS relative to patients with isolated 1q21+ [Citation53]. Baseline analysis of the phase 3 HOVON24 trial, in which patients with MM received high dose chemotherapy and stem cell transplantation, showed that abnormal chromosome 1p/q and a deletion of chromosome 13/13q were highly associated with poor outcome [Citation58]. Patients with 1q21+ or deletion of 1p had significantly shorter OS (16 and 22 months, respectively) compared with patients with no chromosome 1 abnormalities (62 months; P< 0.001), and OS in those with both 1p/q and 13/13q was even worse [Citation58]. Finally, a retrospective study of 794 patients with MM was designed in part to investigate 1q21+ in separate subgroups defined by a primary cytogenetic abnormality. Overall, >95% were treated with an IMiD or PI, and 44% underwent autologous stem cell transplantation (ASCT) [Citation11]. In a subgroup of these patients, clonal gain(11q) and 1q21+ was associated with an OS of less than 4 years whereas, in the absence of 1q21+, clonal gain(11q) was associated with an OS of over 9 years [Citation11].
Additional adverse impacts of other cytogenetic abnormalities on outcome of 1q21+ patients can be partially explained by primary cytogenetic clonal evolution. Hyperdiploidy (acquiring an extra copy of odd number chromosomes) and IgH translocation are primary genetic events in MM, occurring in ~45% and ~55% MM patients, respectively [Citation59,Citation60]. Co-occurrence of 1q21+ with t(4;14) (FGFR3-MMSET), t(14;16) (MAF) suggests that genes located on chromosome 1q provide additional growth advantage to clones, which have already acquired t(4;14), t(14;16) IgH translocations. Although hyperdiploid MM is considered a favorable prognosis factor, hyperdiploid patients with 1q21+ show poor OS when associated with MYC translocation [Citation61], suggesting that MYC dysregulated clones can acquire growth advantage from chromosome 1q genes [Citation61]. Poor outcome of 1q amplification in comparison to 1q gain suggests that genes located on 1q21 may have a dose effect on MM growth. These genes are likely to provide an additional growth advantage to clones which have acquired multiple genetic abnormalities, especially t(4;14), t(14;16), del(1p), and del(17p).
3. 1q amplification/gain in other cancers
Gain or amplification of 1q21 is not restricted to MM. Amplification of 1q21 is an indicator of prior cancer in patients with MM, including prostate cancer, melanoma, lymphoma, and cervical cancer [Citation62]. In patients with B-cell lymphoma, changes in 1q are detected as secondary cytogenetic abnormalities in around one-quarter of cases and associated with an adverse prognosis [Citation63–65]. Gains in chromosomal regions 1q21–q22 and 13q12–q14 are associated with cisplatin-based chemotherapy drug-resistant phenotypes in patients with ovarian cancer [Citation66]. 1q gain is one of the most frequently detected alterations in hepatocellular carcinoma, and represents one of the most frequent minimal amplified region [Citation67]. 1q gain has also been detected in breast cancer; seven candidate genes (ASPM, CKS1B, CENPF, NEK2, DTL, KIF14, and EXO1) located on chromosome 1q were strongly associated with poor survival in breast cancer patients [Citation68]. Gain of chromosome 1q21 has been frequently noted in pediatric malignancies, including pediatric acute lymphoblastic leukemia, Wilms tumor, neuroblastoma, Ewing sarcoma, and brain tumors such as ependymoma and high-grade gliomas; the presence of this anomaly is usually associated with disease recurrence and poor prognosis [Citation65,Citation68–72]. Finally, 1q21+ and tenascin-C are candidate markers in pediatric posterior fossa ependymoma [Citation73].
4. 1q21+ as a prognostic factor for treatment outcomes in myeloma
4.1. Vincristine, doxorubicin, and dexamethasone (VAD) followed by high-dose chemotherapy/ASCT in NDMM
summarizes survival data for patients with NDMM who were treated with VAD, followed by high-dose chemotherapy with melphalan and ASCT. In one observational study and two subgroup analyses of randomized, controlled trials, 1q21+ prevalence ranged from 33 to 41% [Citation5,Citation12]. Overall, patients with 1q21+ MM had shorter durations of PFS and OS than patients without this cytogenic abnormality. In one study, the poor prognosis of patients with 1q21+ (specifically CKS1B gene amplification) was linked to the deletion of p53, which is associated with disease progression in MM [Citation74]. In another study, 35% of patients with 1q21+ were not randomized to post-ASCT consolidation/maintenance treatment due to disease progression or death vs 9% of patients with normal 1q21 copy number [Citation12]. The fact that 1q21+ was an independent prognostic factor for OS but not for PFS in one study may imply an important role for 1q21+ after relapse than at diagnosis.
Table 1. Impact of 1q21+ on outcomes in NDMM patients receiving VAD followed by high-dose chemotherapy and ASCT
4.2. IMiDs and PIs
4.2.1. IMiD/PI induction followed by high-dose treatment/ASCT in NDMM
presents data from several retrospective observational studies and subgroup analyses of clinical trials showing that patients with 1q21+ have shorter durations of PFS and OS than those without 1q21+. Landmark analyses consistently show that the proportion of patients reaching defined survival endpoints at multiple timepoints is lower in patients with 1q21+. In multivariate analysis, 1q21+ was independently associated with a 65−200% increased risk for progression or death (Hazard ratio [HR] range, 1.65−3.0) and an 88−290% increased risk for shorter OS (HR range, 1.88−3.9) [Citation25,Citation26,Citation54,Citation76,Citation77]. Increased 1q21+ copy number was associated with adverse outcomes that PI therapy with either bortezomib or carfilzomib failed to abate [Citation25,Citation26].
Table 2. Impact of 1q21+ outcomes in NDMM patients receiving IMiDs and PIs followed by HDT/ASCT
In the largest retrospective study, conducted at Mayo Clinic between 2005 and 2018, 1q21+ was detected in 27% of NDMM patients undergoing ASCT [Citation4]. Among patients who received IMiD-, PI-, or IMiD plus PI-based induction therapy, OS was significantly shorter in patients with than without 1q21+ (7.5 years vs 11.1 years; P< 0.001) [Citation4]. Rates of overall response or very good partial response to these induction therapies were similar regardless of 1q21+ status. Median time to next treatment was significantly shorter among patients with 1q21+ compared with those without 1q21+ who underwent ASCT after first-line induction chemotherapy (29.8 vs 37.1 months; P= 0.01).
4.2.2. IMiD/PI treatment in non-transplant NDMM
Results of a population-based study of 347 consecutive NDMM patients demonstrated that 1q21+ is one of the most important poor prognostic chromosomal aberrations. Three-year OS for patients with gain 1q21 was 60% vs 74% for patients with other chromosomal aberrations and 82% for patients with no chromosomal aberrations [Citation78]. The 3-year OS associated with treatment with IMiDs or PIs was 58% for patients with 1q21+, 78% for patients with other chromosomal aberrations, and 78% with no chromosomal aberrations. The same trend was observed in the high dose treatment cohorts, with a 3-year OS of 69% for patients with 1q21+ compared with 84% and 88% for patients with other chromosomal aberrations and no chromosomal aberrations, respectively [Citation78].
These data were supported by the aforementioned retrospective study conducted at the Mayo Clinic, in which 1q21+ was detected in 236 of 795 (30%) patients who did not undergo first-line ASCT [Citation4]. In this subgroup, OS was significantly shorter among patients with 1q21+ than those without 1q21+ (3.7 years vs 6.5 years; P< 0.001) [Citation4]. There was also a significant reduction in the median time to next treatment among patients with than without 1q21+ who received first-line induction chemotherapy only (8.5 vs 13.9 months; P< 0.001) [Citation4].
4.2.3. IMiD/PI induction with or without ASCT in NDMM
Unsurprisingly, similar findings have been observed in studies that did not stratify patients according to receipt of ASCT. In both, the UK National Cancer Research Institute [Citation79] Myeloma XI and Medical Research Council (MRC) Myeloma IX trials intensive or non-intensive therapy was given dependent on the patient’s eligibility for ASCT [Citation79,Citation80]. Meta-analysis of the NCRI Myeloma XI and MRC Myeloma IX trials that recruited 1905 NDMM patients from 2003–2016 demonstrated that the presence of 1q21+ was associated with poorer OS (HR 1.68; P= 2.18 × 10−14) [Citation13]. In the UK MRC Myeloma XI trial specifically, 178 patients (19% with 1q21+) received induction therapy with either thalidomide, lenalidomide, or carfilzomib and lenalidomide in combination with dexamethasone and cyclophosphamide [Citation80]. Subgroup analysis revealed that the median OS was 44.3 months for those with 1q21+ at diagnosis and 47.9 months for those with 1q21+ at relapse vs 67.1 months for those with standard risk cytogenetics (P= 0.007) [Citation80]. Carfilzomib-based induction/consolidation therapy with/without ASCT followed by lenalidomide or carfilzomib-lenalidomine (KR) maintenance compared with carfilzomib-cyclophosphamide-dexamethasone (KCd) showed that KRd showed superiority over KCd in all groups of patients except those with 1q21+ who had worse outcomes regardless of treatment [Citation81]. A recent meta-analysis in 2596 NDMM patients receiving PI or IMiD therapy from three phase 3 trials revealed that gain or amplification of 1q21 was negatively prognostic for PFS and OS, independently of R-ISS [Citation82]. There was no significant survival difference observed between gain or amplification of 1q21 [Citation82].
In a real-world administration database analysis that used NDMM data over a 7-year period (2011 − 2018), 844 of 3578 (24%) had 1q21+ [Citation83]. This subgroup was more likely to receive therapy with IMiD and PI combinations than those without (41% vs 34%; P< 0.001) [Citation83]. After adjusting for other high-risk cytogenetic abnormalities, median OS was significantly lower for patients with than without 1q21+ (46.7 months vs 70.1 months). In multivariable analysis that also adjusted for age, sex, race/ethnicity, type of first-line therapy, and use of ASCT, 1q21+ remained an independent predictor of poor prognosis and was deemed to have an adverse impact on survival [Citation83].
4.2.4. IMiD/PI treatment in RRMM
Results of the phase 3, randomized, double-blind, placebo-controlled TOURMALINE-MM1 study of 722 patients with RRMM that ixazomib in combination with lenalidomide-dexamethasone (IRd) conferred a 40% longer period of PFS compared with placebo-Rd (HR 0.74; 95% confidence interval [CI]: 0.59–0.94; P= 0.01), with limited additional toxicity attributed to the triplet regimen [Citation84]. Survival among patients with high-risk cytogenetic abnormalities was an additional secondary endpoint in TOURMALINE-MM1 [Citation84]. Improvements in PFS were detected in favor of IRd vs placebo-Rd in the 25% of patients with high-risk cytogenetics (HR 0.54; 95% CI: 0.32–0.92; P= 0.021) and remaining patients with standard-risk cytogenetics (HR 0.64; 95% CI: 0.46–0.89; P= 0.007), and consistent PFS benefit was detected across subgroups with individual high-risk cytogenetic abnormalities, including patients with del(17p) (HR 0.60; 95% CI: 0.29–1.24) [Citation21]. However, statistical significance for a PFS benefit in favor of IRd vs placebo-Rd was marginal for the 56% of patients in the ‘expanded high-risk’ cohort, defined as presence of high-risk cytogenetic abnormalities and/or 1q21+ (HR 0.66; 95% CI: 0.47–0.93) and was not detected in the 31% of patients with 1q21 amplification alone (HR 0.78; 95% CI: 0.49–1.24) [Citation21]. Thus, ixazomib appears less efficacious in patients with 1q21 amplification than in patients with standard-risk cytogenetics and even other high-risk cytogenetics.
4.3. Monoclonal antibodies
4.3.1. Elotuzumab
Elotuzumab is a monoclonal antibody (mAb) that interferes with the MM cell-surface marker signaling lymphocytic activation molecule family member 7 (SLAMF7) [Citation85–87]. SLAMF7 is a glycoprotein expressed on the surface of plasma cells and natural killer (NK) cells. SLAMF7 expression was detected in more than 95% of bone marrow myeloma but not on normal tissues [Citation85,Citation88]. SLAMF7 gene is located on the long arm of chromosome 1 at 1q23–24 [Citation89]. Elotuzumab exerts an anti-myeloma effect by NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC) [Citation90]. NK cell activation is also induced by elotuzumab through SLAMF7 and CD16 [Citation91]. Elotuzumab was approved in combination with lenalidomide and dexamethasone for patients with RRMM based on the phase 3 ELOQUENT-2 trial (NCT01239797) [Citation87,Citation92]. ELOQUENT-2 recruited 646 patients with RRMM, of whom 48% had 1q21+, 32% had del(17p), and 9% had t(4;14) [Citation92]. In the overall population, median PFS in the elotuzumab plus lenalidomide and dexamethasone group was 19.4 months compared with 14.9 months in the lenalidomide and dexamethasone. Among patients with 1q21+ in ELOQUENT-2, the risk for progression or death was lower in the elotuzumab arm than the control arm (HR 0.75; 95% CI: 0.56–0.99), but this did not translate into improved OS (HR 0.81; 95% CI: 0.61−1.06) [Citation92,Citation93] ().
Table 3. Impact of 1q21+ on outcomes in RRMM patients receiving therapeutic monoclonal antibodies
The combination of elotuzumab plus pomalidomide and dexamethasone was tested against pomalidomide and dexamethasone alone, an effective doublet regimen in MM refractory to lenalidomide and a PI, in the randomized, open-label, phase 2 ELOQUENT-3 trial [Citation94]. PFS was longer in the elotuzumab plus pomalidomide and dexamethasone group than the pomalidomide plus dexamethasone alone group (10.3 vs 4.7 months), which translated into significantly lower risk of progression or death in the elotuzumab combination arm (10.3 vs 4.7 months; HR 0.54; 95% CI: 0.34−0.86; P= 0.008). Statistical analysis of patients with vs without del(17p), t(4;14) or t(14;16) (HR 0.52 [95% CI: 0.22−1.25] vs 0.56 [95% CI: 0.27–1.14]), and with vs without 1q21+ (HR 0.56 [95% CI: 0.29–1.09] vs 0.43 [95% CI: 0.15−1.24]), failed to demonstrate a clear PFS benefit from the addition of elotuzumab although the sample sizes were small [Citation94].
The addition of elotuzumab to lenalidomide, bortezomib, and dexamethasone (RVd) in induction and maintenance therapy did not improve patient outcomes in the randomized, phase 2, SWOG-1211 trial of high-risk NDMM () [Citation95]. High-risk MM was defined by one of the following: HRCA, t(14;16), t(14;20), del(17p), or 1q21+. Of 100 enrolled patients, 47% had 1q21+, 37% had del17p, and 11% had t(14;16). No difference was observed between the RVd-elotuzumab arm and RVd arm regarding PFS (31 months vs 34 months; HR 0.968; 80% CI: 0.697–1.344; P= 0.45) or OS (68 months vs not reached; HR 1.28; 80% CI: 0.82–2.00; P= 0.478). The SWOG-1211 study also explored the PFS outcomes for the 1q21+ subsets and found that addition of elotuzumab did not improve results in 1q21+ patients; the median PFS was 41 months (95% CI: 22–not reached) for the RVd group vs 32 months (95% CI: 18–not reached) for the RVd-elotuzumab group. The SWOG-1211 study also analyzed response rates (partial response or better) in RVd vs RVd-elotuzumab group and observed no difference in response in 1q21+ patients in both groups [Citation95].
4.3.2. Daratumumab
Daratumumab is a human mAb that binds to an epitope on CD38, a type II transmembrane glycoprotein with signal-transduction and ectoenzyme functions that is expressed at high levels on MM cells and low levels on normal lymphoid and myeloid cells [Citation96,Citation97]. Daratumumab has US Food and Drug Administration (FDA) approval in the newly diagnosed and relapsed setting [Citation97]. A meta-analysis of daratumumab clinical trials including CASSIOPEIA, MAIA, and ALCYONE (for NDMM), and CASTOR and POLLUX (for RRMM) showed that the addition of daratumumab to backbone regimens led to a clear PFS benefit in standard risk NDMM and in both standard risk and high-risk patients with RRMM [Citation98]. High-risk was defined in this meta-analysis according to method used in each individual study. A second meta-analysis of daratumumab clinical trials added the CANDOR trial to the aggregation of RRMM data and defined high-risk cytogenetics as the presence of t(4;14), t(14;16), or del(17p) irrespective of the method used in the study and the proportion of cells exhibiting the cytogenetic abnormality [Citation99]. The meta-analysis revealed that adjunctive daratumumab was associated with improved PFS in patients with standard-risk (pooled HR 0.45; 95% CI: 0.37–0.54; P< 0.001) and high risk NDMM (pooled HR 0.67; 95% CI: 0.47–0.95; P= 0.02) as well as standard risk (pooled HR 0.38; 95% CI: 0.26–0.56; P< 0.001) and high risk RRMM (pooled HR 0.45; 95% CI: 0.30–0.67; P< 0.001) [Citation99]. However, both meta-analyses provided no insight into the impact of 1q21+ on the efficacy of adjunctive daratumumab in these patient populations.
In a single prospective observational study, the prognostic impact of 1q21+ and GEP70 risk score at initial presentation and prior to daratumumab therapy was assessed in 81 patients with RRMM () [Citation100]. Daratumumab was given in combination therapy in 80.5% (most frequently in combination with pomalidomide 58%) and as single agent in 19.5%. Median PFS was 0.5 years in the 1q21+ cohort vs 2.1 years in the non-1q21+ cohort and median OS was 0.9 years in the 1q21+ cohort vs not reached in the non-1q21+ cohort. Multivariate analysis revealed that GEP70 score and 1q21 status at initial presentation were independently associated with inferior PFS (P< 0.05) while only GEP70 score was statistically associated with poor OS (P< 0.05): gain 1q showed a trend for poor OS (P= 0.06). The worst outcome was seen for patients who were positive for 1q21+ and classified as GEP70 high risk prior to daratumumab initiation (PFS and OS of 0.3 years and 0.8 years, respectively), whereas the median PFS and OS were not reached by patients without 1q21+ and GEP70 low‐risk status. This observation might suggest that daratumumab may not be able to overcome the poor prognosis associated with 1q2+, but further clinical studies are needed. Overexpression of complement regulatory proteins CD55 and CD59 have been implicated in daratumumab resistance [Citation101]. Interestingly, CD55 is located on chromosome 1q32.2 () and hypothesized to contribute to daratumumab resistance in 1q21+ patients [Citation100].
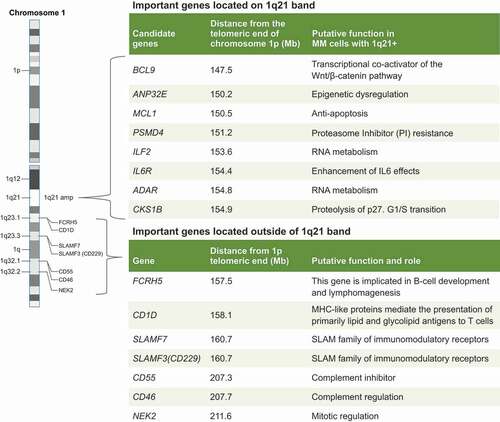
Figure 3. Targeting 1q specific vulnerabilities: Important gene located on chromosome 1q21 band and on 1q arm.
4.3.3. Isatuximab
Isatuximab is a humanized mAb that has FDA approval in the relapsed setting [Citation102]. Isatuximab targets a unique epitope of the CD38 receptor resulting in a distinct mode of action [Citation103,Citation104]. Specifically, isatuximab inhibits CD38 ectoenzyme activity and elicits innate immune responses, namely, antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis [Citation103,Citation104]. Isatuximab also directly induces target-cell killing through apoptotic mechanisms in the absence of cross-linking agents [Citation103,Citation105].
Subgroup analysis of the phase 3 ICARIA-MM trial was conducted to assess the efficacy of adding isatuximab to a backbone of pomalidomide and dexamethasone in RRMM patients (≥2 prior lines of therapy) with high-risk cytogenetics () [Citation106]. Isatuximab-based combination therapy showed a clear benefit in 1q21+ patients regardless of the presence of other high-risk cytogenetic abnormalities [del(17p), t(4;14), and t(14;16)]. Of the 41.7% (128/307) of patients with 1q21+, 26.1% patients had gain of 1q21 (3 copies in at least 30% of cells) and 15.6% had amplification of 1q21 (≥4 copies in at least 30% of cells). Patients with both gain and amplification of 1q21 exhibited improved outcomes under isatuximab treatment regardless of the other high-risk cytogenetic abnormalities () [Citation106]. In patients with 1q21+, median PFS was longer (9.5 vs 3.8 months; HR 0.399; 95% CI: 0.254–0.627) and ORR was higher (53.9% vs 17.3%) in the isatuximab plus pomalidomide and dexamethasone arm than in the pomalidomide and dexamethasone arm [Citation106]. These outcomes compared favorably when judged against the subgroup without 1q21+ ().
Eighty-five of 307 patients (27.7%) had isolated gain 1q21 in ICARIA-MM, including 56 patients (36.4%) in the isatuximab arm and 29 (19.0%) patients in the control arm [Citation106]. For this cohort, median PFS in the isatuximab arm was significantly longer than that in the control arm (11.2 vs 4.6 months; HR 0.50; 95% CI: 0.28–0.88), and similar to the median PFS observed in the isatuximab arm in patients without gain 1q21. The ORR for patients with isolated gain 1q21 was also higher in the isatuximab arm than the control arm (53.6% vs 27.6%; P= 0.0116) and in those without gain 1q21 irrespective of the presence of other high-risk cytogenetic abnormalities (68.4% vs 43.5%; P= 0.0115) [Citation106].
Similar subgroup analysis performed in patients with isolated 1q21 amplification also showed improved PFS in the isatuximab arm relative to the control arm (8.9 vs 2.3 months; HR 0.55; 95% CI: 0.21–1.43). Presence of additional high-risk cytogenetic abnormalities did not affect the positive impact of isatuximab treatment (8.9 vs 2.3 months; HR 0.49; 95% CI: 0.24–0.99). Similar to the isolated gain 1q21 subgroup, the isolated 1q21 amplification subgroup exhibited better ORR (52.2% vs 11.1%; P= 0.0182) upon addition of isatuximab. This ORR was in favor of isatuximab was not affected when 1q21 amplification occurred in the presence of other high-risk cytogenetic abnormalities (51.9% vs 9.5%; P= 0.0111) [Citation106].
The positive impact of isatuximab on 1q21+ was supported by the phase 3 IKEMA study, which investigated the addition of isatuximab to carfilzomib and dexamethasone in patients with relapsed RRMM after at least 1 prior line of therapy. This study included 75 of 179 (41.0%) patients with 1q21 gain in the isatuximab combination arm and 52 of 123 (42.3%) patients with 1q21 gain in the control arm in the presence and absence of other high-risk cytogenetic abnormalities. A clear PFS benefit was observed for patients with gain 1q21 (HR 0.569; 95% CI: 0.330–0.981) [Citation108,Citation109], which was maintained when the analysis was performed on patients with isolated gain 1q21 without other high-risk cytogenetic abnormalities (HR 0.462; 95% CI: 0.219–0.972) or with other high-risk cytogenetic abnormalities (HR 0.678; 95% CI: 0.299–1.537). Moreover, patients with 1q21 amplification (HR 0.531; 95% CI: 0.150–1.878) displayed a similar trend [Citation108]. The favorable outcomes associated with isatuximab based combination therapy in patients with 1q21+ observed in ICARIA and IKEMA suggest that isatuximab based combination therapy can ameliorate the adverse prognostic impact 1q21+.
Downregulation of CD38 on the MM cell surface can be mediated by the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. Inhibiting the JAK-STAT pathway increased the surface expression of CD38 and enhanced the daratumumab-mediated killing of MM cells [Citation110]. Schmidt and colleagues proposed an interesting hypothesis that 1q21+ mediated STAT3 overactivation might contribute to daratumumab resistance by reducing CD38 surface expression [Citation9], but currently available data suggest that CD38 receptor density is not predictive of response for either daratumumab or isatuximab [Citation111,Citation112]. Therefore, antigen downregulation seems to be an unlikely mechanism of resistance.
5. Inclusion of 1q into future high-risk stratification
1q21+ is more prevalent than any cytogenetic abnormality in MM [Citation3], detected in up to half of all patients on initial presentation [Citation4–8,Citation14,Citation18]. The number of MM cells with 1q gain and absolute copy number of 1q increases throughout the disease continuum [Citation8], and the consistent poor prognosis conferred over this time infers that 1q21+ is involved in the pathophysiology of progression and drug resistance. Moreover, GEP indicates that a proliferation subgroup (PR) dominates at relapse, characterized by increases in 1q copy number and cells with 1q gain [Citation42]. Numerous levels of evidence on the efficacy of chemotherapy and combinations of IMiDs and PIs alone and with elotuzumab or daratumumab clearly show that presence of 1q21+ is associated with an adverse prognosis (). Thus, despite tremendous therapeutic advancement over the last 20 years, patients harboring 1q21+ are not benefitting with existing therapies.
These findings underscore the importance of subgrouping in MM according to cytogenetics and clinical presentations to complement established prognostic factors (e.g. age, comorbidities) in order to obtain a more precise estimation of prognosis. Clearly, there is a need for including 1q21+ as a routine marker for high-risk MM, which will enable future delivery of risk-adapted therapy. For example, extramedullary disease, an aggressive proliferative MM phenotype, has an early onset among 1q21+ patients and the frequency of 1q21+ amplification is also higher in extramedullary disease [Citation113,Citation114].
At present, the Mayo Stratification for Myeloma And Risk-adapted Therapy (mSMART) algorithm has incorporated 1q21+ (along with four other high-risk cytogenetic abnormalities) in their definition of high-risk disease [Citation115]. The revised ISS do not incorporate 1q21+ as a marker of high-risk disease, as it combines elements of tumor burden and disease biology (presence of three high risk cytogenetic abnormalities or elevated lactate dehydrogenase level) to create a unified prognostic index [Citation116]. Using an iFISH cutoff of 10%, meta-analysis of GMMG HD4, GMMG MM5, and UK NCRI Myeloma XI trial data revealed that 1q21+ is independently prognostic of the R-ISS; hence, 1q21+ could be used to refine R-ISS−based risk predictions [Citation82].
Another cytogenetic prognostic index predicting survival was developed and validated to avoid potential prognostic misclassification of the IMWG definition of high-risk NDMM [i.e. t(4;14), del(17p), t(14;16), t(14;20), hypodiploidy, and 1q21+] [Citation3,Citation117,Citation118]. Within the IMWG, patients with three unfavorable markers of del(17p), 1q21+, and any IgH translocation on chromosome 14 have an OS of approximately 9 months compared with 23 months in patients who have two of the three adverse markers and 38 to 44 months patients with just one of these abnormalities in isolation [Citation79]. However, cytogenetic and clinical data were collected from patients enrolled in four different randomized clinical trials implemented by the Intergroupe Francophone du Myélome, and who were followed long enough to identify good prognostic factors. The final validated prognostic index is based on a weighted score of six cytogenetic abnormalities, namely, t(4;14), del(17p), 1q21+, del(1p32), trisomy 5, and trisomy 21, and can improve the classification of NDMM in high-risk groups [Citation118]. Note that t(14;16) does not feature in this prognostic index even though it remains part of the R-ISS definition [Citation118].
6. Targeting of 1q gain/amplification specific vulnerabilities in MM
In the 1q21 amplicon, several genes have been associated with aggressive phenotypes () including CKS1B (associated with G1-S transition), PSMD4 (resistance to PI), ANP32E (epigenetic regulation), IL6R (promotion of cell growth), ADAR and ILF2 (RNA metabolism), MCL-1 (apoptosis control), those encoding SLAM family molecules, FCRL5 (encoding FcRH5), and CD1D (cluster differentiation 1D) [Citation10,Citation38,Citation86,Citation119]. Therapeutic targeting of candidate genes in the 1q21 amplicon might benefit patients who do not respond to current treatment [Citation10].
Amp1q and del(13) are associated with upregulation of G1/S specific cyclin D2 (CCND2) and MCL1 overexpression, which represent novel targets for experimental drugs [Citation120]. The B-cell lymphoma-2 (BCL-2) protein family members are inherent mediators of the apoptosis pathway and whether or not a cell undergoes apoptosis is dependent on the expression of the pro-survival proteins BCL-2, BCL-XL, and MCL-1, as well as the availability of a number of proapoptotic proteins [Citation121]. As one of the genes located on 1q21, MCL-1 is overexpressed in MM, and associated with relapse and shorter survival [Citation122]. Plasma cells from patients with MM with 1q21 amplification are highly sensitive to MCL-1 inhibition, especially if 1q21 amplification is associated with increased levels of β-microglobulin [Citation121]. Preclinical and clinical investigation on MCL-1 inhibitors in MM are currently ongoing [Citation123].
The MM transcriptome is aberrantly hyper edited because of ADAR1 overexpression, conferring enhanced growth and proliferation of MM cells, as well as reduced responsiveness to both standard and novel therapy [Citation124]. Overexpression of ADAR1 is an independent risk factor of poor prognosis [Citation124]. As knockdown of ADAR1 reduces malignant regeneration of high-risk MM in patient-derived xenografts, it is plausible that selective inhibition of ADAR1 may obviate progression and relapse [Citation125] ().
Several cell-surface molecular markers are located on the 1q arm a number of which are upregulated upon 1q21 copy number gain and play a role in MM pathogenesis [Citation85,Citation86,Citation126] (). Although targeting these cell surface markers has shown a broad antimyeloma effect, clinical data supporting amplification, or gain of 1q specific vulnerability are lacking [Citation85,Citation86,Citation126]. SLAM family members located on chromosome 1q arm include SLAMF2, SLAMF3 (CD229, LY9), SLAMF6, and SLAMF7 (CS1) [Citation86]. Of these, SLAMF7 is the best studied target in MM, with the anti-SLAMF7 monoclonal antibody elotuzumab approved for RRMM [Citation87]. Several chimeric antigen receptor (CAR) T cell (CAR-T) products targeting SLAMF7 and SLAMF3 are at various stages of development in MM [Citation127]. The cell lineage marker FCRH5 is overexpressed in high-risk MM patients and associated with 1q21+ [Citation86]. An FCRH5XCD3 bispecific T- cell engager (cevostamab) showed good antitumor activity in a phase 1 study (NCT03275103). CD1D expression is highly expressed in premalignant and early myeloma but lost in advanced MM [Citation119]. Efforts to target CD1D via CAR19-invariant NK T-cells in hematologic malignancies are currently underway [Citation128]. CD46 is a negative regulator of the innate immune system and there is a strong correlation between CD46 expression and 1q21+ [Citation129]. An anti-CD46 directed antibody-drug conjugate has shown excellent antiproliferative activity against myeloma cell lines with little effect on normal cells [Citation129]. Whether or not targeting a 1q-linked cell surface marker will provide a 1q-specific therapeutic effect in MM remains an unanswered question. Understanding the biology and surface expression of 1q-linked cell surface markers is an important step in the development of novel therapeutic targets in MM.
7. Conclusions
1q21+ is a cytogenetic abnormality occurring in around 40% of patients with MM. The frequency of 1q21+ increases as MM progresses. MM patients with 1q21+ tend to present with higher tumor burden, greater end-organ damage, and more co-occurring high-risk cytogenetic abnormalities than patients without 1q21+. Presence of 1q21+ predisposes to increased risk of drug resistance, disease progression, and death.
The precise molecular mechanisms of acquiring 1q21+ and its effect on MM pathophysiology are yet to be fully elucidated. 1q21+ can occur as isochromosomes of 1q, JT1q, tandem duplications of 1q12-21, trisomy of chromosome 1, or a combination of all of these events. These rearrangements lead to dysregulation of the expression of many genes on the 1q21 amplicon crucial to the control of MM cell proliferation, differentiation, and survival. The prognostic power of 1q21+ in MM appears to be modulated by the differential expression of these molecular targets in concert with the number and type of concomitant high-risk cytogenetic aberrations.
Patients with MM who harbor 1q21+ require novel pharmacotherapies to overcome the negative impact of 1q21+ on outcomes although carfilzomib may partially overcome the resistance associated with bortezomib. Current standard of MM care using IMiD- and PI-based induction modalities mitigates the negative effect of some high-risk genetic abnormalities but not 1q21+. There are multiple levels of evidence showing that 1q21+ is a poor prognostic marker in patients who receive IMiD- and PI-based induction regardless of receipt of high dose treatment and ASCT.
Monoclonal antibodies are a relatively recent addition to the arsenal of MM therapy. Currently, three mAbs have been approved in MM, including the anti-CD38 daratumumab and isatuximab, and the anti-SLAMF7 elotuzumab. The addition of either daratumumab and elotuzumab to IMiD/PI based combination therapy is associated with improved OS in MM but the evidence showing a benefit in 1q21+ patients is lacking. Elotuzumab improved PFS but not OS among 1q21+ patients in the ELOQUENT-2 study (elotuzumab-Rd vs Rd) [Citation92,Citation93] but in ELOQUENT-3 (elotuzumab-Pd vs Pd) and SWOG-1211 (elotuzumab-RVd vs RVd), elotuzumab failed to show a PFS benefit in the 1q21+ subgroup [Citation94,Citation95]. The reason for this inconsistency was not known. Isatuximab has shown a clear PFS benefit along with improved response rate in 1q21+ patient segment and this benefit was observed irrespective of the existence of other cytogenetic abnormalities and in both 1q21 gain and 1q21 amplification patients. Further study of isatuximab in a larger patient population will help determine the place of this drug in the MM treatment paradigm.
Continued identification and characterization of 1q21+ molecular targets are needed to improve the prognostics and treatment of MM. Integration of validated prognostic markers associated with 1q21+ will help distinguish patient subsets who will benefit from specific treatments and be spared from unnecessary treatment. The identification of 1q21+ prognostic markers may help guide treatment decisions concerning patients with other types of cancer, as 1q21+ is not restricted to MM pathophysiology.
8. Expert opinion
The addressed topic intersects genetic and clinical information on the recalcitrant nature of 1q21+ MM. The studies reviewed included biomedical research, observational studies, clinical trials, meta-analysis, and real-world database analysis. The data represent the current state of the science in 1q21+ MM, and proffers research opportunities in the pursuit of personalized medicine.
It is interesting to speculate on the intimate role 1q21+ plays in the progression of MM to a multidrug resistant phenotype and how this information may be applied prognostically. As MM proceeds along a disease continuum from MGUS to RRMM, manifestation of 1q21+ in tumor subclones represents a broad surrogate marker for aggressive, recalcitrant disease. The precise prognostic power of 1q21+ is blurred because 1q21+ is a collective term for a vast array of genetic dysregulation that occurs on 1q at various stages of disease, which is confounded further by the consequences of co-occurring high-risk genetic abnormalities. Separating 1q21+ into gain or amplification of 1q21 may increase prognostic resolution, although a unified definition for 1q21 amplification has not been devised. Determining if 1q21+ is a cause or consequence of MM disease will require deeper insights into the genetic and molecular basis of MM biology. This will be achieved by correlating outcomes with advanced genomic profiling and protein expression analyses, augmented by specific pharmacotherapies with diverse mechanisms of action that can precisely dissect key interactions.
The unique challenges involved in the treatment of patients with 1q21+ MM must be addressed in the clinical trial setting, using standardized definitions for 1q21+, clone size, and endpoints. Consensus is required on the adoption of a paradigm that supports control, relief, or elimination of disease manifestations that are present at the time treatment is initiated, and/or preventing or delaying relapse. All of these trial criteria may be reworked as more becomes known about 1q21+ tumor biology, regarding specific vulnerabilities of 1q21+, as well as mechanisms of relapse and drug resistance.
In the RRMM setting, subgroup analyses of two phase 3 clinical trials revealed that isatuximab‐based combination therapy of patients with 1q21+ was associated with improved PFS relative to control; while in the IKEMA study (isatuximab plus carfilzomib and dexamethasone) mature PFS data are awaited, the median PFS of patients with isolated 1q gain in the ICARIA study (isatuximab plus pomalidomide and dexamethasone) was similar to median PFS of patients without 1q gain or the median PFS in the intent-to-treat population [Citation106,Citation108]. Similar data with daratumumab in the 1q21+ patient population is currently lacking [Citation99].
In the next 5 years, the results of ongoing and future research and trials should also focus on the therapeutic impact and treatment decisions associated with 1q21+ and other cytogenetic abnormalities in MM.
Article highlights
The addressed topic intersects genetic and clinical information on the recalcitrant nature of 1q21+ MM. The studies reviewed included biomedical research, observational studies, clinical trials, meta-analysis, and real-world database analysis.
The data represent the current state of the science in 1q21+ MM and proffer important research opportunities in the pursuit of personalized medicine.
The unique challenges involved in the treatment of patients with 1q21+ MM must be addressed in the clinical trial setting, using standardized definitions for 1q21+, clone size, and endpoints.
Consensus is required on the adoption of a paradigm that supports control, relief, or elimination of disease manifestations that are present at the time treatment is initiated, and/or preventing or delaying relapse.
Declaration of interest
Kamlesh Bisht is an employee of Sanofi and may hold shares and/or stock options in the company; Shaji K Kumar reports receiving grants and other from BMS/Celgene, grants and other from Takeda, grants and other from Abbvie, grants and other from Roche, grants from Medimmune, grants from Tenebio, grants from Carsgen, personal fees from Oncopeptides, grants and other from Janssen, personal fees from Beigene, personal fees from Antengene, outside the submitted work. Ivan Spicka reports receiving personal fees, advisory board and speakers bureau for Amgen, Celgene, Janssen, Sanofi, BMS and Takeda; Philippe Moreau reports receiving consultancy fees and honoraria from Janssen, BMS, Celgene, Amgen, Sanofi and Abbvie; Tom Martin reports receiving research funding from Sanofi, Amgen, Seattle Genetics, JNJ and Janssen; Consultancy fees–Legend Biotech; Luciano Costa reports personal fees from Sanofi, during the conduct of the study; personal fees from Sanofi, grants and personal fees from Janssen, grants and personal fees from Amgen, grants and personal fees from BMS, GSK and oncopetides outside the submitted work; Joshua Richter reports receiving personal fees from Sanofi, Karyopharm, Janssen, BMS/Celgene, Secura Bio, Oncopeptides, X4 Pharmaceuticals, Adaptive Biotechnologies, Takeda, and Astra Zeneca, outside the submitted work; Taro Fukao, Sandrine Mace and Helgi van de Velde are employees of Sanofi and may hold shares and/or stock options in the company.
The author(s) have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
One peer reviewer has done scientific talks and advisory board work for Janssen, BMS-Celgene, Amgen, and Takeda, and has also received grants from BMS-Celgene and Sanofi. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
Acknowledgments
Medical writing support was provided by John Clarke, PhD, and Malcolm Darkes, PhD, on behalf of Elevate Medical Affairs, contracted by Sanofi Genzyme for publication support services.
Additional information
Funding
References
- Biran N, Jagannath S, Chari A. Risk stratification in multiple myeloma, part 1: characterization of high-risk disease. Clin Adv Hematol Oncol. 2013;11(8):489–503.
- Sawyer JR, Tian E, Walker BA, et al. An acquired high-risk chromosome instability phenotype in multiple myeloma: jumping 1q syndrome. Blood Cancer J. 2019;9(8):62.
- Sonneveld P, Avet-Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the international myeloma working group. Blood. 2016;127(24):2955–2962.
- Abdallah N, Greipp P, Kapoor P, et al. Clinical characteristics and treatment outcomes of newly diagnosed multiple myeloma with chromosome 1q abnormalities. Blood Adv. 2020;4(15):3509–3519.
- Avet-Loiseau H, Attal M, Campion L, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol. 2012;30(16):1949–1952.
- Cremer FW, Bila J, Buck I, et al. Delineation of distinct subgroups of multiple myeloma and a model for clonal evolution based on interphase cytogenetics. Genes Chromosomes Cancer. 2005;44(2):194–203.
- Fonseca R, Van Wier SA, Chng WJ, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20(11):2034–2040.
- Hanamura I, Stewart JP, Huang Y, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108(5):1724–1732.
- Schmidt TM, Fonseca R, Usmani SZ. Chromosome 1q21 abnormalities in multiple myeloma. Blood Cancer J. 2021;11(4):83.
- Hanamura I. Gain/amplification of chromosome arm 1q21 in multiple myeloma. Cancers (Basel). 2021;13(2):256.
- Locher M, Steurer M, Jukic E, et al. The prognostic value of additional copies of 1q21 in multiple myeloma depends on the primary genetic event. Am J Hematol. 2020;95(12):1562–1571.
- Nemec P, Zemanova Z, Greslikova H, et al. Gain of 1q21 is an unfavorable genetic prognostic factor for multiple myeloma patients treated with high-dose chemotherapy. Biol Blood Marrow Transplant. 2010;16(4):548–554.
- Shah V, Sherborne AL, Walker BA, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia. 2018;32(1):102–110.
- An G, Li Z, Tai Y-T, et al. The impact of clone size on the prognostic value of chromosome aberrations by fluorescence in situ hybridization in multiple myeloma. Clin Cancer Res. 2015;21(9):2148–2156.
- Berry NK, Dixon-McIver A, Scott RJ, et al. Detection of complex genomic signatures associated with risk in plasma cell disorders. Cancer Genet. 2017;218-219:1–9.
- Bolli N, Genuardi E, Ziccheddu B, et al. Next-generation sequencing for clinical management of multiple myeloma: ready for prime time? Front Oncol. 2020;10:189.
- Zang M, Zou D, Yu Z, et al. Detection of recurrent cytogenetic aberrations in multiple myeloma: a comparison between MLPA and iFISH. Oncotarget. 2015;6(33):34276–34287.
- Chiecchio L, Dagrada GP, Cabanas ED, et al. Gain of 1q21 does not predict for immediate progression in MGUS. Blood. 2009;114(22):123.
- Merz M, Hielscher T, Hoffmann K, et al. Cytogenetic abnormalities in monoclonal gammopathy of undetermined significance. Leukemia. 2018;32(12):2717–2719.
- Mikulasova A, Smetana J, Wayhelova M, et al. Genomewide profiling of copy-number alteration in monoclonal gammopathy of undetermined significance. Eur J Haematol. 2016;97(6):568–575.
- Avet-Loiseau H, Bahlis NJ, Chng WJ, et al. Ixazomib significantly prolongs progression-free survival in high-risk relapsed/refractory myeloma patients. Blood. 2017;130(24):2610–2618.
- Shaughnessy J. Amplification and overexpression of CKS1B at chromosome band 1q21 is associated with reduced levels of p27Kip1 and an aggressive clinical course in multiple myeloma. Hematology. 2005;10(Suppl 1):117–126.
- An G, Xu Y, Shi L, et al. Chromosome 1q21 gains confer inferior outcomes in multiple myeloma treated with bortezomib but copy number variation and percentage of plasma cells involved have no additional prognostic value. Haematologica. 2014;99(2):353–359.
- Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33(33):3911–3920.
- Neben K, Lokhorst HM, Jauch A, et al. Administration of bortezomib before and after autologous stem cell transplantation improves outcome in multiple myeloma patients with deletion 17p. Blood. 2012;119(4):940–948.
- D’Agostino M, Ruggeri M, Aquino S, et al. Impact of gain and amplification of 1q in newly diagnosed multiple myeloma patients receiving carfilzomib-based treatment in the Forte trial. 62nd American Society of Hematology Virtual Scientific meeting, December 5-8, 2020 [Abstract 1331].
- Zhan F, Colla S, Wu X, et al. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood. 2007;109(11):4995–5001.
- Shi L, Wang S, Zangari M, et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget. 2010;1(1):22–33.
- Treon SP, Maimonis P, Bua D, et al. Elevated soluble MUC1 levels and decreased anti-MUC1 antibody levels in patients with multiple myeloma. Blood. 2000;96(9):3147–3153.
- Inoue J, Otsuki T, Hirasawa A, et al. Overexpression of PDZK1 within the 1q12-q22 amplicon is likely to be associated with drug-resistance phenotype in multiple myeloma. Am J Pathol. 2004;165(1):71–81.
- Sawyer JR, Tricot G, Lukacs JL, et al. Genomic instability in multiple myeloma: evidence for jumping segmental duplications of chromosome arm 1q. Genes Chromosomes Cancer. 2005;42(1):95–106.
- Legartova S, Krejci J, Harnicarova A, et al. Nuclear topography of the 1q21 genomic region and Mcl-1 protein levels associated with pathophysiology of multiple myeloma. Neoplasma. 2009;56(5):404–413.
- Walker BA, Leone PE, Chiecchio L, et al. A compendium of myeloma-associated chromosomal copy number abnormalities and their prognostic value. Blood. 2010;116(15):e56–65.
- Shaughnessy JD Jr., Qu P, Usmani S, et al. Pharmacogenomics of bortezomib test-dosing identifies hyperexpression of proteasome genes, especially PSMD4, as novel high-risk feature in myeloma treated with total therapy 3. Blood. 2011;118(13):3512–3524.
- Zhou W, Yang Y, Xia J, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23(1):48–62.
- Marchesini M, Ogoti Y, Fiorini E, et al. ILF2 is a regulator of RNA splicing and DNA damage response in 1q21-amplified multiple myeloma. Cancer Cell. 2017;32(1):88–100 e6.
- Wu C, Yang T, Liu Y, et al. ARNT/HIF-1beta links high-risk 1q21 gain and microenvironmental hypoxia to drug resistance and poor prognosis in multiple myeloma. Cancer Med. 2018;7(8):3899–3911.
- Teoh PJ, Chung TH, Chng PYZ, et al. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematologica. 2020;105(5):1391–1404.
- Pawlyn C, Morgan GJ. Evolutionary biology of high-risk multiple myeloma. Nat Rev Cancer. 2017;17(9):543–556.
- Fabris S, Ronchetti D, Agnelli L, et al. Transcriptional features of multiple myeloma patients with chromosome 1q gain. Leukemia. 2007;21(5):1113–1116.
- Shaughnessy JD Jr., Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284.
- Zhan F, Huang Y, Colla S, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028.
- Sawyer JR, Tricot G, Mattox S, et al. Jumping translocations of chromosome 1q in multiple myeloma: evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood. 1998;91(5):1732–1741.
- Sawyer JR, Tian E, Heuck CJ, et al. Jumping translocations of 1q12 in multiple myeloma: a novel mechanism for deletion of 17p in cytogenetically defined high-risk disease. Blood. 2014;123(16):2504–2512.
- Fournier A, Florin A, Lefebvre C, et al. Genetics and epigenetics of 1q rearrangements in hematological malignancies. Cytogenet Genome Res. 2007;118(2–4):320–327.
- Lange K, Gadzicki D, Schlegelberger B, et al. Recurrent involvement of heterochromatic regions in multiple myeloma-a multicolor FISH study. Leuk Res. 2010;34(8):1002–1006.
- Sawyer JR, Tian E, Thomas E, et al. Evidence for a novel mechanism for gene amplification in multiple myeloma: 1q12 pericentromeric heterochromatin mediates breakage-fusion-bridge cycles of a 1q12 approximately 23 amplicon. Br J Haematol. 2009;147(4):484–494.
- Black JC, Manning AL, Van Rechem C, et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell. 2013;154(3):541–555.
- Black JC, Atabakhsh E, Kim J, et al. Hypoxia drives transient site-specific copy gain and drug-resistant gene expression. Genes Dev. 2015;29(10):1018–1031.
- Xu J, Xu T, Yang Y, et al. The paradoxical prognostic role of 1q21 gain/amplification in multiple myeloma: every coin has two sides. Leuk Lymphoma. 2020;61(10):2351–2364.
- Li X, Chen W, Wu Y, et al. 1q21 gain combined with high-risk factors is a heterogeneous prognostic factor in newly diagnosed multiple myeloma: a multicenter study in China. Oncologist. 2019;24(11):e1132–e1140.
- Grzasko N, Hus M, Chocholska S, et al. 1q21 amplification with additional genetic abnormalities but not isolated 1q21 gain is a negative prognostic factor in newly diagnosed patients with multiple myeloma treated with thalidomide-based regimens. Leuk Lymphoma. 2012;53(12):2500–2503.
- Grzasko N, Hus M, Pluta A, et al. Additional genetic abnormalities significantly worsen poor prognosis associated with 1q21 amplification in multiple myeloma patients. Hematol Oncol. 2013;31(1):41–48.
- Varma A, Sui D, Milton DR, et al. Outcome of multiple myeloma with chromosome 1q gain and 1p deletion after autologous hematopoietic stem cell transplantation: propensity score matched analysis. Biol Blood Marrow Transplant. 2020;26(4):665–671.
- Schmidt TM, Barwick BG, Joseph N, et al. Gain of chromosome 1q is associated with early progression in multiple myeloma patients treated with lenalidomide, bortezomib, and dexamethasone. Blood Cancer J. 2019;9(12):94.
- Walker BA, Mavrommatis K, Wardell CP, et al. A high-risk, double-hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33(1):159–170.
- Baysal M, Demirci U, Umit E, et al. Concepts of double hit and triple hit disease in multiple myeloma, entity and prognostic significance. Sci Rep. 2020;10(1):5991.
- Wu KL, Beverloo B, Lokhorst HM, et al. Abnormalities of chromosome 1p/q are highly associated with chromosome 13/13q deletions and are an adverse prognostic factor for the outcome of high-dose chemotherapy in patients with multiple myeloma. Br J Haematol. 2007;136(4):615–623.
- Rajan AM, Rajkumar SV. Interpretation of cytogenetic results in multiple myeloma for clinical practice. Blood Cancer J. 2015;5(10):e365.
- Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat Rev Clin Oncol. 2018;15(7):409–421.
- Weinhold N, Kirn D, Seckinger A, et al. Concomitant gain of 1q21 and MYC translocation define a poor prognostic subgroup of hyperdiploid multiple myeloma. Haematologica. 2016;101(3):e116–9.
- Lamont EB, Yee AJ, Goldberg SL, et al. Associations between amplification (1q) and prior cancer in a real-world de novo myeloma cohort. JNCI Cancer Spectr. 2021;5(1): pkaa111
- Garcia JL, Hernandez JM, Gutierrez NC, et al. Abnormalities on 1q and 7q are associated with poor outcome in sporadic Burkitt’s lymphoma. A cytogenetic and comparative genomic hybridization study. Leukemia. 2003;17(10):2016–2024.
- Le Baccon P, Leroux D, Dascalescu C, et al. Novel evidence of a role for chromosome 1 pericentric heterochromatin in the pathogenesis of B-cell lymphoma and multiple myeloma. Genes Chromosomes Cancer. 2001;32(3):250–264.
- Davidsson J, Andersson A, Paulsson K, et al. Tiling resolution array comparative genomic hybridization, expression and methylation analyses of dup(1q) in Burkitt lymphomas and pediatric high hyperdiploid acute lymphoblastic leukemias reveal clustered near-centromeric breakpoints and overexpression of genes in 1q22-32.3. Hum Mol Genet. 2007;16(18):2215–2225.
- Kudoh K, Takano M, Koshikawa T, et al. Gains of 1q21-q22 and 13q12-q14 are potential indicators for resistance to cisplatin-based chemotherapy in ovarian cancer patients. Clin Cancer Res. 1999;5(9):2526–2531.
- Chen L, Chan TH, Guan XY. Chromosome 1q21 amplification and oncogenes in hepatocellular carcinoma. Acta Pharmacol Sin. 2010;31(9):1165–1171.
- Muthuswami M, Ramesh V, Banerjee S, et al. Breast tumors with elevated expression of 1q candidate genes confer poor clinical outcome and sensitivity to Ras/PI3K inhibition. PLoS One. 2013;8(10):e77553.
- Hing S, Lu YJ, Summersgill B, et al. Gain of 1q is associated with adverse outcome in favorable histology Wilms’ tumors. Am J Pathol. 2001;158(2):393–398.
- Lo KC, Ma C, Bundy BN, et al. Gain of 1q is a potential univariate negative prognostic marker for survival in medulloblastoma. Clin Cancer Res. 2007;13(23):7022–7028.
- Puri L, Saba J. Getting a clue from 1q: gain of chromosome 1q in cancer. J Cancer Biol Res. 2014;2:1053.
- Segers H, van den Heuvel-Eibrink MM, Williams RD, et al. Gain of 1q is a marker of poor prognosis in Wilms’ tumors. Genes Chromosomes Cancer. 2013;52(11):1065–1074.
- Araki A, Chocholous M, Gojo J, et al. Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol Commun. 2016;4(1):88.
- Chang H, Qi C, Yi QL, et al. p53 gene deletion detected by fluorescence in situ hybridization is an adverse prognostic factor for patients with multiple myeloma following autologous stem cell transplantation. Blood. 2005;105(1):358–360.
- Chang H, Qi X, Trieu Y, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br J Haematol. 2006;135(4):486–491.
- Bock F, Lu G, Srour SA, et al. Outcome of patients with multiple myeloma and CKS1B gene amplification after autologous hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22(12):2159–2164.
- Shah GL, Landau H, Londono D, et al. Gain of chromosome 1q portends worse prognosis in multiple myeloma despite novel agent-based induction regimens and autologous transplantation. Leuk Lymphoma. 2017;58(8):1823–1831.
- Nahi H, Vatsveen TK, Lund J, et al. Proteasome inhibitors and IMiDs can overcome some high-risk cytogenetics in multiple myeloma but not gain 1q21. Eur J Haematol. 2016;96(1):46–54.
- Boyd KD, Ross FM, Chiecchio L, et al. A novel prognostic model in myeloma based on co-segregating adverse FISH lesions and the ISS: analysis of patients treated in the MRC myeloma IX trial. Leukemia. 2012;26(2):349–355.
- Croft J, Ellis S, Sherborne AL, et al. Copy number evolution and its relationship with patient outcome-an analysis of 178 matched presentation-relapse tumor pairs from the myeloma XI trial. Leukemia. 2021;35(7):2043–2053.
- Gay F, Mina R, Rota-Scalabrini D, et al. Carfilzomib-based induction/consolidation with or without autologous transplant (ASCT) followed by lenalidomide (R) or carfilzomib-lenalidomide (KR) maintenance: efficacy in high-risk patients. J Clin Oncol. 2021;39(15 Suppl):8002.
- Weinhold N, Salwender HJ, Cairns DA, et al. Chromosome 1q21 abnormalities refine outcome prediction in patients with multiple myeloma - a meta-analysis of 2,596 trial patients. Haematologica. 2021. DOI:https://doi.org/10.3324/haematol.2021.278888
- Giri S, Huntington SF, Wang R, et al. Chromosome 1 abnormalities and survival of patients with multiple myeloma in the era of novel agents. Blood Adv. 2020;4(10):2245–2253.
- Moreau P, Masszi T, Grzasko N, et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;374(17):1621–1634.
- Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–2784.
- Ishibashi M, Morita R, Tamura H. Immune functions of signaling lymphocytic activation molecule family molecules in multiple myeloma. Cancers (Basel). 2021;13(2):279.
- EMPLICITI (elotuzumab) for injection. Prescribing information. Bristol-Myers Squibb Company. 2018. Available at: https://packageinserts.bms.com/pi/pi_empliciti.pdf [Cited 2021 May 11th].
- Kumaresan PR, Lai WC, Chuang SS, et al. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol Immunol. 2002;39(1–2):1–8.
- Boles KS, Stepp SE, Bennett M, et al. 2B4 (CD244) and CS1: novel members of the CD2 subset of the immunoglobulin superfamily molecules expressed on natural killer cells and other leukocytes. Immunol Rev. 2001;181(1):234–249.
- Tai YT, Dillon M, Song W, et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112(4):1329–1337.
- Pazina T, James AM, MacFarlane A, et al. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology. 2017;6(9):e1339853.
- Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621–631.
- Dimopoulos MA, Lonial S, White D, et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10(9):91.
- Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379(19):1811–1822.
- Usmani SZ, Hoering A, Ailawadhi S, et al. Bortezomib, lenalidomide, and dexamethasone with or without elotuzumab in patients with untreated, high-risk multiple myeloma (SWOG-1211): primary analysis of a randomised, phase 2 trial. Lancet Haematol. 2021;8(1):e45–54.
- Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. 2015;373(13):1207–1219.
- DARZALEX (daratumumab) injection for intravenous use. Prescribing information. Janssen Biotech, Inc. 2021. Available at: https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/DARZALEX-pi.pdf [Cited 2021 May 11th].
- Premkumar V, Pan S, Lentzsch S, et al. Use of daratumumab in high risk multiple myeloma: a meta-analysis. eJHaem. 2020;1(1):267–271.
- Giri S, Grimshaw A, Bal S, et al. Evaluation of daratumumab for the treatment of multiple myeloma in patients with high-risk cytogenetic factors: a systematic review and meta-analysis. JAMA Oncol. 2020;6(11):1759–1765.
- Mohan M, Weinhold N, Schinke C, et al. Daratumumab in high-risk relapsed/refractory multiple myeloma patients: adverse effect of chromosome 1q21 gain/amplification and GEP70 status on outcome. Br J Haematol. 2020;189(1):67–71.
- Nijhof IS, Casneuf T, van Velzen J, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. 2016 Aug 18;128(7):959–970.
- Sarclisa. Summary of product characteristics April 2021 [2021 Jul 28]. Available from: https://products.sanofi.us/Sarclisa/sarclisa.pdf
- Deckert J, Wetzel MC, Bartle LM, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. 2014;20(17):4574–4583.
- Martin TG, Corzo K, Chiron M, et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells. 2019;8(12):1522.
- Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23(15):4290–4300.
- Harrison SJ, Perrot A, Alegre A, et al. Subgroup analysis of ICARIA-MM study in relapsed/refractory multiple myeloma patients with high-risk cytogenetics. Br J Haematol. 2021;194(1):120–131.
- Garcìa JB, Eufemiese RA, Storti P, et al. Role of 1q21 in multiple myeloma: from pathogenesis to possible therapeutic targets. Cells. 2021;10(6).
- Spicka I, Moreau P, Martin TG, et al. Isatuximab plus carfilzomib and dexamethasone in relapsed multiple myeloma patients with high-risk cytogenetics: IKEMA subgroup analysis [Abstract 8042]. J Clin Oncol. 2021;39(15_suppl): 8042.
- Moreau P, Dimopoulos MA, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet. 2021;397(10292):2361–2371.
- Ogiya D, Liu J, Ohguchi H, et al. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: therapeutic implications. Blood. 2020;136(20):2334–2345.
- Krejcik J, Frerichs KA, Nijhof IS, et al. Monocytes and granulocytes reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res. 2017;23(24):7498–7511.
- Richardson PG, Facon T, Bensinger WI, et al. Predictive biomarkers with isatuximab plus pomalidomide and dexamethasone in relapsed/refractory multiple myeloma. Blood Cancer J. 2021;11(3):55.
- Qu X, Chen L, Qiu H, et al. Extramedullary manifestation in multiple myeloma bears high incidence of poor cytogenetic aberration and novel agents resistance. Biomed Res Int. 2015;2015:787809.
- Biran N, Malhotra J, Bagiella E, et al. Patients with newly diagnosed multiple myeloma and chromosome 1 amplification have poor outcomes despite the use of novel triplet regimens. Am J Hematol. 2014;89(6):616–620.
- Rajkumar SV. Multiple myeloma: 2020 update on diagnosis, risk-stratification and management. Am J Hematol. 2020;95(5):548–567.
- Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol. 2015;33(26):2863–2869.
- Lancman G, Tremblay D, Barley K, et al. The effect of novel therapies in high-molecular-risk multiple myeloma. Clin Adv Hematol Oncol. 2017;15(11):870–879.
- Perrot A, Lauwers-Cances V, Tournay E, et al. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J Clin Oncol. 2019;37(19):1657–1665.
- Spanoudakis E, Hu M, Naresh K, et al. Regulation of multiple myeloma survival and progression by CD1d. Blood. 2009;113(11):2498–2507.
- Ziccheddu B, Biancon G, Bagnoli F, et al. Integrative analysis of the genomic and transcriptomic landscape of double-refractory multiple myeloma. Blood Adv. 2020;4(5):830–844.
- Slomp A, Moesbergen LM, Gong JN, et al. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 2019;3(24):4202–4214.
- Wuilleme-Toumi S, Robillard N, Gomez P, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia. 2005;19(7):1248–1252.
- Bolomsky A, Vogler M, Kose MC, et al. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J Hematol Oncol. 2020;13(1):173.
- Teoh PJ, An O, Chung TH, et al. Aberrant hyperediting of the myeloma transcriptome by ADAR1 confers oncogenicity and is a marker of poor prognosis. Blood. 2018;132(12):1304–1317.
- Lazzari E, Mondala PK, Santos ND, et al. Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat Commun. 2017;8(1):1922.
- Li J, Stagg NJ, Johnston J, et al. Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell. 2017;31(3):383–395.
- Bruno B, Wasch R, Engelhardt M, et al. European myeloma network perspective on CAR T-cell therapies for multiple myeloma. Haematologica. 2021;106(8):2054–2065.
- Papoutselis M, Spanoudakis E. Navigating the role of CD1d/invariant natural killer T-cell/glycolipid immune axis in multiple myeloma evolution: therapeutic implications. Clin Lymphoma Myeloma Leuk. 2020;20(6):358–365.
- Sherbenou DW, Aftab BT, Su Y, et al. Antibody-drug conjugate targeting CD46 eliminates multiple myeloma cells. J Clin Invest. 2016;126(12):4640–4653.