
1. Introduction
Interstitial lung diseases (ILDs) include more than 100 respiratory rare diseases that affect the lung parenchyma, with a variable component of inflammation and/or fibrosis, which leads to progressive dyspnea and respiratory failure [Citation1–10]. Pulmonary fibrosis is present in several ILDs, which is irreversible by definition and may progress independently of the origin. Idiopathic pulmonary fibrosis (IPF) is the most lethal and frequent fibrotic ILD. However, many other non-IPF fibrotic ILDs may present a similar disease behavior to IPF despite the initial treatment, which is currently known as progressive pulmonary fibrosis (PPF) [Citation1,Citation2]. The most frequent symptoms and signs in IPF and other fibrotic ILDs include dyspnea, dry cough, and inspiratory crackles, and, in some cases, finger clubbing [Citation2,Citation3]. Natural history differs among patients but involves a decline in lung function that worsens quality of life and eventually leads to death. It occurs in middle-aged and older adults, and the estimated mean survival before the use of anti-fibrotic drugs in IPF was 3–5 years from diagnosis [Citation4]. The most common non-IPF fibrotic ILDs include fibrotic hypersensitivity pneumonitis (F-HP), unclassifiable ILD (uILD), connective tissue disease associated ILD (CTD-ILD), smoking related interstitial fibrosis (SRIF), and fibrotic nonspecific interstitial pneumonia (fNSIP), although less frequently other ILDs such as sarcoidosis and pneumoconiosis may also progress to the fibrotic progressive form [Citation11,Citation12] (, ). Although the advances in the diagnostic and therapeutic approaches have improved patient survival and prognosis, the delay in diagnosis remains frequent and one of the main unmet patient needs.
Figure 1. Fibrotic ILDs at risk of progression. IPF is the paradigm of fibrotic progression, sooner or later these patients progress. Non-IPF fibrotic ILD involves many ILDs with variable fibrotic component that are at risk of progression depending on different factors, including the type of ILD.
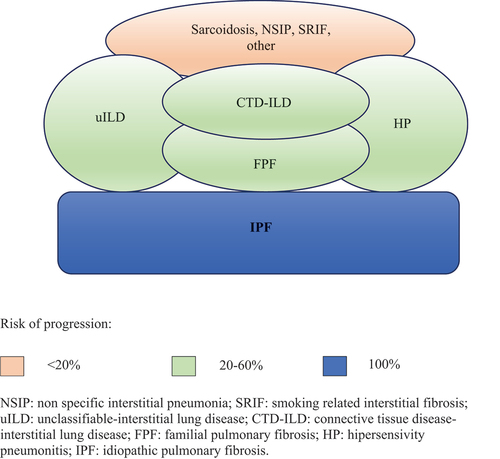
Table 1. Diagnostic criteria for identifying progressive pulmonary fibrosis (PPF) in non-IPF fibrotic ILDs.
2. Rapid diagnosis and treatment
The confident diagnosis in ILD requires a multidisciplinary committee approach that evaluates clinical, radiological, serological and, in some cases, cito-histological features [Citation1–10]. However, patients with an ILD in advanced stages are not candidate for invasive procedures due to a high risk of morbi-mortality, and the final diagnosis is usually probable or unclassifiable. On the other hand, the initial treatment depends on the type of ILD diagnosed, but other conditions such as the patient performance status and comorbidities are also considered. Therefore, patients evaluated for diagnosis after years of onset of symptoms frequently present severe fibrotic ILD and impaired quality of life, which makes the confident diagnosis and targeted treatment, impossible.
Early intervention is crucial to improve ILD prognosis. Based on three studies (n = 473), up to 18–32% of patients with non-IPF fibrotic ILDs develop PPF within 61–80 months from the onset of symptoms [Citation6,Citation13–15]. However, most of these ILDs were diagnosed once the fibrotic component was established and then, the management of the initial trigger is not affordable. The irreversible lung damage caused by fibrotic progression underscores the importance of early treatment. Late IPF diagnosis is consistently associated with worse prognosis. In a single-center prospective study, the delay in patient referral to a tertiary care center increased the mortality risk independently of age and lung function [Citation16].
Antifibrotic drugs, nintedanib and pirfenidone, have been shown to slow disease progression in a broad spectrum of IPF and PPF patients through clinical trials [Citation2,Citation17–20]. Treating IPF patients from diagnosis is desirable because disease outcome is unpredictable and fatal events such as severe acute exacerbation can appear at any stage [Citation2,Citation3,Citation21]. Importantly, real world data from a multi-center, prospective, observational registry suggest a beneficial effect of anti-fibrotic therapy on IPF survival (Hazard Ratio (HR) 0.56, 95% CI 0.34–0.92, p = 0.022) and transplant-free survival independently of baseline disease severity [Citation19]. Furthermore, non-IPF fibrotic ILD patients with criteria of PPF () also benefit from nintedanib treatment [Citation22]. Therefore, early initiation of treatment is essential. But even more important is the holistic treatment of fibrotic ILD patients. This integral and multidisciplinary therapeutic approach not only looks at improving quality of life but also to avoid risk factors for fibrotic progression such as gastro-esophageal reflux, respiratory infections, and smoking habit, and even modifying the natural history of the disease in case that the cause or inducer may be removed [Citation2,Citation3,Citation23]. In addition, early intervention could increase the efficacy of therapies such as pulmonary rehabilitation; patients with less deteriorated lung function achieve greater benefits from this non-pharmacological approach than patients with more severe disease [Citation3,Citation24].
3. The relevance of early identification
One of the main reasons for the delay in ILD diagnosis is the difficulty in identifying these types of diseases. Even in subjects with connective tissue diseases, in which ILD can be a systemic manifestation, the evaluation of the lung through high resolution computed tomography (HRCT) is often performed once respiratory symptoms are present. In case of rheumatoid arthritis (RA), ILD is one of the main causes of mortality, especially the usual interstitial pneumonia (UIP) pattern [Citation23]. Other factors associated with a higher mortality in RA-ILD are age, male sex, severity of fibrosis, and the activity and duration of RA [Citation24–28]. Therefore, anticipating RA-ILD outcome could optimize the therapeutic approach, including the timely referral to lung transplant [Citation29]. However, recent studies have demonstrated that the median time to ILD identification after appearance of respiratory symptoms in RA patients is 14.36 ± 15.3 months [Citation29]. The recommendations in the diagnosis of systemic sclerosis (SSc) include the screening of ILD through HRCT from diagnosis. Similarly, a recent Spanish consensus rheumatological-pneumological position paper has proposed to perform chest HRCT in those RA patients at higher risk of ILD (old males with smoking history and high RA activity) [Citation30]. Therefore, the early identification of ILD involvement in CTDs is currently fostered, especially in those systemic diseases that more frequently associate lung involvement.
Classically, subjects regularly exposed to inorganic or organic inhaled occupational dust such as coal, silica, asbestos, mold, or bird droppings, were recommended to be evaluated for respiratory screening. Similarly, pneumotoxic drugs, such as amiodarone or bleomycin, and thoracic radiotherapy would require an active screening of lung damage. However, the identification of ILD in these cases is also commonly preceded by clinical symptoms, probably after years of disease onset. Avoiding the exposure as soon as some radiological HRCT sign is present could decrease the induced lung damage [Citation31]. Therefore, more preventive strategies are needed to early identify those cases at risk for induced fibrotic ILD.
Finally, risk factors associated with ILD development are not only limited to those induced or associated ILD cases. Some relevant predisposition and environmental factors have been identified in idiopathic interstitial pneumonias, including IPF. The presence of family aggregation exponentially increases the risk of IPF development in first degree relatives [Citation32]. Furthermore, HRCT scan identifies subclinical ILD in relatives of sporadic IPF cases [Citation33]. Therefore, the screening of ILD in familial cases has been proposed, and it is currently implemented in some countries for the clinical practice [Citation34].
Genetic predisposing factors in adult patients (usually older than 50 years), such as MUC5B polymorphism or telomerase related gene variants, have been also associated with the development of IPF. However, using genetic information for screening ILD is more complex and difficult to implement worldwide due to technical and skills requirements. Early identification of those cases at higher risk for developing IPF or presenting early interstitial changes would avoid or prevent other modifiable factors that could increase the probability of disease development or fibrotic progression such as smoking habit, dust exposures, viral infections, and gastroesophageal reflux [Citation35].
For all of these reasons, accurate diagnosis of ILD is crucial for management and prognosis but can be challenging even for experienced clinicians. Expert multidisciplinary discussion (MDD) is considered the gold standard for ILD diagnosis [Citation2].
4. ILD patient journey: identification and referral
From the onset of symptoms to diagnosis of pulmonary fibrosis, the patient journey involves delays at each stage of the diagnostic pathway [Citation36]. The diagnostic journey usually starts with patients presenting to their primary care physicians with initial symptoms of cough or dyspnea on exertion. These nonspecific symptoms, combined with the heterogeneity and rarity of pulmonary fibrosis, as well as requirement for multiple diagnostic investigations, results in a prolonged time to diagnosis with potential delays related to patient factors and health-care systems [Citation37]. Time to diagnosis from the onset of symptoms varies depending on the studies, but the common median is 2.1 years (IQR 0.9–5.0) [Citation38]. Longer time to diagnosis is associated with worse outcomes in IPF [Citation39,Citation40], causes delayed treatment, leads to more extensive fibrosis and affects patients’ emotional well-being [Citation41]. Most of these delays are avoidable. There is a particular need to raise awareness of pulmonary fibrosis in the general population.
A European IPF Patient Charter was developed to identify unmet needs of IPF patients across Europe [Citation42]. There was an overall agreement regarding the need for an early and accurate diagnosis; the need for better access to treatment, holistic, and palliative care; and the need to raise awareness of the disease severity and its chronic nature among patients, health-care professionals, and general population [Citation42]. Similar results were obtained in an in-depth survey from European countries [Citation43]. A general complaint among IPF patients was the delay of diagnosis and the lack of detailed information delivered to the patient in a sensitive and empathetic manner [Citation42,Citation43]. Furthermore, patient knowledge and satisfaction were higher in those treated in reference centers [Citation43]. Additionally, patients reported a negative impact of IPF on the quality of life in personal autonomy, personal relationships, and financial difficulties [Citation38]. Hence, early holistic therapeutic approach would not only preserve lung function but also translate into a better quality of life for IPF patients. Further, patients’ experiences highlight the need for understandable information concerning the diagnostic tests performed, differential diagnosis, final diagnosis, and treatments as well as peer support groups. Improving several aspects of the diagnostic pathway is therefore warranted to minimize delays and improve patient satisfaction [Citation44].
5. First steps for an ILD rapid diagnosis
Although ILD may be identified in subclinical cases by incidental discovery through radiological chest images performed for another reason or by active screening in subjects at high-risk (familial screening, lung evaluation in CTD study, or preventive occupational programs), in most cases, the respiratory symptoms precede the diagnosis, and the initial assessment is usually performed in primary care centers. Therefore, for a rapid diagnosis two relevant concerns required have been worked; 1) ILD awareness among medical community at primary care centers, especially focused on the early identification by general practitioners (GPs) and/or radiologists and 2) Organization of a rapid ILD diagnostic circuit, facilitating the connection between GPs and ILD Units. If specific attention is devoted to educational interventions to GPs and radiologists on basic notions on the main ILDs and the rapid circuit is organized, the diagnostic delay may decrease.
In many countries, the primary care setting has been devoid of pre- and post-graduate educational interventions focused on basic knowledge of ILD [Citation45]. A Portuguese study analyzed the level of awareness on basic ILD diagnostic and management aspects among primary care physicians to perceive possible weaknesses. Globally, 69% of questions were correctly answered but only 21.9% of GPs considered themselves to have a satisfactory self-perceived level of knowledge on ILD. Except for sarcoidosis (p = 0.017) and some isolated questions on other ILDs, no significant differences were found between physicians younger and older than 45 years [Citation42]. IPF questions had the worst performance and only 48.5% of GPs recognized the importance of velcro-crackles at auscultation for suggesting fibrotic ILD. Typically, inspiratory crackles in fibrotic ILD patients are discontinuous, short sounds similar to those heard when slowly separating Velcro, even present in the early stages of fibrosis it may be heard at the subaxilar thoracic line of auscultation when the patient takes slow, deep breaths [Citation43]. Unlike the crackles occasionally heard in healthy elderly subjects, which disappear after several deep breaths, these fine crackles are heard consistently at the end of inspiration and originate from the basal areas of the lung [Citation43]. Training the ears to differentiate this type of crackles from those heard in patients with heart failure or traction bronchiectasis is crucial for optimizing the sensitivity and specificity of this relevant fibrotic sign. Among other relevant patient signs, finger clubbing has been described in 25–50% of patients with IPF, it is easily recognized since fingertips and fingernails become rounder but may be also present in other respiratory conditions [Citation45]. On the other hand, although the respiratory symptoms are similar to other more common diseases, some differential features may be perceived; dyspnea is progressive and, on exertion and cough, when present, is dry and may increase with exercise or laughing. Both symptoms worsen despite common treatments for bronchial diseases (bronchodilators, mucolytic or anti-histaminic drugs). Considering individual risk factors together with symptoms and signs is essential for suspecting fibrotic ILD [Citation46] (). Family aggregation, the existence of some CTD or pneumotoxic drug use, exposure to organic or inorganic inhaled dust, or a recent viral infection (including COVID19), are individually strong enough to perform a chest radiological study. Other associated risk factors that should be globally considered are age (>40 years), smoking history and the presence of gastroesophageal reflux (with or without symptoms). The initial complementary studies at the primary care center include forced spirometry, chest X-Ray and/or computed tomography (CT). Although not available in all primary care centers, the chest CT is the most sensitive complementary test for ILD and allows identification of mild interstitial changes even in subclinical subjects. The frequent use and easy availability of chest X-Ray makes this procedure the first study to perform in these patients; however, a normal chest X-Ray is not able to discriminate mild fibrotic ILD. The spirometry may show a decrease in the forced vital capacity (FVC), with a non-obstructive altered ventilatory pattern. However, the appropriate technical performance of the spirometry is out most important. Once an ILD patient is identified or suspected, the possibility of referring the patient directly to an ILD Unit would decrease the number of unnecessary complementary tests and treatments, decreasing the stress for the patient and reducing the timelines to the final diagnosis and treatment ().
Figure 2. Proposed patient referral circuit for rapid diagnosis and continuum care.
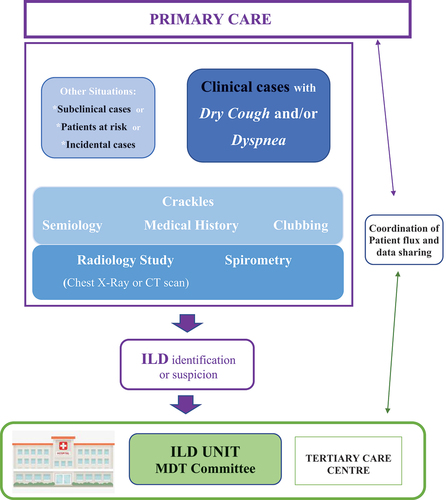
Table 2. Clinical features that help to suspect fibrotic ILDs.
6. Discussion
Primary care physicians may play a key role for improving ILD diagnosis and disease outcomes, suspecting and/or identifying ILD and referring patients to the ILD Unit [Citation47]. These ILD Units coordinate the MDD committee that involves expert disciplines including pulmonologists, radiologists, pathologists, thoracic surgeons and even rheumatologists, and/or internal medicine for evaluating ILDs in the context of a systemic disease. The discussion in these expert committees is the best way to promote communication between different specialists in order to make a quick and efficient diagnosis of ILD and early treatment [Citation48].
Early identification and timely referral of an ILD patient may modify the disease’s natural history, even saving lives in some ILDs. Thinking about the ILD option by GPs and radiologists is crucial for timely diagnosis and integral treatment, reducing risks, complementary tests and optimizing patient journey. Institutional strategies for allowing or enhancing the interaction between primary care centers and ILD centers may improve patient journey and holistic care.
Diagnostic delay is a common problem for patients with fibrotic ILD that stems from several factors including the low incidence of these rare diseases, a lack of awareness, insidious onset and nonspecific symptoms, which represents a major challenge for the suspicion and early identification [Citation46].
The time elapsed from symptom onset to the final ILD diagnosis can be troubled by delays and misdiagnosis during the evaluation of the patient by non-expert physicians that may initially suspect and treat other common respiratory diseases such as asthma or chronic obstructive pulmonary disease (COPD) [Citation49]. On the other hand, the rapid diagnostic circuit from primary care centers to ILD units may associate non-ILD cases such as bronchiectasis, infections, or heart failure. Therefore, a rapid diagnostic process could involve a minority of patients with other causes of interstitial lung infiltrates.
Prompt diagnosis of ILD is important to ensure that patients receive appropriate care and support and to enable them to be considered for lung transplantation or clinical trials. Primary care centers have a key role to play in expediting the diagnosis of ILD by referring middle-aged/elderly patients with unexplained chronic dyspnea and cough who do not meet the diagnostic criteria for other diseases to an ILD center or pulmonologist with expertise in this group of disorders [Citation46]. The best strategy for achieving this purpose is currently unknown, and probably more than one option will be possible depending on the country and even on the region. Since greater benefits are achieved with early actions in patients with ILD, it is necessary to improve early detection and management of ILD by raising awareness and educating health-care professionals and general population and placing management strategies that facilitate quick referral to specialized centers and early and accurate treatment of ILD patients [Citation50]. An effective initial working group for setting the proper strategy would include the different professionals and institutions involved, including health-care system decision-making representatives and patient-representatives from patient associations, foundations, and federations.
7. Conclusions
Early identification and timely referral of an ILD patient may modify the disease’s natural history, even saving lives in some ILDs. Thinking about the ILD option by GPs and radiologists is crucial for timely diagnosis and integral treatment, reducing risks, complementary tests and optimizing patient journey. Institutional strategies for allowing or enhancing the interaction between primary care centers and ILD centers may improve patient journey and holistic care.
Declaration of interest
G Bermudo Peloche has been paid for attending educational activities (Boehringer Ing) in the last past 3 years. M Molina-Molina has received grants and payments for scientific advice (outside this work) from Boehringer Ing, Roche, Ferrer, Chiesi, Veracyte in the past 3 years. M Roman-Rodriguez has been paid to make lectures and educational activities from many different pharmaceutical companies including AstraZeneca, Boehringer Ingelheim, Chiesi, FAES, Gebro, GSK, Menarini and Pfizer: and his institution has received grants for research from Astra Zeneca in the past 3 years. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Stanel S, Rivera-Ortega P. Present and future perspectives in early diagnosis and monitoring for progressive fibrosing interstitial lung diseases. Front Med. 2023;10:1114722. doi: 10.3389/fmed.2023.1114722
- Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205(9):e18–e47. doi: 10.1164/rccm.202202-0399ST
- National Clinical Guideline Centre (UK). Diagnosis and management of suspected idiopathic pulmonary fibrosis: idiopathic pulmonary fibrosis. London: Royal College of Physicians (UK); 2013. Nice Clinical Guidelines, n63.
- Khor YH, Ng Y, Barnes H, et al. Prognosis of idiopathic pulmonary fibrosis without anti-fibrotic therapy: a systematic review. Eur Respir Rev. 2020; 29(157):190158. doi: 10.1183/16000617.0158-2019
- Faverio P, Piluso M, De Giacomi F, et al. Progressive fibrosing interstitial lung diseases: prevalence and characterization in two Italian referral centers. Respiration. 2020;99(10):838–845. doi: 10.1159/000509556
- Guler SA, Winstone TA, Murphy D, et al. Does systemic sclerosis–associated interstitial lung disease burn out? Specific phenotypes of disease progression. Ann Am Thorac Soc. 2018;15(12):1427–1433. doi: 10.1513/AnnalsATS.201806-362OC
- Nasser M, Larrieu S, Si-Mohamed S, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. 2021;57(2):2002718. doi: 10.1183/13993003.02718-2020
- Richeldi L, Launders N, Martinez F, et al. The characterisation of interstitial lung disease multidisciplinary team meetings: a global study. ERJ Open Res. 2019;5(2):00209–2018. doi: 10.1183/23120541.00209-2018
- Simpson T, Barratt SL, Beirne P, et al. The burden of progressive fibrotic interstitial lung disease across the UK. Eur Respir J. 2021;58(1):2100221. doi: 10.1183/13993003.00221-2021
- Copeland CR, Lancaster LH. Management of progressive fibrosing interstitial lung diseases (PF-ILD). Front Med. 2021;8:743977. doi: 10.3389/fmed.2021.743977
- Rajan SK, Cottin V, Dhar R, et al. Progressive pulmonary fibrosis: an expert group consensus statement. Eur Respir J. 2023;61(3):2103187. doi: 10.1183/13993003.03187-2021
- Dhooria S, Agarwal R, Sehgal IS, et al. Spectrum of interstitial lung diseases at a tertiary center in a developing country: a study of 803 subjects. PLoS One. 2018;13(2):e0191938. doi: 10.1371/journal.pone.0191938
- Cottin V. Treatment of progressive fibrosing interstitial lung diseases: a milestone in the management of interstitial lung diseases. Eur Respir Rev. 2019;28(153):190109. doi: 10.1183/16000617.0109-2019
- Reiseter S, Gunnarsson R, MogensAaløkken T, et al. Progression and mortality of interstitial lung disease in mixed connective tissue disease: a long-term observational nationwide cohort study. Rheumatology. 2018;57(2):255–262. doi: 10.1093/rheumatology/kex077
- Zamora-Legoff JA, Krause ML, Crowson CS, et al. Progressive decline of lung function in rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheumat. 2017;69(3):542–549. doi: 10.1002/art.39971
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184(7):842–847. doi: 10.1164/rccm.201104-0668OC
- Richeldi L, Cottin V, Du Bois RM, et al. Nintedanib in patients with idiopathic pulmonary fibrosis: combined evidence from the TOMORROW and INPULSIS® trials. Respir med. 2016;113:74–79. doi: 10.1016/j.rmed.2016.02.001
- Kolb M, Richeldi L, Behr J, et al. Nintedanib in patients with idiopathic pulmonary fibrosis and preserved lung volume. Thorax. 2017;72(4):340–346. doi: 10.1136/thoraxjnl-2016-208710
- Jo HE, Glaspole I, Grainge C, et al. Baseline characteristics of idiopathic pulmonary fibrosis: analysis from the Australian idiopathic pulmonary fibrosis registry. Eur Respir J. 2017;49(2):1601592. doi: 10.1183/13993003.01592-2016
- Raman L, Stewart I, Barratt S, et al. Nintedanib for non-IPF progressive pulmonary fibrosis: 12-month outcome data from a real-world multicentre observational study. ERJ Open Res. 2023;9(2):00423–2022. doi: 10.1183/23120541.00423-2022
- Nalysnyk L, Cid-Ruzafa J, Rotella P, et al. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–361. doi: 10.1183/09059180.00002512
- Flaherty KR, Wells AU, Cottin V, et al. INBUILD trial investigators. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–1727. doi: 10.1056/NEJMoa1908681
- Kim E, Elicker BM, Maldonado F, et al. Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. 2010;35(6):1322–1328. doi: 10.1183/09031936.00092309
- Zamora- Legoff JA, Krause ML, Crowson CS, et al. Progressive decline of lung function in rheumatoid arthritis–associated interstitial lung disease. Arthritis Rheumat. 2017;69(3):542–549. doi: 10.1002/art.39971
- Dawson J. Predictors of progression of HRCT diagnosed fibrosing alveolitis in patients with rheumatoid arthritis. Ann Rheumatic Dis. 2002;61(6):517. doi: 10.1136/ard.61.6.517
- Jacob J, Hirani N, van Moorsel CHM, et al. Predicting outcomes in rheumatoid arthritis related interstitial lung disease. Eur Respir J. 2019;53(1):1800869. doi: 10.1183/13993003.00869-2018
- Cano-Jimenez E, VazquezRodriguez T, Martín-Robles I, et al. Diagnostic delay of associated interstitial lung disease increases mortality in rheumatoid arthritis. Sci Rep. 2021;11(1):9184. doi: 10.1038/s41598-021-88734-2
- Wells AU, Brown KK, Flaherty KR, et al. What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J. 2018;51(5):1800692. doi: 10.1183/13993003.00692-2018
- Michalski JE, Schwartz DA. Genetic risk factors for idiopathic pulmonary fibrosis: insights into Immunopathogenesis. J Inflamm Resp. 2021 Jan 5;13:1305–1318. doi: 10.2147/JIR.S280958
- Narváez J, Aburto M, Seoane-Mato D, et al. Criterios de cribado de la enfermedad pulmonar intersticial difusa asociada a la artritis reumatoide: propuesta de expertos basada en metodología Delphi. Reumatología Clínica. 2023;19(2):74–81. doi: 10.1016/j.reuma.2021.12.006
- Reynolds C, Feary J, Cullinan P. Occupational contributions to interstitial lung disease. Clin Chest Med. 2020;41(4):697–707. doi: 10.1016/j.ccm.2020.08.015
- García-Sancho C, Buendía-Roldán I, Fernández-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir med. 2011 Dec;105(12):1902–1907.
- Doyle TJ, Hunninghake GM, Rosas I. Subclinical interstitial lung disease: why you should care. Am J Respir Crit Care Med. 2012 Jun 1;185(11):1147–1153.
- Borie R, Kannengiesser C, Antoniou K, et al. European Respiratory Society statement on familial pulmonary fibrosis. Eur Respir J. 2023 Mar 16;61(3):2201383. doi: 10.1183/13993003.01383-2022
- Zaman T, Lee J. Risk factors for the development of idiopathic pulmonary fibrosis: a review. Curr Pulmonol Rep. 2018 Dec;7(4):118–125. doi: 10.1007/s13665-018-0210-7
- Hoyer N, Prior TS, Bendstrup E, et al. Risk factors for diagnostic delay in idiopathic pulmonary fibrosis. Respir Res. 2019;20(1):103. doi: 10.1186/s12931-019-1076-0
- Vasakova M, Mogulkoc N, Sterclova M, et al. Does timeliness of diagnosis influence survival and treatment response in idiopathic pulmonary fibrosis? Real-world results from the EMPIRE registry. Eur Respir J. 2017;50. doi: 10.1183/1393003.congress-2017.PA4880
- Lamas DJ, Kawut SM, Bagiella E, et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184(7):842–847. doi: 10.1164/rccm.201104-0668OC
- Pritchard D, Adegunsoye A, Lafond E, et al. Diagnostic test interpretation and referral delay in patients with interstitial lung disease. Respir Res. 2019;20(1):253. doi: 10.1186/s12931-019-1228-2
- Bonella F, Wijsenbeek M, Molina-Molina M, et al. European idiopathic pulmonary fibrosis patient charter: a missed opportunity. Eur Respir J. 2016;48(1): 283–284. doi: 10.1183/13993003.00604-2016
- Schoenheit G, Becattelli I, Cohen AH. Living with idiopathic pulmonary fibrosis: an in depth qualitative survey of European patients. Chron Respir Dis. 2011;8(4):225–231. doi: 10.1177/1479972311416382
- Silva M, Fernandes A, Pereira AR, et al. Awareness towards the main ILD among primary care physicians. Multidis Res Med. 2022;17:848. doi: 10.4081/mrm.2022.848
- Cottin V, Cordier JF. Velcro crackles: the key for early diagnosis of idiopathic pulmonary fibrosis? Eur Respir J. 2012;40(3):519–521. doi: 10.1183/09031936.00001612
- van der Sar IG, Jones S, Clarke DL, et al. Patient reported experiences and delays during the diagnostic pathway for pulmonary fibrosis: a multinational European survey. Front Med. 2021;8:711194. doi: 10.3389/fmed.2021.711194
- Borchers AT, Chang C, Keen CL, et al. Idiopathic pulmonary fibrosis—an epidemiological and pathological review. Clin Rev Allergy Immunol 2011; 40(2):117–134. doi: 10.1007/s12016-010-8211-5
- Spagnolo P, Ryerson CJ, Putman R, et al. Early diagnosis of fibrotic interstitial lung disease: challenges and opportunities. Lancet Respir Med. 2021;9(9):1065–1076. doi: 10.1016/S2213-2600(21)00017-5
- D Zibrak JD, Price D. Interstitial lung disease: raising the index of suspicion in primary care. 2014;24(1):14054 NPJ Prim Care Respir Med. doi: 10.1038/npjpcrm.2014.54
- Bermudo G, Suárez-Cuartin G, Rivera-Ortega P, et al. Different faces of idiopathic pulmonary fibrosis with preserved forced vital capacity. Archivos de Bronconeumología. 2022 Feb;58(2):135–141.
- Adegunsoye A. Diagnostic delay in idiopathic pulmonary fibrosis: where the rubber meets the road. Ann ATS. 2019 Mar;16(3):310–312. doi: 10.1513/AnnalsATS.201812-883ED
- Molina-Molina M, Aburto M, Acosta O, et al. Importance of early diagnosis and treatment in idiopathic pulmonary fibrosis. Expert Rev Respir Med. 2018;12(7):537–539. doi: 10.1080/17476348.2018.1472580