
ABSTRACT
Introduction: Many women seek treatment to alleviate menopausal vasomotor symptoms (VMS). Numerous women use combination compounded hormone therapy (CHT) to achieve the benefits of estrogen/progesterone for endometrial protection. TX-001HR is a combination of bioidentical 17β-estradiol (E2) and progesterone (P4) in a single capsule designed for continuous daily use to treat moderate to severe VMS.
Areas covered: This drug profile describes the efficacy and safety of 4 doses of this E2/P4 (mg/mg: 1/100, 0.5/100, 0.5/50, 0.25/50) for treating moderate to severe VMS in menopausal woman with a uterus.
Expert opinion: In REPLENISH (NCT01942668), the two highest doses of TX-001HR significantly reduced VMS frequency and severity at 4 and 12 weeks versus placebo (co-primary endpoints); all doses met the primary endpoint of endometrial safety. Rates of amenorrhea were high and improved over time; the Menopause Quality of Life and Medical Outcomes Study-Sleep instruments improved with E2/P4. TX-001HR was well tolerated and had no clinically significant impact on vital signs, metabolic or coagulation parameters, or breast safety. The combination bioidentical E2/P4 capsule (1 mg/100 mg dose was FDA-approved as Bijuva in October 2018) may provide a safe, effective, rigorously studied alternative for women with a uterus who prefer CHT for relief of VMS.
1. Introduction
It is estimated that 50–80% of menopausal women have moderate to severe vasomotor symptoms (VMS) [Citation1,Citation2]. More than half of menopausal women have been shown to consult with a healthcare provider for symptoms, with VMS of hot flashes and night sweats being the most frequent symptoms discussed [Citation3]. The Study of Women’s Health Across the Nation (SWAN) found that frequent VMS persist a median of 4.5 years after the final menstrual period [Citation4]. Current treatment guidelines for VMS in menopausal women recommend hormone therapy (HT) as first-line therapy [Citation5,Citation6]. Women with a uterus using systemic estrogen require an adequate dose or duration of a progestogen for endometrial protection from estrogen stimulation [Citation5,Citation6].
After the Women’s Health Initiative (WHI) trial identified potential risks with HT containing synthetic progestin [Citation7], an increasing demand for bioidentical HT containing estradiol (E2) and progesterone (P4) was observed [Citation8] as many women perceive bioidentical HT to be more ‘natural’ and safer [Citation9–Citation12]. Since no US Food and Drug Administration (FDA)-approved bioidentical combination products were available at that time, many women opted to take unapproved, unregulated compounded HT (CHT; commonly advertised as bioidentical). As of 2016, an estimated 33 million prescriptions were filled annually in the US for CHT [Citation8]. However, due to the lack of quality control, unestablished safety and efficacy, and low level of regulatory oversight, medical societies [Citation5,Citation6] and the FDA [Citation13] recommend against the use of CHT. TX-001HR was developed to address the demand for a well-studied, formulation of combined bioidentical E2 and bioidentical P4 in a single, oral capsule that would be reviewed and approved by regulatory authorities to ensure the safety and efficacy of the formulation as an appropriate treatment that could be prescribed for women suffering from moderate to severe VMS.
TX-001HR was studied in a phase 3, randomized, controlled study called REPLENISH, in menopausal women with a uterus. The 1 mg E2/100 mg P4 dose of TX-001HR was approved by the FDA as Bijuva (TherapeuticsMD, Boca Raton, FL) in October 2018 [Citation14]. This manuscript will review the clinical development of TX-001HR as the first combined, bioidentical, E2/P4, oral formulation for the treatment of moderate to severe VMS in menopausal women with a uterus.
1.1. Current FDA-approved treatment options
Current FDA-approved treatment options for women desiring HT include combination of synthetic hormones and off-label use of separate formulations of bioidentical E2 and bioidentical P4. The most commonly prescribed estrogens for HT are E2, conjugated equine estrogens (CEE), and synthetic conjugated estrogens, which can be delivered orally, transdermally, or vaginally [Citation5]. Meta-analysis of trials that evaluated FDA-approved estrogens found no significant difference in effectiveness between E2 and CEE in treating VMS [Citation15]. A progestogen is required for women with a uterus to prevent endometrial hyperplasia and increased risk of endometrial cancer due to chronic unopposed exposure to estrogens [Citation5]. Synthetic progestins such as norethindrone acetate, medroxyprogesterone acetate (MPA), and drospirenone can be used to antagonize the effect of estrogens on the endometrium [Citation5,Citation16]. However, these synthetic progestins have been associated with risks of breast cancer [Citation17–Citation20] and venous thromboembolism [Citation21–Citation23], while P4 has not, leading to a high demand for HT containing bioidentical P4. Women who wanted to take bioidentical P4 prior to the approval of 1 mg E2/100 mg P4 likely needed to use two separate medications with potentially two different delivery routes (whether approved or compounded). Additionally, no previous, randomized, controlled studies have demonstrated endometrial protection using a continuous combined oral or transdermal E2 in combination with P4 regimen. Studies conducted have shown that oral combination products have better adherence than taking two concurrent medications [Citation24,Citation25].
2. Formulation and pharmacokinetics
The ultimate goal of HT is to provide symptomatic relief of menopausal symptoms without increasing the risk of endometrial cancer due to estrogenic stimulation. An obvious approach to this goal was to combine E2 and P4; however creating an effective oral combination of these hormones was challenging due to differences in their structure and low aqueous solubility [Citation26], and required a lipid drug carrier. The ratio of E2 to P4 within each capsule (as much as 1:100 for the 1 mg E2/100 mg P4 dose) presented significant formulation challenges in order to maintain E2 in solution while having a homogeneous P4 suspension within the same lipid. The interactions between the aqueous gelatin of the capsule wall and its lipid interior added to the complexity of developing a stable formulation. Furthermore, TX-001HR was formulated without peanut oil [Citation27], a potential allergen and an ingredient in currently available P4 products [Citation28].
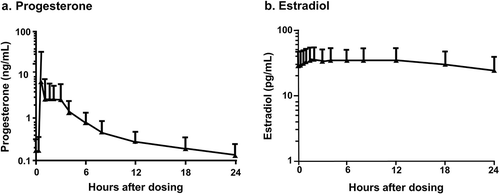
Pharmacokinetics (PK) of the 1 mg E2/100 mg P4 and 0.5 mg E2/100 mg P4 doses of TX-001HR in healthy postmenopausal women were assessed in a phase 1, open-label, parallel-group, randomized trial [Citation29]. The mean Cavg for E2 at day 7 was 38.1 pg/mL for the 1 mg E2/100 mg P4 dose and 29.2 pg/mL for the 0.5 mg E2/100 mg P4 dose. The mean Cmax for P4 ranged from 4.4 to 11.3 ng/mL and the mean P4 Cavg was 0.77 ng/mL for the 1 mg E2/100 mg P4 dose and 0.53 ng/mL for the 0.5 mg E2/100 mg P4 dose at day 7 [Citation29]. While these P4 levels are lower than luteal-phase levels in ovulatory women, these values are significantly higher than the generally undetectable baseline levels of postmenopausal women and, as demonstrated in the REPLENISH trial, are sufficient to ensure endometrial protection [Citation30]. Both TX-001HR doses were delivered consistently with steady states for P4 and E2 levels being reached within 1 week [Citation29]. Stable serum levels during steady state () for P4 were 0.12–0.18 ng/mL for the two E2/P4 formulations containing 100 mg P4; for E2, they were 28.4–34.2 pg/mL for 1 mg E2 and 22.7–24.2 pg/mL for the 0.5 mg E2 [Citation29]. Another phase 1, open-label, randomized, crossover trial found that the bioavailability of a single dose of 1 mg E2/100 mg P4 increased approximately 2-fold with food [Citation31].
Figure 1. Mean steady-state serum (a) progesterone (P4) and (b) estradiol (E2) concentrations following daily oral administration of 1 mg E2/100 mg P4 (no baseline adjustment) at day 7.
3. Efficacy and other endpoints: the REPLENISH study
3.1. Study design
The REPLENISH trial (NCT01942668; www.clinicaltrials.gov) was a multicenter, 12-month, phase 3, prospective, randomized, double-blind, placebo-controlled trial that evaluated the efficacy and safety of four daily doses of TX-001HR for the treatment of moderate to severe VMS in menopausal women with a uterus. The protocol and its amendments, participant consent form, and recruitment materials were approved by a central or local institutional review boards.
The main inclusion and exclusion criteria from REPLENISH are shown in . Women with moderate to severe hot flushes (≥7/day or ≥50/week) were included in a VMS substudy and were randomized 1:1:1:1:1 to 1 of 4 daily doses of E2/P4 (E2/P4 [mg/mg]: 1/100, 0.5/100, 0.5/50, and 0.25/50), or placebo for 12 months. Women not meeting VMS substudy eligibility were randomized 1:1:1:1 to the same active E2/P4 doses only as part of the primary safety endpoint analysis of endometrial hyperplasia.
Table 1. Main inclusion and exclusion criteria in the REPLENISH study.
3.2. Study outcomes
The primary safety endpoint of REPLENISH was the incidence of endometrial hyperplasia with TX-001HR doses at 12 months. Participants had endometrial biopsies at screening and at the end of the treatment period (12 months) or early study termination to assess endometrial histology. All biopsies were processed and evaluated centrally by two pathologists (screening biopsies) or three pathologists (post baseline biopsies). Each biopsy was classified as Category 1 (non-endometrial malignancy/non-hyperplasia), Category 2 (endometrial hyperplasia with or without atypia), or Category 3 (endometrial malignancy). The final biopsy classification required pathologist consensus. If the two primary pathologists disagreed, the third pathologist’s interpretation was incorporated and the diagnosis was based on the majority. If the three pathologists’ interpretations were different, the most severe was made the final diagnosis. The presence or absence of endometrial polyps was also noted for each biopsy.
The four co-primary efficacy endpoints were mean changes in frequency and severity of moderate to severe VMS from baseline to weeks 4 and 12 with TX-001HR doses versus placebo; changes at other time points were secondary efficacy outcomes. All participants completed diaries through week 12 to assess hot flush frequency and severity. The severity of VMS was defined as: mild (sensation of heat without sweating); moderate (sensation of heat with sweating, able to continue activity); or severe (sensation of heat with sweating, causing cessation of activity). All women completed a daily bleeding/spotting diary throughout the trial. Analysis of bleeding/spotting was based on participants who took at least one dose of study medication and who had at least one post-baseline bleeding/spotting diary entry. The percentage of women with no bleeding was calculated by cycle and for consecutive cycles. Cumulative no bleeding and cumulative amenorrhea rates were defined as the percentage of women who reported consecutive cycles of these outcomes for a given cycle time.
A secondary efficacy outcome of the study was mean changes in Clinical Global Impression (CGI) at weeks 4, 8, and 12, which was also used to determine clinical meaningful thresholds for VMS reductions [Citation32]. Women in the VMS substudy provided a CGI response by answering the following question: ‘Rate the total improvement, whether or not in your judgment it is due entirely to drug treatment. Compared to your condition at admission to the study, how much has it changed?’ Participants with CGI ratings of much improved or very much improved were considered to have a ‘clinically meaningful’ response, those with ratings of minimally improved as having a ‘minimally improved’ response, while those with ratings of no change, much worse, and very much worse were grouped into ‘no change or worse response.’ The ‘clinically meaningful’ category was used to define a clinically important difference (CID) and the ‘minimally improved’ category was used to define minimal CID (MCID) in thresholds for VMS reductions at weeks 4 and 12.
Other secondary efficacy outcomes included the Menopause-Specific Quality of Life (MENQOL) and the Medical Outcomes Study (MOS)-Sleep questionnaires completed at baseline, week 12, and months 6 and 12. MENQOL is a self-administered, validated questionnaire that assesses changes in quality of life over one month, and has 30 questions, divided into 4 domains: physical, vasomotor, psychosocial, and sexual [Citation33]. The MOS-Sleep questionnaire is a self-completed instrument that measures six dimensions of sleep over the past 4 weeks [Citation34]. A total score is calculated for the MOS-Sleep questionnaire, and item scores are averaged together to create seven additional scores (sleep disturbance, snoring, awakening from sleep short of breath or headache, sleep adequacy, sleep somnolence, sleep problems index I, and sleep problems index II) [Citation34].
Other secondary safety endpoints included recording adverse events (AEs) and treatment emergent adverse events (TEAEs) occurring through 15 days after the last study dose at each visit and assessed for severity and relationship to study medication. Serious AEs were collected through 30 days after the last study dose. Lipids, glucose, and coagulation parameters were measured at baseline, at week 12, and at months 6, 9, and 12, and vital signs were recorded at each study visit. All were summarized using descriptive statistics. Shifts in laboratory values and vital signs, as well as potentially clinically important (PCI) findings were also summarized. Participants had mammograms at screening and a study end (month 12 or early discontinuation). Mammograms were assessed using the Breast Imaging and Reporting and Database System (BI-RADS), a universal classification system [Citation35]. BI-RADS scores of 1 and 2 are defined as ‘negative’ and ‘benign’ respectively; a BI-RADS score of 3 indicates ‘probably benign’ with 6-month follow up or continued surveillance recommended and a score of 4 is ‘suspicious for malignancy’ and tissue diagnosis is recommended [Citation35].
3.3. Study demographics and disposition
A total of 1835 women were randomized to the study and took at least one capsule of study drug (safety population). The endometrial safety population had 1255 women and 766 were enrolled in the VMS substudy. The modified intent-to-treat (MITT)-VMS population consisted of 726 participants who took at least one dose of study drug, had sufficient data on VMS frequency and severity at baseline and on treatment ().
Figure 2. Subject disposition in the REPLENISH trial.
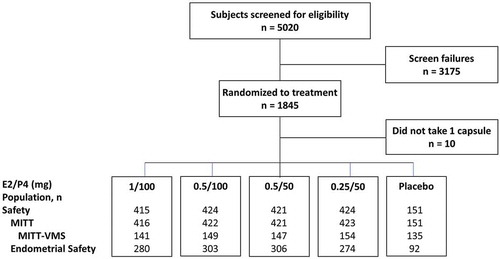
Women in the MITT-VMS population had a mean age of 55 years (range, 40–65) and a mean BMI of 27 kg/m2 (range, 14.0–34.5) at study entry (). Approximately two-thirds of the women were White (67%) and one-third were African American (31%). The frequency of moderate to severe VMS ranged from 72.1 to 77.0 per week or 10.3 to 11.0 per day. Demographics for the safety population were similar (data not shown).
Table 2. Participant demographics and baseline characteristics of the modified intent-to-treat (MITT)-VMS population.
3.4. Vasomotor symptoms (VMS)
The 1 mg E2/100 mg P4 and 0.5 mg E2/100 mg P4 doses met the four co-primary efficacy endpoints of statistically significant reductions in the frequency and severity of VMS versus placebo. Significant reductions in VMS frequency were observed by week 3 for 1 mg E2/100 mg P4, by week 4 for 0.5 mg E2/100 mg P4, and by week 6 for 0.5 mg E2/50 mg P4 () [Citation30]. All doses were significantly better than placebo in reducing the mean number of moderate to severe VMS at week 12 (p ≤ 0.002) [Citation30].
Figure 3. Weekly reduction in VMS (a) frequency and (b) severity in MITT-VMS population. For (a) p < 0.05 versus placebo from *weeks 3–12; †weeks 4–12; ‡weeks 6–12; for (b) p < 0.05 versus placebo from *weeks 3–12; †weeks 7, 9–12; ‡weeks 6, 7, 9. Weekly severity score = [(number of mild hot flushes for 7 days) x 1 + (number of moderate hot flushes for 7 days) x 2 + (number of severe hot flushes for 7 days) x 3]/(total number of mild, moderate and severe hot flushes over 7 days). E2: 17β-estradiol; MITT: modified intent-to-treat; P4: progesterone; VMS: vasomotor symptoms. Reprinted with permission from Lobo et al [Citation30].
![Figure 3. Weekly reduction in VMS (a) frequency and (b) severity in MITT-VMS population. For (a) p < 0.05 versus placebo from *weeks 3–12; †weeks 4–12; ‡weeks 6–12; for (b) p < 0.05 versus placebo from *weeks 3–12; †weeks 7, 9–12; ‡weeks 6, 7, 9. Weekly severity score = [(number of mild hot flushes for 7 days) x 1 + (number of moderate hot flushes for 7 days) x 2 + (number of severe hot flushes for 7 days) x 3]/(total number of mild, moderate and severe hot flushes over 7 days). E2: 17β-estradiol; MITT: modified intent-to-treat; P4: progesterone; VMS: vasomotor symptoms. Reprinted with permission from Lobo et al [Citation30].](/cms/asset/ebd68eba-5c26-48b8-ab84-cdaea1876dc1/ierj_a_1637731_f0003_oc.jpg)
The 1 mg E2/100 mg P4 and 0.5 mg E2/100 mg P4 doses also significantly reduced the severity of VMS by week 3 and this was maintained through week 12. Women taking 0.5 mg E2/50 mg P4 had significantly reduced VMS severity at week 7, and then at weeks 9–12. The lowest dose (0.25 mg E2/50 mg P4) resulted in inconsistent improvement in VMS severity, which was expected () [Citation30].
3.5. Clinical global impression (CGI)
Significantly more women taking TX-001HR felt their symptoms were ‘much or very much improved’ at week 4 (50–63%) and week 12 (73–82%) than those taking placebo (33% at week 4, 53% at week 12; all, p < 0.01) [Citation32]. At week 4, the MCID and CID thresholds were defined as weekly reductions in moderate to severe VMS frequency of ≥15 and ≥36, respectively [Citation32]. At week 4, significantly more women taking TX-001HR met the MCID (75–85%) and CID (46–59%) thresholds than with placebo (64% and 33%, respectively; all, p < 0.05) [Citation32]. At week 12, weekly reductions in moderate to severe VMS frequency were ≥25 for the MCID and ≥39 for the CID. All 4 doses of TX-001HR had significantly more women who met the MCID (82–88%) and CID (68–73%) versus placebo (69% and 52%, respectively) at week 12 (all, p < 0.05) [Citation32].
3.6. Menopause-specific quality of life (MENQOL) questionnaire
Women in the MITT-VMS population had significantly greater mean improvements in the MENQOL overall score at 12 weeks with all E2/P4 doses versus placebo (all, p < 0.05; ) [Citation36]. The MENQOL overall scores also significantly improved versus placebo at months 6 and 12 for most doses [Citation36]. The MENQOL vasomotor domain scores also significantly improved from baseline in all E2/P4 groups versus placebo at all time points (all, p < 0.01; )) [Citation36]. Further, all E2/P4 doses improved hot flushes, night sweats, and sweating scores within MENQOL.
Figure 4. Mean reduction from baseline in Menopause-specific Quality of Life (MENQOL) (a) overall and (b) vasomotor domain scores in MITT-VMS population and in Medical Outcome Study (MOS)-Sleep (c) total and (d) Sleep Problems Index I scores in MITT population. *p < 0.05; †p < 0.01; ‡p < 0.001 vs placebo. Sleep Problems Index I based on the following questions: How often during the past 4 weeks did you: Get enough sleep to feel rested upon waking; Awaken short of breath or with a headache; Have trouble falling asleep; Awaken and have trouble falling asleep again; Have trouble staying awake during the day; Get the amount of sleep you needed? E2, 17β-estradiol; MITT, modified intent-to-treat; P4, progesterone; VMS: vasomotor symptoms. Figure 4(a,b) adapted with permission from Simon et al [Citation36] Figure 4(c,d) adapted with permission from Kagan et al [Citation37].
![Figure 4. Mean reduction from baseline in Menopause-specific Quality of Life (MENQOL) (a) overall and (b) vasomotor domain scores in MITT-VMS population and in Medical Outcome Study (MOS)-Sleep (c) total and (d) Sleep Problems Index I scores in MITT population. *p < 0.05; †p < 0.01; ‡p < 0.001 vs placebo. Sleep Problems Index I based on the following questions: How often during the past 4 weeks did you: Get enough sleep to feel rested upon waking; Awaken short of breath or with a headache; Have trouble falling asleep; Awaken and have trouble falling asleep again; Have trouble staying awake during the day; Get the amount of sleep you needed? E2, 17β-estradiol; MITT, modified intent-to-treat; P4, progesterone; VMS: vasomotor symptoms. Figure 4(a,b) adapted with permission from Simon et al [Citation36] Figure 4(c,d) adapted with permission from Kagan et al [Citation37].](/cms/asset/bcc83a26-7cc5-42a7-84a7-24146c504b44/ierj_a_1637731_f0004_oc.jpg)
The MITT population had similar improvement in MENQOL overall and vasomotor domain scores (all, p < 0.05). One of the MENQOL questions addresses vaginal dryness during intercourse. Significant improvements in vaginal dryness were seen at week 12 with most doses of E2/P4 versus placebo in women who were age 55 and older (all, p < 0.01). No consistent significant differences between the E2/P4 groups and placebo were observed for the physical, sexual, and psychosocial domains of MENQOL in either the MITT or MITT-VMS population [Citation36].
3.7. Medical outcomes study (MOS)-Sleep questionnaire
There was significantly more improvement in the MOS-Sleep total score at week 12 among women taking the 1 mg E2/100 mg P4, 0.5 mg E2/100 mg P4, and 0.5 mg E2/50 mg P4 compared with those taking placebo (all, p < 0.05) [Citation37]. All doses of TX-001HR improved the MOS-Sleep total score at months 6 and 12 significantly more than placebo ()). All doses of TX-001HR significantly improved the Sleep Problems Index I subscale from baseline to week 12 (all, p < 0.05) and these improvements were maintained to months 6 (all, p < 0.01) and 12 (all, p ≤ 0.001) ()) [Citation37]. Women taking the 0.5 mg E2/100 mg P4 and 0.5 mg E2/50 mg P4 doses also had significant improvement on the MOS-Sleep somnolence subscale at month 12 compared with those taking placebo [Citation37]. Lastly, while drowsiness has been reported as a side effect of P4 [Citation28], few women in the safety population reported somnolence as a TEAE (TX-001HR doses: 0.2%–1.2%; placebo: 0%) [Citation37].
4. Safety and tolerability
4.1. Overall safety
A similar number of participants in the E2/P4 groups reported at least one TEAE (68–72%), while 52% of women who took placebo reported a TEAE [Citation30]. Most TEAEs in all groups were mild to moderate. There were 110 (6.0%) women who experienced at least one severe TEAE: 26 (6.3%) in 1 mg E2/100 mg P4 group, 30 (7.1%) in 0.5 mg E2/100 mg P4, 26 (6.2%) in 0.5 mg E2/50 mg P4, 24 (5.7%) in 0.25 mg E2/50 mg P4, and 4 (2.6%) in placebo. Breast tenderness was the most common related TEAE reported in the study (); most AEs were considered mild (72 of 96 [75%]); 22 (23%) were considered moderate and 2 (2%) were considered severe [Citation30]. Serious AEs were consistent with what would be expected in women of similar age, health, and menopausal status. Treatment-related serious AEs () include one venous thrombotic event in a woman with a family history of deep vein thrombosis (DVT) and six cases of breast cancer.
Table 3. Treatment-related TEAEs occurring in ≥3% in any treatment arm and more commonly than placebo and treatment-related serious AEs (safety population).
4.2. Endometrial safety
All E2/P4 doses met the primary safety endpoint for endometrial hyperplasia with an incidence rate of ≤1% with an upper limit of the 1-sided 95% confidence interval (CI) being ≤4% [Citation30], as recommended by the FDA [Citation38]. The incidence of proliferative endometrium at month 12 ranged from 0.3% (0.5 mg E2/50 mg P4) to 2.9% (1 mg E2/100 mg P4); no cases were observed in the placebo group [Citation30]. No endometrial malignancies were reported during the study [Citation30].
4.3. Bleeding and spotting
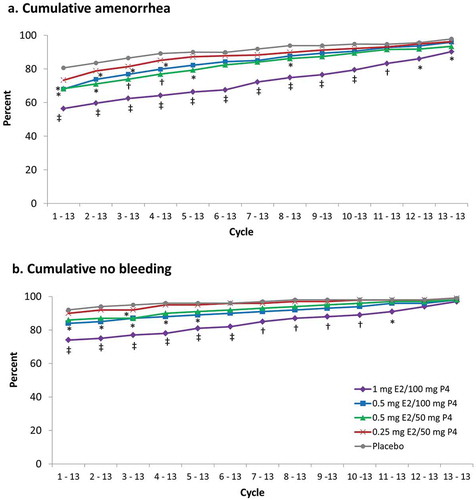
Cumulative amenorrhea rates from cycle 1 to 13 ranged from 56.1% (1 mg E2/100 mg P4 group, p < 0.001 versus placebo) to 73.1% in the 0.25 mg E2/50 mg P4 group (). The 1-year amenorrhea rates were high for all groups (≥90%). Similar results were observed for cumulative no bleeding (). There was no difference in rates of no bleeding at cycle 13 between the treatment groups and placebo, all were >97% [Citation30].
Figure 5. Cumulative (a) amenorrhea and (b) no bleeding in safety population. *p < 0.05; †p ≤ 0.01; ‡p < 0.001 vs placebo. Cycles are 28 days in length. E2, 17β-estradiol; P4, progesterone.
4.4. Breast safety
The rates of abnormal mammograms at study end were similar between all TX-001HR doses (range, 1.7–3.7%) and placebo (3.1%) [Citation39]. A total of 6 women who took TX-001HR experienced an AE of breast cancer during the study (incidence 6/1684 = 0.36%: 2 [0.5%] with 1.0 mg/100 mg, 2 [0.5%] with 0.5 mg/100 mg, 1 [0.2%] with 0.5 mg/50 mg, 1 [0.2%] with 0.25 mg/50 mg, and 0% with placebo).
4.5. Cardiovascular disease
A serious treatment-related AE of VTE occurred during the study in a woman taking 0.5 mg E2/50 mg P4; this woman had a family history of DVT () [Citation40]. Three cases of coronary heart disease were also reported. One woman (0.5 mg E2/50 mg P4) had unstable angina, one (1 mg E2/100 mg P4) had angina and coronary artery disease, and one had stress cardiomyopathy; none of these were considered related to treatment [Citation40].
4.6. Lipids and glucose
There were no clinically meaningful changes in mean levels for total cholesterol, LDL-cholesterol, or triglycerides [Citation30]. One woman (1 mg E2/100 mg P4) experienced an AE of increased blood glucose that led to study discontinuation, otherwise mean fasting glucose levels at 12 months had no clinically significant change from baseline.
4.7. Coagulation factors
There were no clinically meaningful changes in any coagulation parameters evaluated. Antithrombin activity, factor XIV and protein S levels tended to decrease from baseline, but these decreases were not clinically significant. Likewise, mean activated partial thromboplastin time, prothrombin time, fibrinogen levels and prothrombin international normalized ratio did not change by 12 months [Citation40].
4.8. Vital signs
Systolic blood pressure (BP), diastolic BP and body weight did not have clinically meaningful changes with E2/P4 from baseline to month 12 [Citation41]. PCI changes with E2/P4 were observed in 24 (2.0%) women for weight change (change of ≥15% and ≥20 lbs), 9 (0.8%) for systolic BP (change of ≥20 mm Hg to above ≥160 mm Hg or below ≤90 mg Hg), and 35 (3.0%) for diastolic BP (change of ≥15 mm Hg to above ≥90 mm Hg or below ≤60 mg Hg). No overall increase in weight was observed with E2/P4 and few women reported weight gain (1.4–2.6%) as a treatment-related TEAE. Hypertension as a treatment-related TEAE was also low with E2/P4 (0.2–1.2%). Discontinuation rates were <1% for weight gain and hypertension in all treatment groups [Citation41].
5. Conclusions
The first, single capsule of combined E2/P4 was evaluated in the randomized, placebo-controlled, double-blind, phase 3 REPLENISH trial, in which the 1 mg E2/100 mg P4 and 0.5 mg E2/100 mg P4 doses met the four co-primary efficacy endpoints while providing endometrial safety with up to one year of use. Serum levels of E2 achieved were stable and adequate for VMS relief, and importantly, P4 levels protected from estrogen-induced endometrial stimulation. The reductions in VMS frequency were also clinically meaningful as per the CGI analysis and positive effects on menopause-related quality of life and sleep parameters were found. Women using TX-001HR experienced low levels of bleeding and spotting with high rates of amenorrhea that improved with continued use. TX-001HR was well tolerated and had no clinically significant impact on lipids, glucose, or coagulation parameters. TX-001HR was studied in REPLENISH, a randomized, controlled trial, in which safety and efficacy were evaluated. The 1 mg E2/100 mg P4 dose was approved by FDA in October 2018 for the treatment of moderate to severe VMS, and provides an alternative to CHT, for which safety and efficacy have not been rigorously studied and proven.
6. Expert opinion
Given the widespread use of CHT by menopausal women to alleviate VMS, the newly approved, single-capsule E2/P4 (1 mg E2/100 mg P4) formulation may be a safe and effective treatment option given the desire of many women to use bioidentical hormones. Efficacy, safety, and PK data reviewed here show that TX-001HR met both the VMS efficacy and endometrial safety outcomes with adequate levels of E2 and P4 achieved. In addition to meeting statistical significance for reducing the frequency and severity of VMS, women found the improvements in VMS while taking E2/P4 to be clinically meaningful, as supported by the CGI and MENQOL. Sleep disruption, common in menopausal women with VMS, also improved. Rigorous data are in contrast to CHT, for which efficacy and safety have not been rigorously studied, the critical ratio of E2 to P4 is unknown, and cases of endometrial hyperplasia/cancer have been reported [Citation42–Citation44]. Endometrial protection from E2 with continuous P4 shown in this randomized, controlled trial is noteworthy as REPLENISH was the first rigorous trial to demonstrate this. Endometrial safety data from the phase 3 REPLENISH study may help guide clinicians regarding the continuous use of E2/P4 for their patients with moderate to severe VMS, as 100 mg P4 is shown to adequately protect the endometrium with 1 mg of E2.
Overall, no unexpected safety signals were identified, and the safety and tolerability of E2/P4 capsules were comparable to transdermal HT formulations [Citation45,Citation46]. Additionally, TX-001HR had no clinically meaningful impact on lipids, glucose, coagulation parameters, liver function tests, systolic or diastolic BP, or weight. Mean values for lipids and coagulation factors also remained within the normal limits. This is in contrast to the triglyceride increase observed with all CEE-containing regimens evaluated in the PEPI trial [Citation47]. While the lipid carrier required for formulating E2/P4 in the same capsule may contribute to the lack of impact on lipid and coagulation parameters, further evidence would be needed to confirm this hypothesis.
Epidemiological studies show that HT containing progesterone does not increase the risk of breast cancer as has been shown for some synthetic progestins [Citation17–Citation20]. The REPLENISH study found low rates of abnormal mammograms in TX-001HR users; these low rates were similar to placebo, and consistent with the incidence in US screening mammograms (5–6%) [Citation48]. The rate of abnormal mammograms was also lower than the 9.4% observed after one year with CEE+MPA in the WHI [Citation49]. Additionally, the breast cancer rate in REPLENISH (0.36%) after one year was comparable to that from the Surveillance, Epidemiology, and End Results (SEER) data (0.29%) [Citation50]. In contrast, HT containing synthetic progestogen has been associated with an increased incidence of abnormal mammograms and breast cancer [Citation18,Citation49,Citation51].
Progesterone has not been associated with an increased risk for VTE, in contrast to other synthetic progestins [Citation21–Citation23]. One VTE occurred in the REPLENISH trial in a woman who had a family history of DVT [Citation40]. The VTE rate in REPLENISH (1/1684) compares favorably with that from the WHI (3.5 per 1000 person-years for CEE+MPA and 1.7 per 1000 for placebo) [Citation52]. A population-based, case-control study reported a greater risk of VTE associated with current use of oral CEE than with current oral E2 use (OR 2.08; 95% CI 1.02–4.27, p = 0.045) [Citation53]. The French ESTHER (Estrogen and THromboEmbolism Risk) Study, a case-control study of VTE among postmenopausal women, suggested micronized P4 may be safe with respect to thrombotic risk [Citation54].
Few cardiovascular AEs were observed in the REPLENISH trial, and similar to VTE, differences in HT formulations have been reported. A summary of two studies of younger menopausal women (ages 50–59) reported an annual estimate of 2–3/1000 coronary heart disease (CHD) events/year in US women with placebo group, and had no CHD events in the HT groups (CEE alone or CEE+MPA) [Citation55]. Myocardial infarction incidence trended higher (p = NS) with CEE vs estradiol (OR 1.87, 95% CI 0.91–3.84, p = 0.09) in this case-control study [Citation53].
Bleeding/spotting with HT is an important tolerability issue and a frequent reason for HT discontinuation. Cumulative amenorrhea rates from cycle 1 to 13 in the REPLENISH trial improved over time, and less bleeding was reported with E2/P4 (56% to 73%) than reported separately with currently available CEE/MPA, E2/norethisterone acetate, and E2/drospirenone (23% to 49% [Citation56–Citation58]). Women may be more satisfied and discontinue E2/P4 less than other HT formulations, based on its bleeding profile. Convenience in a continuous regimen of one capsule may also enhance adherence, which may also improve bleeding as well as efficacy.
Advances in the field will impact the way health care providers prescribe menopausal HT, including guidelines for HT prescribing. Some women and health care providers are wary of taking and prescribing HT, respectively, since publication of the WHI results. In turn, many women prefer and are prescribed a bioidentical CHT, not rigorously reviewed by the FDA. The 1 mg E2/100 mg P4 dose described here was approved by the FDA in October 2018 and as such, is an approved bioidentical HT that can be an option for women looking for a more ‘natural’ HT. Further research will provide evidence for the safety of taking E2/P4, and with corroborated safety, increased use of this approved HT by postmenopausal women will likely occur in the upcoming years such that fewer women seeking VMS treatment will be exposed to potential risks with compounded bioidentical HT.
Physicians should be able to feel confident that their patients are getting the medications and doses they prescribe, an assurance that is difficult to achieve with CHT. Women deserve therapies that are safe, effective, and provide clinically meaningful treatment of symptoms with maximum convenience. The FDA-approved single, oral capsule of 1 mg E2/100 mg P4 for continuous use, should provide the qualities that patients and their health care professionals expect from a VMS treatment, especially for those who prefer to use or prescribe bioidentical HT [Citation14].
Article Highlights
A combined, bioidentical, oral, 17β-estradiol (E2; 1 mg)/progesterone (P4; 100 mg) capsule was approved for the treatment of moderate to severe vasomotor symptoms in postmenopausal women with a uterus
This E2/P4 formulation is the first time the two bioidentical hormones have been combined in one capsule due to biochemical challenges
The REPLENISH study found that combined E2/P4 statistically and clinically significantly improved menopausal vasomotor symptoms
Continuous use of P4 contained in the TX-001HR formulation was shown to protect the endometrium from estradiol stimulation in the randomized, controlled, phase 3, REPLENISH trial; these data cannot be transferred to other E2 + P4 products
Combined E2/P4 (TX-001HR) had a good uterine bleeding profile that improved over time
Various doses of E2/P4 significantly improved quality of life and sleep outcomes in postmenopausal women
Declaration of interest
DF Archer serves as a consultant for Abbvie, Actavis, Agile Therapeutics, Bayer Healthcare, Endoceutics, Exeltis, InnovaGyn, Merck, Pfizer, Radius Health, Sermonix, Shionogi, Teva Women’s Healthcare, and TherapeuticsMD; and has received research support from Actavis, Bayer Healthcare, Endoceutics, Glenmark, Merck, Radius Health, Shionogi, and TherapeuticsMD. B Bernick and S Mirkin are employees of TherapeuticsMD with stock/stock options. B Bernick is also a Board member of TherapeuticsMD.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
The authors have disclosed to the journal editor that a scientific accuracy check of the paper was carried out by the sponsor.
Acknowledgments
The authors appreciate the medical writing assistance provided by Elizabeth Jennison, MD and Kathleen Ohleth, PhD of Precise Publications, LLC.
Additional information
Funding
References
- Woods NF, Mitchell ES. Symptoms during the perimenopause: prevalence, severity, trajectory, and significance in women’s lives. Am J Med. 2005;118(Suppl 12B):14–24.
- Gold EB, Colvin A, Avis N, et al. Longitudinal analysis of the association between vasomotor symptoms and race/ethnicity across the menopausal transition: study of women’s health across the nation. Am J Public Health. 2006;96(7):1226–1235.
- Williams RE, Kalilani L, DiBenedetti DB, et al. Healthcare seeking and treatment for menopausal symptoms in the United States. Maturitas. 2007;58(4):348–358.
- Avis NE, Crawford SL, Greendale G, et al. Duration of menopausal vasomotor symptoms over the menopause transition. JAMA Intern Med. 2015;175(4):531–539.
- [No authors listed]. The 2017 hormone therapy position statement of The North American Menopause Society. Menopause. 2017; 24(7): 728–753.
- Baber RJ, Panay N, Fenton A, et al. 2016 IMS recommendations on women’s midlife health and menopause hormone therapy. Climacteric. 2016;19(2):109–150.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the women’s health initiative randomized controlled trial. JAMA. 2002;288(3):321–333.
- Pinkerton J, Constantine G. Compounded non-FDA‒approved menopausal hormone therapy prescriptions have increased: results of a pharmacy survey. Menopause. 2016;23(4):359–367.
- MacLennan AH, Gill TK, Broadbent JL, et al. Continuing decline in hormone therapy use: population trends over 17 years. Climacteric. 2009;12(2):122–130.
- American College of Obstetricians and Gynecologists. Compounded bioidentical menopausal hormone therapy. Fertil Steril. 2012;98(2):308–312.
- Iftikhar S, Shuster LT, Johnson RE, et al. Use of bioidentical compounded hormones for menopausal concerns: cross-sectional survey in an academic menopause center. J Womens Health (Larchmt). 2011;20(4):559–565.
- Boothby LA, Doering PL, Kipersztok S. Bioidentical hormone therapy: A review. Menopause. 2004;11(3):356–367.
- US Food and Drug Administration. FDA takes action against compounded menopause hormone therapy drugs; [cited 2015 Aug 11]. Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116832.htm
- BijuvaTM (estradiol and progesterone) capsules, for oral use Prescribing Information. Boca Raton, FL: TherapeuticsMD; 2018.
- Gaudard AM, Silva de Souza S, Puga ME, et al. Bioidentical hormones for women with vasomotor symptoms. Cochrane Database Syst Rev. 2016;8:CD010407.
- Mirkin S. Evidence on the use of progesterone in menopausal hormone therapy. Climacteric. 2018;21(4):346–354.
- Fournier A, Berrino F, Riboli E, et al. Breast cancer risk in relation to different types of hormone replacement therapy in the E3N-EPIC cohort. Int J Cancer. 2005;114(3):448–454.
- Fournier A, Berrino F, Clavel-Chapelon F. Unequal risks for breast cancer associated with different hormone replacement therapies: results from the E3N cohort study. Breast Cancer Res Treat. 2008;107(1):103–111.
- Cordina-Duverger E, Truong T, Anger A, et al. Risk of breast cancer by type of menopausal hormone therapy: a case-control study among post-menopausal women in France. PLoS One. 2013;8(11):e78016.
- Yang Z, Hu Y, Zhang J, et al. Estradiol therapy and breast cancer risk in perimenopausal and postmenopausal women: a systematic review and meta-analysis. Gynecol Endocrinol. 2017;33(2):87–92.
- Canonico M, Oger E, Plu-Bureau G, et al. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation. 2007;115(7):840–845.
- Canonico M, Fournier A, Carcaillon L, et al. Postmenopausal hormone therapy and risk of idiopathic venous thromboembolism: results from the E3N cohort study. Arterioscler Thromb Vasc Biol. 2010;30(2):340–345.
- Olie V, Plu-Bureau G, Conard J, et al. Hormone therapy and recurrence of venous thromboembolism among postmenopausal women. Menopause. 2011;18(5):488–493.
- Sutton SS, Hardin JW, Bramley TJ, et al. Single- versus multiple-tablet HIV regimens: adherence and hospitalization risks. Am J Manag Care. 2016;22(4):242–248.
- Coca A, Agabiti-Rosei E, Cifkova R, et al. The polypill in cardiovascular prevention: evidence, limitations and perspective - position paper of the European Society of Hypertension. J Hypertens. 2017;35(8):1546–1553.
- Pickar JH, Bon C, Amadio JM, et al. Pharmacokinetics of the first combination 17beta-estradiol/progesterone capsule in clinical development for menopausal hormone therapy. Menopause. 2015;22(12):1308–1316.
- Mirkin S, Amadio JM, Bernick BA, et al. 17β-Estradiol and natural progesterone for menopausal hormone therapy: REPLENISH phase 3 study design of a combination capsule and evidence review. Maturitas. 2015;81(1):28–35.
- Prometrium® (Progesterone, USP) Prescribing Information. Catalent Pharma Solutions. St. Petersburg, FL; 2011.
- Lobo RA, Liu J, Stanczyk FZ, et al. Estradiol and progesterone bioavailability for moderate to severe vasomotor symptom treatment and endometrial protection with the continuous-combined regimen of TX-001HR (oral estradiol and progesterone capsules). Menopause. 2019; 26(7):720–727.
- Lobo RA, Archer DF, Kagan R, et al. A 17β-estradiol-progesterone oral capsule for vasomotor symptoms in postmenopausal women: a randomized controlled trial. Obstet Gynecol. 2018;132(1):161–170.
- Lobo RA, Stanczyk FZ, Constantine G, et al. Progesterone bioavailability for preventing endometrial stimulation with a continuous-combined regimen of TX-001HR (oral estradiol and micronized progesterone capsules). Presented at IMS 16 World Congress on Menopause; 2018, June 6–9, 2018, Vancouver, Canada. Climacteric.
- Constantine GD, Revicki DA, Kagan R, et al. Evaluation of clinical meaningfulness of estrogen plus progesterone oral capsule (TX-001HR) on moderate to severe vasomotor symptoms. Menopause. 2019;26(5):513–519.
- Hilditch JR, Lewis J, Peter A, et al. A menopause-specific quality of life questionnaire: development and psychometric properties. Maturitas. 1996;24:161–175.
- Spritzer KL, Hays RD MOS Sleep Scale: A manual for use and scoring; 2003 November.
- American College of Radiology. ACR practice parameter for the performance of ultrasound-guided precutaneous breast interventional procedures; 2016 [cited 2018 Jan 23]. Available from: https://www.acr.org/-/media/ACR/Files/Practice-Parameters/US-GuidedBreast.pdf
- Simon JA, Kaunitz A, Kroll R, et al. Oral 17β-estradiol-progesterone (TX-001HR) and quality of life in postmenopausal women with vasomotor symptoms. Menopause. 2019;26(5):506–512.
- Kagan R, Constantine G, Kaunitz AM, et al. Improvement in sleep outcomes with a 17β-estradiol-progesterone oral capsule (TX-001HR) for postmenopausal women. Menopause. 2019;26(6):622–628.
- US Department of Health and Human Services (FDA). Draft guidance for industry: estrogen and estrogen/progestin drug products to treat vasomotor symptoms and vulvar and vaginal atrophy symptoms–recommendations for clinical evaluation; January 2003. [cited 2018 Dec 21]. Available from: http://www.fda.gov/downloads/Drugs/DrugSafety/informationbyDrugClass/UCM135338.pdf
- Archer DF, Pickar JH, Graham S, et al. Incidence of abnormal mammograms with oral, combined 17β-estradiol and progesterone capsules. Endocr Rev. 2018;39:2. Supplement 1.
- Lobo RA, Liu J, Kaunitz AM, et al. Effects of single-capsule 17β-estradiol/progesterone (TX-001HR) on metabolic parameters and cardiovascular outcomes in menopausal women of the REPLENISH trial. Menopause. 2018;25(12):1484–1485.
- Archer DF, Pickar JH, Graham S, et al. Effects of single-capsule 17β-estradiol/progesterone (TX-001HR) on weight and blood pressure in menopausal women of the REPLENISH trial. Menopause. 2018;25(12):1496–1497.
- Eden JA, Hacker NF, Fortune M. Three cases of endometrial cancer associated with “bioidentical” hormone replacement therapy. Med J Aust. 2007;187(4):244–245.
- Davis R, Batur P, Thacker HL. Risks and effectiveness of compounded bioidentical hormone therapy: a case series. Journal of Women's Health (Larchmt). 2014;23(8):642–648.
- Dezman VL, Gersak MZ, Gersak K. 2015. Two case of atypical endometrial hyperplasia associated with “bioidentical” hormone replacement therapy: IGCS-0084 Uterine Cancer, including sarcoma. Int J Gynecol Cancer. 2015;25(Suppl 1):71.
- Climara Pro® (estradiol/Levonorgestrel transdermal system) Prescribing Information. 3M Drug Delivery Systems. Northridge, CA; 2007.
- CombiPatch® (estradiol/norethindrone acetate transdermal system) Prescribing Information. Novartis Pharmaceuticals Corporation. East Hanover, NJ; 2005.
- Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) trial. JAMA. 1995;273(3):199–208.
- Monticciolo DL, Caplan LS. The American College of Radiology’s BI-RADS 3 classification in a nationwide screening program: current assessment and comparison with earlier use. Breast J. 2004;10(2):106–110.
- Chlebowski RT, Hendrix SL, Langer RD, et al. Influence of estrogen plus progestin on breast cancer and mammography in healthy postmenopausal women: the women’s health initiative randomized trial. JAMA. 2003;289(24):3243–3253.
- National Cancer Institute. SEER cancer statistics review 1975-2014; cancer of the female breast; [cited 2017 Jul 20]. Available from: https://seer.cancer.gov/csr/1975_2014/results_merged/sect_04_breast.pdf
- Banks E, Reeves G, Beral V, et al. Hormone replacement therapy and false positive recall in the million women study: patterns of use, hormonal constituents and consistency of effect. Breast Cancer Res. 2006;8(1):R8.
- Cushman M, Kuller LH, Prentice R, et al. Estrogen plus progestin and risk of venous thrombosis. JAMA. 2004;292(13):1573–1580.
- Smith NL, Blondon M, Wiggins KL, et al. Lower risk of cardiovascular events in postmenopausal women taking oral estradiol compared with oral conjugated equine estrogens. JAMA Intern Med. 2014;174(1):25–31.
- Canonico M, Plu-Bureau G, PY S. Progestogens and venous thromboembolism among postmenopausal women using hormone therapy. Maturitas. 2011;70(4):354–360.
- Lobo RA. Evaluation of cardiovascular event rates with hormone therapy in healthy, early postmenopausal women: results from 2 large clinical trials. Arch Intern Med. 2004;164(5):482–484.
- Prempro™/Premphase® (conjugated estrogens/medroxyprogesterone acetate tablets) Prescribing Information. Wyeth Pharmaceuticals Inc. Philadelphia, PA; 2008.
- Activella® (estradiol/norethindrone acetate) tablets. Novo Nordisk Inc. Princeton, NJ; 2006.
- Angeliq® (drospirenone and estradiol) tablets, for oral use Prescribing Information. Bayer Healthcare. Whippany, NJ; 2005.