
ABSTRACT
Introduction
Cannabis is an increasingly popular recreational and medicinal drug in the USA. While cannabis is still a Schedule 1 drug federally, many states have lifted the ban on its use. With its increased usage, there is an increased possibility for potential drug-drug interactions (DDI) that may occur with concomitant use of cannabis and pharmaceuticals.
Area covered
This review focuses on the current knowledge of cannabis induced DDI, with a focus on pharmacokinetic DDI arising from enzyme inhibition or induction. Phase I and phase II drug metabolizing enzymes, specifically cytochrome P450s, carboxylesterases, and uridine-5’-diphosphoglucuronosyltransferases, have historically been the focus of research in this field, with much of the current knowledge of the potential for cannabis to induce DDI within these families of enzymes coming from in vitro enzyme inhibition studies. Together with a limited number of in vivo clinical studies and in silico investigations, current research suggests that cannabis exhibits the potential to induce DDI under certain circumstances.
Expert opinion
Based upon the current literature, there is a strong potential for cannabis-induced DDI among major drug-metabolizing enzymes.
1. Introduction
Since ancient times, cannabis has been used for a variety of different health conditions and it continues to be one of the most widely used psychoactive substances both globally and in the USA [Citation1,Citation2]. In addition to its recreational use, cannabis is used for a variety of medicinal applications, including the treatment of anxiety, depression, epilepsy, multiple sclerosis, chronic pain, nausea/vomiting from chemotherapy, and a number of other conditions [Citation3–9].
Cannabis indica and Cannabis sativa are the two most common and well-known species of the cannabis plant, and extracts from both species contain cannabinoids of therapeutic interest, such as (-)-trans-Δ9-tetrahydrocannabinol (THC), cannabidiol (CBD), and cannabinol (CBN) [Citation10–14]. However, the classification of cannabis as monotypic (one species) or polytypic (multiple species) is still a debate [Citation15,Citation16]. In the 1960s and 1990s, early research into the therapeutic potential of cannabis yielded the identification of over 500 cannabis constituents [Citation17,Citation18], of which 150 were classified as cannabinoids [Citation19]. C. sativa, tall and narrow-leafed, tends to have a higher THC content as compared to C. indica, which is shorter and wide-leafed and has a higher proportion of CBD [Citation20,Citation21]. Most commercial cannabis-containing products currently on the market are a mix of cannabis from both varieties of C. sativa and C. indica [Citation22,Citation23], as the ratio of THC and CBD varies considerably between strains [Citation24].
THC is the main psychoactive component of cannabis and is responsible for the known euphoriant effect. Due to specialized breeding programs and targeted hybridization efforts, the levels of THC (and thus cannabis potency) in modern commercial plants have increased significantly over the last 30 years, from 2% on average in 1970 to more than 20% in some varieties currently in production [Citation25,Citation26]. CBD and CBN are non-intoxicating constituents and lack euphoric properties; however, both display pharmacological properties and have been classified as anxiolytics, muscle relaxants, anticonvulsants, and anti-inflammatories [Citation27–30].
While the majority of modern cannabis use is recreational, cannabis and cannabis-derived substances are also used for medical purposes. In fact, three standardized cannabinoid-based pharmaceutical products have been approved by the US Food and Drug Administration (FDA) for medicinal use. Dronabinol (marketed as Marinol) and nabilone (marketed as Cesamet) are synthetic analogs of THC used to treat anorexia related with weight loss in patients with HIV, as well as for nausea and vomiting caused by chemotherapy [Citation31–33]. Recently, the FDA approved the first CDB-based medication (Epidiolex) for treating two types of pediatric epilepsy, Lennox-Gastaut and Dravet syndrome [Citation6,Citation34,Citation35]. Another drug approved outside the USA is nabiximols (Sativex), a 1:1 mix of THC and CBD used to relieve neuropathic pain and spasticity in multiple sclerosis patients [Citation8,Citation36].
Despite being classified as a Schedule I drug under the Controlled Substances Act, cannabis consumption for medical purposes is legal in 37 US states, three territories, and the District of Columbia [Citation37]. Cannabis decriminalization became a movement in the late 1960s [Citation38], and as of May 2022, 19 states within the USA have legalized both medicinal and recreational cannabis and recent changes in many state cannabis laws have resulted in a rapid increase in its accessibility and subsequent usage [Citation37]. An estimated 200 million people, or nearly 4% of the world’s population, used cannabis in 2019 [Citation39], and globally they were approximately 13 million cannabis dependent people in 2010 [Citation1,Citation2]. Furthermore, cannabis use has significantly increased in the US over the past 17 years, rising from approximately 11% in 2002 to 18% in 2019 [Citation40]. In 2020, 49.6 million people over the age of 12 used cannabis in the US [Citation41].
As cannabis acceptance and use grows globally, so does the potential for cannabis constituents to interact with conventional medications. Cannabinoids may interact with other therapeutics through pharmacodynamic or pharmacokinetic mechanisms via the inhibition or induction of metabolizing enzymes or drug transporters, leading to potential adverse drug events (ADE) and drug-drug interactions (DDI). The risk of cannabis-drug interactions (CDI) is potentially even higher due to the fact that individuals who use cannabis to treat chronic diseases or conditions often take other psychoactive drugs concurrently [Citation42]. As a result of increased cannabis use, new state laws regarding cannabis, the development of new cannabis-based pharmacotherapies, and the possibility of DDI with other prescribed medications, the risk of adverse effects and toxicity from CDI has increased in recent years and will continue to play a role in drug safety in the foreseeable future.
The major cannabinoids present in cannabis undergo metabolism primarily through cytochrome P450 (CYP) and UDP-glucuronosyltransferase (UGT) enzymes, and these same enzymes are often involved in the biotransformation of other drugs. Recent studies have been conducted to evaluate the effect of major cannabinoids (THC, CBD, and CBN) and their metabolites on major drug-metabolizing enzymes. This review will provide an overview of cannabis constituents, cannabis pharmacokinetics and pharmacology in humans, as well as potential cannabis-drug interactions involving phase I and phase II enzymes, with a particular focus on the major circulating metabolites of THC and CBD.
2. Overview of cannabis
2.1. Phytocannabinoids
The cannabis plant contains more than 500 constituents [Citation43], including cannabinoids, terpenes, flavonoids, alkaloids, and other minor compounds [Citation44]. The term ‘cannabinoids’ refers to a group of chemical compounds that share a similar structural and/or functional properties with THC and bind to cannabinoid receptors [Citation45]. Phytocannabinoids, also known as plant-derived cannabinoids, are meroterpenoids made of 21 or 22 atoms and a pentyl or propyl side chain [Citation46,Citation47].
In the plant, the precursors Δ9-tetrahydrocannabinol acid (THCA) and cannabidiolic acid (CBDA) are decarboxylated via light and high temperature to produce THC and CBD [Citation46,Citation48]. In addition to the major cannabinoids, cannabis contains other minor phytocannabinoids such as cannabigerol (CBG), cannabichromene (CBC), and cannabidivarin (CBDV), as well as other minor constituents [Citation46,Citation49]. Another important cannabinoid is Δ8-THC, which is similar to Δ9-THC but has greater stability and fewer intoxicating effects [Citation50], and whose recreational usage has grown in recent years [Citation51]. Δ8-THC is present in minimal amounts in the cannabis plant, but it can be synthesized from CBD extracted from hemp using a strong acid and heat [Citation51]. All phytocannabinoids undergo hepatic and extra-hepatic metabolism upon human consumption, and some of their metabolites retain significant affinity towards cannabinoid receptors.
2.2. Pharmacokinetics and administration
It is important to fully understand phytocannabinoid pharmacokinetic profiles, particularly their metabolic pathways, if a full understanding of the potential for CDI is to be achieved. The following section will discuss the pharmacokinetic properties of the major cannabinoids THC and CBD, and their metabolites.
2.2.1. Absorption
Cannabis is mainly administered via inhalation (e.g. smoking or vaping), orally (e.g. edibles or prescription cannabis), or by other routes (e.g. sublingual or transdermal) [Citation52]. The pharmacokinetic profile of major cannabinoids varies based on the individual user, route of administration, dose, and type of use (acute vs. chronic). As a general rule, the consumption of cannabis via the inhalation route leads to higher levels of cannabinoids, a quicker onset of action, and greater bioavailability.
Δ9-THC, CBD, CBN, and several of their major metabolites including 11-hydroxy-∆9-tetrahydrocannabinol (11-OH-THC), 11-nor-9-carboxy-Δ9- tetrahydrocannabinol (THC-COOH), 11-nor-9-carboxy-Δ9-tetrahydrocannabinol-glucuronide (THC-COOH-Gluc), 7-hydroxy-CBD (7-OH-CBD), and 7-carboxy-CBD (CBD-COOH), are the cannabinoids whose pharmacokinetic and pharmacodynamic profiles have been the most well-studied [Citation53]. Following inhalation, THC and CBD are detected in the plasma within seconds, and the maximum plasma concentration (Cmax) is attained within 1.2–30 minutes [Citation54–58]. The bioavailability of inhaled THC and CBD ranges from 10–35% and 11–45%, respectively, and is dependent upon on the dose, the number of inhalations, breath hold, depth of inhalation, puff duration, and acute or chronic use [Citation55,Citation58–60]. In one study, the bioavailability of THC for frequent users was 23–27%, as compared to 10–14% for occasional users [Citation61]. The low observed bioavailability of THC may be due to incomplete absorption, since approximately 30% of THC is lost during the pyrolysis process and up to 50% is lost in side stream (non-inhaled) smoke [Citation62].
In contrast, absorption of THC and CBD following oral ingestion is slower, and the time to reach maximum plasma concentration (Tmax) varies from 1–5 hours [Citation63–68]. In addition, the oral bioavailability for THC (4–12%) and CBD (13–19%) is reduced due to first pass metabolism and poor solubility [Citation53,Citation55,Citation59,Citation67,Citation69].
Several studies have shown that the Cmax and the area under the curve (AUC) for major cannabinoids which reflects the extent of absorption, varies by the route of administration [Citation53,Citation55,Citation65]. The average plasma concentrations of THC after a 10 mg oral dose range from 1.1–12.7 ng/ml, whereas the Cmax values of THC in six healthy subjects after smoking a cannabis cigarette containing 3.55% THC averaged 162 ng/ml [Citation53,Citation70]. Taylor et al. [Citation65] showed that the mean Cmax of CBD after oral consumption (1500–6000 mg) was 292–782 ng/ml (0.928–2.49 µM), whereas a 10 mg oral CBD resulted in a mean Cmax of 2.47 ng/ml [Citation71]. Furthermore, after smoking a marijuana cigarette containing 19 mg of deuterium-labeled CBD, the Cmax was reported to be 110 ng/ml (at 3 minutes post dose) [Citation55].
Circulating THC metabolites found in the plasma of cannabis smokers include 11-OH-THC, THC-COOH, and THC-COOH-Gluc. Like THC, 11-OH-THC is considered an active cannabinoid as it interacts directly with cannabinoid receptors in the brain (both CB1 and CB2); interaction with CB1 produces their psychotropic effects [Citation58,Citation72]. 11-OH-THC, shows a similar pharmacokinetic profile as THC, reaching peak levels between 5–15 minutes [Citation70,Citation73]. Other major THC metabolites observed in the urine or feces of cannabis smokers are the inactive metabolites, THC-COOH, and THC-COOH-Gluc, which peak more slowly than the active cannabinoids but remain in the blood at high and stable levels [Citation53,Citation57,Citation61].
After cannabis inhalation, the level of THC-COOH and THC-COOH-Gluc are approximately 2.0 and 7.6 -fold higher, respectively, as compared to THC [Citation57,Citation63,Citation74]; after oral consumption, their levels are approximately 2.5- and 40-fold higher, respectively, than THC [Citation57,Citation63,Citation74]. The plasma concentration of 11-OH-THC is much lower after smoking compared to oral administration [Citation61,Citation75].
CBD and its metabolites exhibit a similar pharmacokinetic profile when compared to THC. The main CBD metabolites detected in the plasma after cannabis inhalation include
7-hydroxy-CBD (7-OH-CBD) and 7-carboxy-CBD (CBD-COOH). In the urine the main excretion products are parent CBD and glucuronidated CBD (CBD-Gluc) [Citation53,Citation76,Citation77]. Similar to THC, the inactive metabolite of CBD, CBD-COOH, is the major circulating product in plasma, followed by the active metabolite 7-OH-CBD [Citation65]. Not unexpectedly, the oral absorption of CBD is influenced by the consumption of fatty foods, with recent studies showing that the CBD AUC increases by 4.2-fold after consumption of CBD (1500 mg) after a high-fat meal vs. in a fasting state [Citation65].
2.2.2. Distribution
Cannabinoids are highly lipophilic, lipid-soluble, compounds (logP: THC, 6.97; CBD, 5.79; CBN, 4.38) [Citation78,Citation79]. Therefore, they sequester in fatty tissues and penetrate highly vascularized tissue such as adipose, heart, brain, liver, lungs, and spleen, resulting in their rapid decrease in plasma concentration [Citation80]. For instance, the active metabolite, 11-OH-THC, is present in the brain at higher levels as compared to THC [Citation58,Citation81,Citation82]. Recent in vivo studies in adult rats demonstrated a greater CBD accumulation in adipose tissue, as compared to muscle and liver following oral gavage [Citation83]. Similar patterns of accumulation have been observed with THC in humans [Citation84,Citation85]. The volume of distribution (Vd) of THC and CBD in adults varies from 2.5–10 L/kg, assuming a 70 kg body weight [Citation53,Citation58]. The majority of THC in the blood (90%) is distributed in the plasma and the remaining 10% in red blood cells [Citation68,Citation86]. Cannabinoids display lower blood concentrations compared to plasma, indicating low partition into erythrocytes. The mean (range) human blood to plasma ratio is 0.63 (0.3–1.7) for THC, 0.60 (0.1–1.3) for 11-OH-THC, and 0.55 (0.2–1.5) for THC-COOH [Citation68,Citation87].
THC and CBD have a protein binding capability of >95%, mainly to lipoproteins [Citation66,Citation87,Citation88]. This means that 1–5% of total THC and CBD concentration is unbound and have pharmacological effects. The metabolites of THC and CBD, as well as their glucuronides bind extensively to albumin [Citation66,Citation87].
2.2.3. Metabolism
Cannabinoid metabolism proceeds mainly through hepatic mechanisms, and to a lesser extent in the brain, small intestine, and lung [Citation58,Citation89]. The two main cannabinoids, THC and CBD, undergo hydroxylation or oxidation by cytochrome P450 (CYP450) enzymes, followed by glucuronidation via UDP-glucuronosyltransferase (UGT) enzymes and excretion in the urine, bile, or feces (). Upon consumption, THC is hydroxylated via CYPs 2C9, and 2C19, to the active metabolite 11-OH-THC [Citation90–92] (see ).
Figure 1. The major metabolic pathway of THC. Abbreviations: CYP: cytochrome P450; UGT: UDP-glucuronosyltransferase.
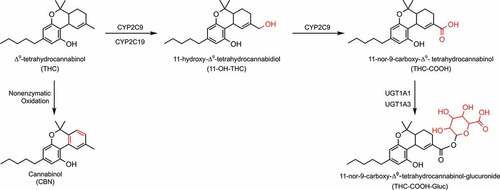
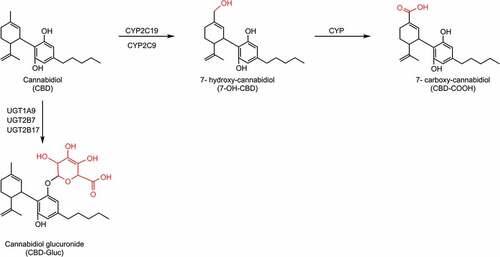
Figure 2. CBD biotransformation pathway by CYP and UGT enzymes. The CYP enzymes responsible for the metabolism of 7-OH-CBD are unknown. Abbreviations: CYP: cytochrome P450; UGT: UDP-glucuronosyltransferase.
Further hydroxylation of 11-OH-THC by CYP2C9 results in the inactive carboxylic acid derivative THC-COOH, which does not interact with the CB1 receptor [Citation93,Citation94] but is a useful biomarker of cannabis exposure [Citation95,Citation96]. THC-COOH formation from 11-OH-THC can also be mediated by alcohol dehydrogenase or aldehyde oxygenase [Citation97]. 11-OH-THC and THC-COOH also undergo phase II metabolism by UGT enzymes [Citation98], with THC-COOH metabolized via UGTs 1A1 and A3 to produce THC-COOH-Gluc, the major urinary metabolite of THC [Citation98,Citation99], and 11-OH-THC glucuronidated by UGTs 1A9 and 1A10 [Citation98].
The minor cannabinoid CBN, a non-enzymatic oxidation byproduct of THC, is commonly detected after prolonged storage of cannabis products, especially at higher temperatures. Due to the fact that CBN has an extra aromatic ring, it is metabolized less extensively than THC [Citation92,Citation100,Citation101]. After oral consumption, CBN is mainly metabolized to the major active metabolite 11-hydroxy-CBN, and a minor metabolite 8-hydroxy-CBN, via hydroxylation catalyzed by CYPs 2C9 and 3A4, respectively [Citation92]. CBN can also undergo direct glucuronidation by hepatic and extrahepatic enzymes such as UGTs 1A9, 1A7, 1A8, and 1A10 [Citation98].
CBD follows the same metabolic scheme as THC and undergoes primarily hepatic metabolism by CYP and UGT enzymes (See ). CBD is metabolized via CYPs 2C19, and 2C9 to an active metabolite 7-OH-CBD, which is further metabolized to the inactive metabolite CBD-COOH [Citation102,Citation103]. Like CBN, CBD also undergoes direct glucuronidation via UGTs 1A9, 2B7, and 2B17 [Citation98].
2.2.4. Elimination
THC is mainly excreted via feces (approximately 65%) and urine (20%) [Citation75]. The urine contains predominantly acidic metabolites such as THC-COOH [Citation104]. In contrast, the metabolites in the feces are both neutral and acidic, with 11-OH-THC (21%) and THC-COOH (28%) being the most prevalent [Citation95,Citation105]. THC is a highly lipid soluble compound and undergoes a high degree of tubular reabsorption, resulting in low levels of THC detected in the urine [Citation58]. The mode of consumption does not appear to effect elimination as THC and its metabolites are excreted in both feces and urine after oral consumption as well as inhalation.
There is variability in the estimated half-lives for THC and CBD in different studies [Citation53,Citation69]. Based on a population pharmacokinetic model, THC was shown to have a short initial half-life (roughly 6 minutes) followed by a terminal half-life of 22 hours [Citation89,Citation106]. Heavy users display a longer elimination half-life (>24 hours), likely due to the slow redistribution from fatty tissues [Citation73,Citation89,Citation107]. The urinary excretion half-life of THC-COOH in occasional cannabis users is 1.3 days but is 5–13 days in frequent users [Citation61,Citation108]. The average plasma clearance was reported to be higher in men (14.9 ± 3.7 L/hour) than in women (11.8 ± 3.0 L/hour), indicating a potential sex-related difference in THC clearance [Citation109].
Unlike THC, the majority of CBD is excreted unchanged through feces (33%) and as both unchanged and glucuronidated CBD in the urine [Citation77,Citation110]. CBD has also been reported to have a long half-life of between 18 to 32 hours after smoking [Citation55,Citation69,Citation111]. Chronic oral administration of CBD displays a half-life of 2–5 days [Citation112].
2.3. Pharmacology
Cannabinoids exert their pharmacological and physiological effects by interacting with multiple receptors within the endocannabinoid system (ECS) [Citation113]. Briefly, this system includes the two main cannabinoid receptors (CB1 and CB2), endocannabinoids and the enzymes responsible for their biosynthesis and degradation [Citation114,Citation115]. Phytocannabinoids bind and activate the G-protein coupled receptors CB1 and CB2 [Citation12,Citation116], which share an amino acid sequence homology of 48% but have unique expression profiles [Citation117,Citation118]. CB1 is found mainly within the central and peripheral nervous system [Citation119], whereas CB2 is predominantly located in immune tissues, specifically in the thymus, bone marrow, and spleen [Citation116,Citation120–122]. The hippocampus, cerebral cortex, amygdala, and cerebellum are among the tissues that express high levels of the CB1 receptor [Citation123,Citation124]. THC partially activates the CB1 and CB2 receptors with binding constant (Ki) values ranging between 5.05–80.3 nM and 3.13–75.3 nM, respectively [Citation72,Citation125–130]. The psychoactive effect of THC is associated with its binding to the CB1 receptor as a partial agonist [Citation72]. As reviewed by Pertwee [Citation72], some studies report that THC also behaves as a CB2 antagonist.
In contrast, CBD has a relatively low affinity for both CB1 and CB2, with Ki values ranging between 1458.5–4900 nM, and 372.4–4200 nM, respectively [Citation72,Citation131]. CBD acts as an inverse agonist of CB2 receptor [Citation132], as a negative allosteric modulator (NAM) of the CB1 receptor [Citation133] and as a positive allosteric modulator (PAM) of the GABA-A receptor [Citation134,Citation135].
Besides binding to CB1 and CB2, phytocannabinoids also modulate other non-cannabinoid receptors (acting as agonist /antagonist/ inverse agonist /allosteric modulator) including G-protein coupled receptor 55 (GPR55) [Citation136], β-adrenoreceptor, µ-opioid receptor [Citation137], the peroxisome proliferator-activated receptors (PPAR2, PPARγ) [Citation138], transient receptor potential channels (TRPM) and serotonin receptor 1A (5-HT1A) [Citation139,Citation140]. THC has well-documented antispasmodic, anti-inflammatory and neuroprotective properties, which are mediated through the activation of receptors such as CB2 and PPARy [Citation72,Citation141]. In addition, CBD can act as an agonist of the serotonin receptor 5-HT1A that modulates anxiety and hunger [Citation140]. Minor phytocannabinoids (e.g. CBN, THCV) bind at both cannabinoid and non-cannabinoid receptors [Citation142]. Further studies are necessary to better understand the mechanism of action of minor phytocannabinoids.
3. Cannabis interactions with metabolism
While cannabis is used more commonly in combination with pharmaceuticals and illicit drugs, the safety of this polypharmacy has yet to be thoroughly investigated. Virtually all xenobiotics that enter the body undergo metabolism, a process that can often be described in two major phases: phase I and phase II. Phase I metabolism consists of the initial transformation of xenobiotics into more polar species, either by adding or exposing polar functional groups. Phase I metabolism is catalyzed by several enzymatic families, with the largest being the cytochrome P450 (CYP) enzyme family. Other major phase I enzyme families include the flavin monooxygenases (FMOs), alcohol dehydrogenases (ADH), carboxylesterases (CES), and epoxide hydrolases (EH).
Phase II metabolism consists of further transformation of xenobiotics into more water-soluble metabolites that can be easily excreted from the body through the addition of various functional groups. Phase II metabolism is comprised of numerous enzymatic families, with the largest being the uridine-5’-glucuronosyltransferases (UGT). Other major phase II enzyme families include sulfotransferases, N-acetyltransferases, glutathione S-transferases, and methyltransferases.
3.1. Cannabis-CYP interactions: in vitro studies
There are 10 major CYP enzymes involved in xenobiotic drug metabolism: CYP3A4, CYP2A6, CYP2D6, CYP2C8, CYP2C9, CYP1A2, CYP2C19, CYP2E1, CYP2B6, and CYP2A6, and these account for 70–80% of all drug metabolism [Citation143]. CYP-mediated metabolism is highly susceptible to DDI and is thought to account for much of the variability in drug response [Citation143,Citation144]. As the risk for DDI is high for the CYP family of enzymes, the FDA has proposed a set of guidelines for the study of the interactions between CYPs and potential perpetrator drugs [Citation145]. This guidance focuses both on the inhibition and induction of CYP enzymes in an in vitro system, and also tasks researchers with the characterization of the DDI potential of metabolites, if they exceed parent drug exposure by 1.25-fold [Citation145]. As several THC and CBD metabolites exceed parent drug exposure by ≥ 1.25-fold, this becomes particularly important when considering the potential inhibitory effects of cannabis. The FDA recommends not only in vitro DDI studies but also static (mechanistic static modeling) and dynamic (physiologically based pharmacokinetic – PBPK) modeling when preliminary data and physiological conditions suggests a potential interaction [Citation145].
A number of in vitro DDI studies have been performed and most are focused on the main cannabinoids, THC, CBD, and CBN; however, more recent studies also incorporate the common metabolites, 11-OH-THC, THC-COOH, THC-COOH-Gluc, 7-OH-CBD, and CBD-COOH. THC has been shown to reversibly inhibit 7-ethoxyresorufin hydroxylation activity of CYP1A/1B1 [Citation146], theophylline and phenacetin hydroxylation activity of CYP1A2 [Citation74,Citation147], coumarin hydroxylation activity of CYP2A6 [Citation148,Citation149], bupropion hydroxylation activity of CYP2B6 [Citation74,Citation148,Citation149], midazolam hydroxylation and diltiazem demethylation activity of CYP3A4 [Citation74,Citation147,Citation150], diclofenac and warfarin hydroxylation activity of CYP2C9 [Citation74,Citation147,Citation148,Citation151] omeprazole and mephenytoin hydroxylation activity of CYP2C19 [Citation74,Citation102,Citation147,Citation148], and dextromethorphan demethylation activity of CYP2D6 [Citation74,Citation147,Citation148,Citation152], with Ki,u (Ki values corrected for non-specific binding of cannabinoids to labware) values ranging from 0.056 µM (CYP2C19) to 0.38 µM (CYP2B6) () [Citation74]. THC has also been shown to be a time-dependent inhibitor of CYP1A1 [Citation146] and CYP2A6[Citation149] in human liver microsomes (HLM), with inactivation values (KI and Kinact) comparable to the Ki values noted above (). Similarly, reversible THC inhibition of selected CYPs was described in individual CYP-overexpressing HEK293 cell lines or baculosomes, again with Ki values comparable to those observed in HLM () [Citation74,Citation146,Citation149,Citation151]. The active THC metabolite 11-OH-THC, has been shown in both HLM and recombinant enzyme systems to reversibly inhibit CYPs 2B6, 2C8, 1A2, 2C9, 2C19, and 2D6; with Ki,u values in the range of 0.057 µM (CYP2C9) to 0.32 µM (CYP2D6) () [Citation74,Citation147,Citation148]. THC-COOH-Gluc was found to inhibit CYPs 2B6, 2C9, and 2D6 in both HLM and recombinant enzyme systems, with Ki,u values ranging from 0.9 µM (CYP2B6) to 3.4 µM (CYP2C9) () [Citation74]. Interestingly, THC-COOH was also studied in these model systems and did not show significant inhibition (IC50 > 50 µM) of any CYP enzyme [Citation74,Citation147].
Table 1. Reversible inhibition of recombinant CYPs and HLM by THC.
Table 2. Inactivation kinetic constants of time dependent inhibition of recombinant CYPs and HLM.
Table 3. Reversible inhibition of recombinant CYPs and HLM by the metabolites of THC and CBD.
CBD also inhibits numerous CYPs in both HLM and recombinant enzyme systems. These include CYPs 1A2, 3A4, 2B6, 2C8, 2C9, 2C19, 2D6, and 2E1, with Ki,u values ranging from 0.021 µM (CYP2E1) to 0.31 µM (CYP2D6) () [Citation74,Citation147–153]. Similar to THC, CBD also exhibits time-dependent inhibition towards CYPs 1A1, 1A2, 1B1, 2C19, and 3A4 in HLM and recombinant enzyme systems, with inactivation values similar to those seen with reversible inhibition () [Citation146–150,Citation152,Citation153]. The CBD metabolite, 7-OH-CBD, reversibly inhibits CYPs 2B6, 2C8, 2C9, 2C19, 2D6 and 3A4 and exhibits time-dependent inhibition against CYP2C19 and CYP3A4, with inactivation values ranging from 0.67 µM (CYP2C19) to 1.0 µM (CYP3A) () [Citation147,Citation148]. CBN also reversibly inhibits CYPs 1A1, 1A2, 1B1, 2A6, 2B6, 2C9, 2C19, and 2E1 in both HLM and recombinant enzyme systems with Ki,u values ranging from 0.063 µM (CYP2E1) to 0.99 µM (CYP2C9) (Supplementary Table 1) [Citation74,Citation146,Citation149,Citation151,Citation153].
Table 4. Reversible inhibition of recombinant CYPs and HLM by CBD.
Very few of the in vitro studies performed to date have performed static modeling or dynamic modeling to further investigate the potential for in vivo DDI between the pharmaceutics and cannabinoids. Earlier studies did not perform these static models, nor did they account for the non-specific binding of the cannabinoids in their experimental systems and adjust their IC50 and Ki values accordingly. Of those studies that have performed in vitro to in vivo extrapolation (IVIVE), all of them have been static modeling. Nasrin et al. [Citation74] found that CBD exhibited the greatest potential for in vivo DDI, with it showing an inhibitory effect for 7 of the major CYPs (1A2, 3A4, 2B6, 2C9, 2C19, 2D6, and 2E1). They also found that THC and 11-OH-THC have the potential for in vivo DDI for CYPs 2C19, 2C9, and 1A2, and CYPs 2B6 and 2C9, respectively [Citation74]. Similarly, Bansal et al. [Citation147] found that CBD and THC have the potential to cause in vivo DDI with CYPs 1A2, 2C9, 2C19, 2D6, and 3A4 and CYPs 1A2, 2C9, and 3A4, respectively. In a follow-up study, the authors considered time-dependent inhibition, and found that CBD and THC had the potential for in vivo DDI with CYPs 1A2, 2C9, 2C19, 2D6, and 3A4, and CYPs 1A2, 2C9, and 3A4, respectively [Citation148].
In contrast, another study found the potential for in vivo DDI with CBD, CBDV, cannabidiolic acid (CBDA), CBG, and CBN only with CYP2C9 and only for drugs with narrow therapeutic windows [Citation153]. It is important to note that this study did not account for the non-specific binding of these cannabinoid to labware and incubational system. Thus, the reported Ki values were likely to be much higher than the more physiologically relevant Ki,u values that would indicate a potential DDI in vivo. Doohan et al. [Citation153], noted the importance of substrate specificity when considering DDI. They showed that certain cannabinoids exhibited greater potency depending on which probe substrate was used in the same in vitro system [Citation153].
3.2. Cannabis-CYP interactions: in vivo studies
Due to the legal and societal issues governing cannabis research, there have been very few in vivo clinical trials undertaken to investigate the potential DDI between cannabis and various drugs. Geffrey et al. [Citation154] investigated the effects of concomitant oral administration of 5–25 mg/kg/day CBD (Epidiolex) with clobazam in patients with epilepsy. They found a significant increase in the plasma concentrations of N-desmethylclobazam (an active metabolite of clobazam) with concomitant use of CBD as compared to baseline (without CBD). These findings were attributed to a CBD-mediated inhibition of CYP2C19, as CYP2C19 is known to be the major enzyme involved in the metabolism of N-desmethylclobazam to the inactive metabolite, 4-hydroxy-N-desmethylclobazam [Citation154,Citation155]. Gaston et al. [Citation156], found similar results after administering 5–50 mg/kg/day of CBD (Epidiolex) again in patients with epilepsy who were taking various antiepileptic drugs including clobazam (19 drugs total). There was a significant increase in N-desmethylclobazam plasma concentrations after concomitant CBD use compared to baseline [Citation156]. Furthermore, they noted increased concentrations of numerous other antiepileptic drugs (rufinamide, eslicarbazepine, zonisamide, and topiramate) in the presence of CBD [Citation156]. Another study investigated the effects that CBD (Epidiolex) had on antiepileptic drugs in patients with Dravet Syndrome, with CBD doses ranging from 5–20 mg/kg/day. There was an increase in N-desmethylclobazam plasma concentration, but no other antiepileptic drug studied were affected (stiripentol, topiramate, valproate, and levetiracetam) [Citation157]. In contrast, a similar study examining the effects of CBD (Epidiolex, 750 mg twice daily) on valproate, clobazam, and stiripentol found an increase in both Cmax and AUC for N-desmethylclobazam and stiripentol, a finding which had not been reported previously [Citation158]. Overall, these studies found similar results with the concomitant use of CBD (Epidiolex) and clobazam where the active metabolite, N-desmethylclobazam, was increased, most likely due to the CBD-mediated inhibition of CY2C19, and a subsequent decrease in N-desmethylclobazam metabolism to 4-hydroxy-N-desmethylclobazam.
A single case study also found that a 37 y male patient taking the antiepileptic drug carbamazepine in combination with smoked cannabis (1–2 joints per day) exhibited an increase in blood carbamazepine levels as compared to the carbamazepine levels in this individual after cessation of cannabis use [Citation159]. Carbamazepine metabolism proceeds mainly through CYP3A4, and the increased carbamazepine levels are consistent with in vitro data showing inhibition of CYP3A4 by CBD, and inhibition of CYP2B6 by both THC and CBD [Citation74,Citation147,Citation148,Citation160,Citation161]. However, within this case study, the measurements used to identify the increase in carbamazepine were within the time frame of an increase in the patient’s dosing regimen. It is thus unclear if the increase in carbamazepine levels were due to an actual pharmacokinetic DDI precipitated by cannabinoids, or simply due to the increase in the dose of carbamazepine. Further studies are needed to elucidate a potential pharmacokinetic DDI between cannabinoids and carbamazepine.
Thai et al. [Citation162], investigated the effects of CBD (Epidiolex) (250 mg once daily vs. 750 mg twice daily) on caffeine (200 mg) pharmacokinetics in a phase I open-label clinical trial. A 95% increase in the plasma AUC of caffeine with coadministration of CBD was observed, along with an increase in Tmax. The authors also examined the effects that CBD had on paraxanthine pharmacokinetics under the same conditions (250 mg once daily vs. 750 mg twice daily) and found that the plasma paraxanthine AUC increased by only 18% [Citation162]. As both caffeine and paraxanthine are substrates for CYP1A2, these results suggest that substrate specificity is an important consideration when investigating cannabinoid mediated DDI in vitro and in vivo.
3.3. Cannabis-CYP induction
Identifying the effects of cannabis on the induction of phase I drug metabolizing enzymes is also an area of research that has been understudied. An early study found that THC induced CYP1A1 mRNA expression in a murine hepatoma cell line (Hepa-1) [Citation163]. Another study found that THC and two of its metabolites (11-OH-THC and 11-COOH-THC) induced CYP2C9 activity, shown by an increase in phenytoin metabolite levels in HLM [Citation90]. This last finding is somewhat contradictory to more recent studies (described above) where THC exhibits an inhibitory effect on CYP2C9 [Citation74,Citation147,Citation148,Citation151,Citation153], a discrepancy potentially explained by differences in experimental conditions including differences in inhibitor concentrations.
3.4. Cannabis-CES interactions
Carboxylesterases (CES) are predominantly expressed in the liver and intestine, with minor levels of expression in other tissues such as the brain [Citation164–168]. The two major CES isoforms are CES1, primarily found in the liver, and CES2, primarily found in the intestine [Citation169,Citation170]. Both of these isoforms play a crucial role in endogenous and xenobiotic metabolism [Citation169].
Qian et al., examined potential DDI between cannabis constituents (THC, 11-OH-THC, THC-COOH, CBD, and CBN) and CES1 using CES-overexpressing HEK293 S9 fractions [Citation171]. They found that the major cannabinoids, THC, CBD, and CBN, potently inhibited CES1 by a mixed competitive – noncompetitive mechanism. The Ki for THC, CBD, and CBN against oseltamivir phosphate biotransformation to oseltamivir carboxylate was 0.54 ± 0.081 µM, 0.97 ± 0.17 µM, and 0.26 ± 0.034 µM respectively, and it was suggested that THC and CBD may inhibit oseltamivir phosphate biotransformation via CES1 in vivo (). Supporting this possibility is the fact that these Ki values did not account for cannabinoid non-specific binding, with the true Ki value (i.e. Ki,u) likely being much lower. Qian et al. [Citation172], also investigated the potential DDI between cannabinoids and another CES1 substrate, methylphenidate [Citation172]. In this study, they found that THC and CBD reversibly inhibited methylphenidate biotransformation to ritalinic acid by CES1 with Ki,u values of 0.031 ± 0.003 µM and 0.091 ± 0.004 µM, respectively () [Citation172]. Physiologically based pharmacokinetic (PBPK) modeling supported this finding and suggested that an in vivo clinical trial is necessary to further characterize this potential DDI with CES1 [Citation172].
Table 5. IC50 and Ki values of cannabinoids against major non-CYP450 enzymes in recombinant enzymes, HKM and HLM.
3.5. Cannabis-UGT interactions: in vitro studies
UGTs are major phase II drug metabolizing enzymes that catalyze the conjugation of UDP-glucuronic acid to endogenous (e.g. bilirubin, steroids) and xenobiotics including many drugs, resulting in more water-soluble metabolites that are more easily excreted from the body in the urine or bile [Citation174]. These enzymes are classified into four main families (UGT1, UGT2, UGT3, UGT8) that exhibit different selectivity toward various UDP-sugars [Citation175]. The UGT1 and UGT2 families utilize UDP-glucuronic acid (UDPGA) as a co-substrate, whereas the UGT3A family uses the alternative sugar donors UDP-N-acetylglucosamine, UDP-glucose, and UDP-xylose [Citation174,Citation176]. Approximately 35% of drugs in clinical use (e.g. acetaminophen, benzodiazepines, opioids, nonsteroidal anti-inflammatory drugs) are glucuronidated by hepatic and extrahepatic UGTs [Citation177].
Unlike CYPs, only a few studies have investigated the potential DDI between cannabinoids and UGT substrates [Citation173]. One such study suggests that CBD and CBN inhibited UGT1A9-mediated ethanol glucuronidation in vitro by 49% and 34%, respectively [Citation178]. CBD showed strong non-competitive inhibition of ethanol glucuronidation in HLM, with IC50 values of 1.17 mg/L () [Citation178]. Interestingly, the UGT2B7-mediated metabolism of ethanol was inhibited 3.3-fold by CBD but activated in a concentration-dependent manner by 2.5-6-fold by CBN [Citation178].
A recent study showed that CBD, THC, and CBN strongly inhibited the glucuronidation activities of UGTs 1A9, 1A6, 2B4, and 2B7 [Citation173]. CBD was reported to strongly inhibit the glucuronidation activity of propofol, furosemide, acetaminophen (UGT1A9), codeine (2B4), serotonin (1A6), and AZT (2B7) in recombinant enzymes, HKM and HLM, with binding-corrected IC50 (i.e. IC50,u) values ranging from 0.073–0.82 µM, 0.22–2.5 µM, and 0.4–1.5 µM, respectively () [Citation173]. IC50,u values of THC toward UGT1A9, 2B4, and 2B7 in recombinant enzymes, HKM, and HLM ranged from 0.33–1.4 µM, 0.54–2.9 µM, and 0.61–2.8 µM, respectively () [Citation173]. Unlike that observed with CBD and THC, CBN only inhibited UGT1A9 and 2B7 in recombinant enzymes, HKM and HLM, with IC50,u values ranging from 0.51–4.2 µM, 0.9–6.9 µM, and 2.8–5.5 µM, respectively () [Citation173]. The major hepatic UGTs are 2B7 (17% of total UGT expression in the liver), 2B4 (16.1%), 2B15 (11.2%), and 1A1 (11%) [Citation179]. In contrast, the three main UGTs expressed in the kidney are UGT1A9 (45% of total renal UGT expression), UGT2B7 (41%), and UGT1A6 (7%) [Citation179]. The IC50,u values for all UGT1A9 substrates were comparable in HKM and UGT1A9 recombinant microsomes but were approximately 3-fold higher in HLM for CBD, THC, and CBN. In contrast, the IC50,u values for the UGT2B7 substrate, AZT, was similar in HLM and in UGT2B7 recombinant enzymes but different in HKM [Citation173]. Together, these studies suggest that THC, CDB, and CBN strongly inhibited major hepatic and renal UGT enzymes.
3.6. Cannabis-UGT interactions: in vivo studies
Recent in vivo studies examined the effect of oral CBD (Epidiolex) on the disposition of midazolam (a CYP3A4 probe substrate) in healthy volunteers [Citation180]. The active metabolite of midazolam, 1-hydroxymidazolam, is further metabolized by UGT enzymes [Citation181]. Administration of midazolam with steady-state levels of Epidiolex resulted in increased plasma concentrations of active 1-hydroxymidazolam (12% and 68% increases in Cmax and AUCtau, respectively), as well as a delay in Tmax (difference in median of 2.2 h) and a 35% increase in t1/2 [Citation180]. The increase in active metabolite exposure could be attributed to CBD-mediated inhibition of UGTs, specifically UGT2B7. However, in studies examining the morphine disposition kinetics with or without concomitant inhaled vaporized cannabis [Citation182], no significant differences were observed in morphine exposure (AUC) between the treatments [Citation182]. Since the major mode of metabolism of morphine is via glucuronidation by UGT2B7 to the inactive 3-glucuronide and the active 6-glucuronide [Citation183], the in vivo effects of CBD and other cannabinoids on UGT2B7 and other UGT enzymes require further characterization.
3.7. Clinical implications
The limited number of in vivo studies investigating cannabis induced DDIs has limited the translation of in vitro identified cannabis induced DDIs into the clinic. This mostly stems from the lack of accurate in vitro to in vivo (IVIVE) extrapolation. Anderson et al [Citation184] conducted in vitro and in vivo studies investigating the inhibitory effect of CBD on citalopram metabolism and found low Ki values for CBD against CYP3A4 and CYP2C19, 0.685 µM and 1.4 µM, respectively. In a small clinical trial (n = 6), they also showed that the introduction of CBD increased citalopram plasma concentrations by 1.7-fold. The Ki values reported by Anderson et al [Citation184] were found to correspond to achievable plasma CBD concentrations after administration of clinical doses of CBD [Citation154,Citation158,Citation185,Citation186]. Based solely on in vitro determined Ki values for a cannabinoid against a given drug, it would not be possible to predict whether a DDI is likely to occur for that drug in vivo, since much would depend on the route of administration, various inter-individual usage patterns (occasional vs chronic user, depth of inhalation, breath hold, dosing, etc.) [Citation61], and the levels of a given cannabinoid in the cannabis product being consumed. Therefore, clinical studies will be required to identify which drugs need special attention when prescribed with cannabis (if they should be prescribed together at all). For the few in vivo studies examining potential cannabis-drug DDIs, DDIs have been described for warfarin, midazolam, clobazam, and other antiepileptics [Citation154,Citation156,Citation159,Citation180,Citation187–191] and clinical guidance needs to be updated. For example, patients on a stable drug regimen that are metabolized by enzymes inhibited by cannabis should undergo careful medical surveillance if they subsequently use cannabis or other cannabis products (e.g. CBD) on a regular basis. For example, routine blood draws should occur to monitor the original drug regimen for tolerability and dosage adjustments, with removal of cannabis or cannabinoid product usage recommended if drug levels are significantly altered. Greater counseling regarding cannabis use or cannabis products is necessary, especially in populations where co-use of cannabis and prescribed medications is common (i.e. chronic pain) [Citation192,Citation193]. For example, a patient taking warfarin was shown to exhibit internal bleeding events when also using cannabis on a regular basis, suggesting that the co-use of cannabis with warfarin should be eliminated [Citation189]. In this case study, the international normalized ratio (INR) was also significantly increased after the initiation of smoking cannabis [Citation189] This pattern of increased INR upon co-usage of warfarin and cannabis (edible and smoking) was also observed in two other case studies [Citation188,Citation190]. However, in recent studies, Thomas et al [Citation194] suggested that THC and CBD doses in medicinal cannabis may be too low to inhibit the enzymes involved in warfarin metabolism. Together, the data suggest that patients on specific drug regimens be carefully monitored by their care provider to decide the best course of action when using cannabis or cannabis product is being considered.
4. Conclusions
The in vitro studies currently in the literature suggest extensive cannabis-CYP and cannabis-UGT interactions. Some of these studies have further utilized IVIVE models to predict a risk for cannabis interactions with the following hepatic CYP enzymes: CYPs 1A2, 2C9, 2C19, and 3A4 by THC; CYPs 1A2, 2B6, 2C9, 2C19, 2D6, 2E1, and 3A4 by CBD; and CYP2B6 and CYP2C9 by 11-OH-THC [Citation74,Citation147,Citation148,Citation153]. In addition, the major cannabinoids (THC, CBD, and CBN) were found to inhibit UGTs 1A9 and 2B7, while CBD alone additionally inhibited UGTs 1A6 and 2B4 [Citation74]. However, IVIVE has not been performed for the UGT enzymes. Data from the few clinical trials available suggests that CYP2C19 [Citation154,Citation156,Citation158] and CYP1A2 [Citation162] are susceptible to inhibition by CBD.
While many studies have demonstrated a potential DDI effect between cannabis and certain CYP enzymes, there are also studies that do not support these interactions. Many of these studies investigated the effect of oral THC on drugs administered intravenously, such as irinotecan, primarily metabolized by CYP3A [Citation195]; docetaxel, metabolized via CYPs 1B1, 2B6, and 3A [Citation195]; cyclophosphamide, metabolized via CYPs 2B6, and 3A [Citation196]; and doxorubicin, primarily metabolized by carbonyl reductase 1 [Citation196]. Each of these studies found no interactions. Similarly, a study that investigated inhaled THC and oral THC (dronabinol) did not find an effect on nelfinavir (a substrate of CYPs 2C19 and 3A4), and indinavir (a substrate of CYP3A) [Citation197]. Given these seemingly disparate results, it is important to understand and control for the many nuances in performing controlled studies on cannabis-drug interactions. Some of the variation observed between studies could arise from dosing differences, substrate specificity and individual variation in genetics and metabolism.
The knowledge of cannabis-enzyme interactions remains limited due to many factors, including a lack of well-controlled, in vivo clinical trials investigating the impact of cannabis consumption on both enzyme inhibition and induction, the wide variety of cannabis strains and potencies, and the inconsistent level of exposure and consumption due to individual variability (route of administration, depth of inhalation, amount consumed/smoked, occasional versus chronic users). Overall, more work is needed to comprehensively examine the risk for cannabis-induced DDI involving drug-metabolizing enzymes. With the wide variety and ever-increasing number of cannabis-derived products on the market, well-controlled studies are needed to elucidate the effect that cannabinoids have on common pharmaceuticals, and it will be important to investigate the effects of chronic cannabis use, substrate specificity, dosing differences, and genetic disposition when investigating potential drug-drug interactions.
5. Expert opinion
Research in this field has provided evidence for cannabis-induced DDI among a significant number of major hepatic drug metabolizing enzymes (DMEs). Additionally, these potential DDI are shown to occur across at least three enzyme families: cytochrome P450s (CYP), UDP-glucuronosyltransferases (UGT), and carboxylesterases. Given that these enzyme families (particularly the CYPs and UGTs) are responsible for the majority of the metabolism of drugs currently on the market, further research into cannabis-induced DDI is needed to fully understand how these interactions might effect a large portion of the population. This knowledge will add to this understudied field of medicine, assist in the safe and efficacious delivery of therapeutics, and increase public health outcomes. As potential DDI are further investigated in vitro, in vivo, and in silico (through physiologically based pharmacokinetic (PBPK) modeling), prescribing practices can be updated to reflect the most current understanding of this polypharmacy. Cannabis is now recreationally and medicinally legal in many states; therefore, the timely generation of robust and relevant clinical guidance is imperative to improve patient safety and avoid adverse drug events (ADE). Specifically, additional clinical studies investigating potential DDIs in vivo will allow for specific clinical guidance to be distributed on cannabis induced DDIs. Improved guidance will help clinicians make the most informed decisions for their patients and their therapeutic regimens. Improved clinical guidance and DDI studies will occur when the stigma surrounding cannabis use decreases, which will ultimately occur when restrictions surrounding cannabis use and research are changed. In addition, the removal of federal restrictions on cannabis use and its classification as a Schedule 1 substance will significantly improve the capacity for federally-funded research on cannabis, which would enable increases in clinical research on cannabis-related DDIs.
An important consideration within the field is the necessity of evaluating every possible drug for a potential cannabis DDI. This is not only tedious and expensive, but also limitless as new drugs are constantly approved. Current best practices assess probe substrates and cannabis for DDI for each of the major DMEs. These DDI are then analyzed and used to issue blanket clinical guidance. However, this approach does not account for the effects of substrate specificity and individual genetic variation, and important DDI may be missed. In the future, an assessment of multiple agents within a drug class may better represent the broad spectrum of pharmaceutics and potential DDI than relying solely on a single probe substrate to demonstrate the DDI potential against a DME. It has become increasingly important to standardize DDI assays and models in vitro to reduce variability generated across labs. Harmonized methodologies will allow for higher throughput of data generation and improve clinical guidance as there should be less conflicting information. Furthermore, current in vitro data have poor IVIVE outcomes (clearance, exposure, DDI potential, etc.). Traditional in vitro DDI studies are not physiologically similar enough to in vivo, leading to the poor IVIVE noted above. Nevertheless, great strides have been made in the field to create more sophisticated in vitro systems to investigate DDIs. These include hepatocyte studies, organ on a chip, and various micro-physiological systems (linking numerous organs on a chip to simulate multiple organ systems in vivo). Further studies aimed at improving IVIVE outcomes should be the goal going forward.
Additionally, the current knowledge of cannabis-enzyme interactions is focused primarily on hepatically-expressed drug metabolizing enzymes. Other tissues including the kidney and intestine [Citation198,Citation199] also express sizeable quantities of DMEs that could affect drug exposure and elimination. Nasrin et al. [Citation173] showed differential inhibition by THC and CBD on UGT isoforms differentially expressed in both the kidney and liver, suggesting that drugs which are renally cleared may be more affected by cannabis exposure. These results highlight the need for studies that broaden the scope of current investigations, incorporating extra-hepatic tissues into further investigations of potential cannabis-induced DDI.
In the not-so-distant future, it will become feasible to make clinical decisions regarding cannabis-induced DDI based upon computer models, rather than performing a costly clinical trial. However, the current static modeling approach used to predict the potential for DDI in humans (as required by the FDA) is limited. The static and mechanistic static modeling equation does not include all relevant biological processes that may affect pharmacokinetic DDI. Incorporating quantitative systems pharmacology (QSP) and PBPK modeling will drastically improve the biological relevance of DDI predictions, as they incorporate whole systems biology and can simulate various and complex pharmacokinetic and pharmacodynamic effects. The field will move to rely solely on QSP and PBPK methods for determining the clinical relevance of potential DDI, as is already becoming more common for NDA approvals with the FDA [Citation200–203]. However, it will be important to expand on current studies with IVIVE models (both static mechanistic and dynamic) in order to better predict the potential for in vivo DDI. Performing in vitro DDIs studies with more complex systems (e.g. organ on a chip, micro-physiological systems) will also enhance the physiological relevance of the generated data and improve IVIVE. The field will continue to progress towards encompassing pharmacogenomic effects, genetic variability, and special populations (i.e. cancer patients who may use cannabis) into these DDI predictions. As models become more sophisticated, and the data used to populate them become more physiologically relevant, it will become standard to perform PBPK and similar modeling for all investigative DDI studies. Based upon these models, targeted in vivo clinical trials can be conducted to determine the conclusive impact of these potential cannabis drug interactions.
Even as in silico data moves to inform this field of research, the classification of cannabis as a Schedule I controlled substance continues to severely limit the accessibility of cannabis to investigators wishing to conduct well-controlled in vivo studies. Furthermore, due to the ballooning diversity of commercial cannabis products, research results may not realistically reflect current cannabis use. Commercially available products today vary by the source strain, THC and CBD content, contaminant documentation, and many other quality components. As cannabis becomes more commercially unregulated, it is extremely important to characterize and/or set quality benchmarks that will standardize the current, on the market, cannabis products. Presently, the amounts of prescribed THC and CBD and the amounts of THC and CBD in NIH-standardized cannabis cigarettes are well below the amount of THC and CBD consumed for recreational and self-medication purposes, which is well-documented to vary widely from individual to individual. Thus, current research may underpredict the potential DDI for severe or chronic use of cannabis.
Article highlights
Several metabolites of THC and CBD are found in the plasma at higher concentrations and for longer periods of time than their parent cannabinoids.
Plasma levels of the active and inactive cannabinoids are in the low micromolar range and vary depending on the mode of consumption, dose, and frequency of use.
THC, CBD, and their metabolites are inhibitors of many major cytochrome P450 and UDP-glucuronosyltransferase enzymes.
The major circulating plasma THC metabolite, THC-COOH-Gluc, inhibited several CYP enzymes including CYP2B6, CYP2C9, and CYP2D6.
CBD strongly inhibited the glucuronidation activity of UGTs 1A9, 2B4, 1A6, and 2B7, with IC50 values in the micromolar range.
Quantitative systems pharmacology and PBPK modelling will likely be a major focus for determining the clinical relevance of potential cannabinoid-drug interactions in the future.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Supplemental Material
Download MS Word (25.3 KB)Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/17512433.2022.2148655
Additional information
Funding
References
- Bonomo Y, Souza JDS, Jackson A, et al. Clinical issues in cannabis use. Br J Clin Pharmacol. 2018 Nov;84(11):2495–2498.
- Degenhardt L, Whiteford HA, Ferrari AJ, et al. Global burden of disease attributable to illicit drug use and dependence: findings from the Global Burden of Disease Study 2010. Lancet. 2013;Vol. 382(9904):1564–1574.
- Atakan Z. Cannabis, a complex plant: different compounds and different effects on individuals. Ther Adv Psychopharmacol. 2012;2(6):241–254.
- Ben Amar M. Cannabinoids in medicine: a review of their therapeutic potential. J Ethnopharmacol. 2006;105(1):1–25.
- Devinsky O, Marmanillo A, Hamlin T, et al. Observational study of medical marijuana as a treatment for treatment-resistant epilepsies. Ann Clin Transl Neurol. 2022;9(4):497–505.
- Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016;15(3):270–278.
- Grotenhermen F, Müller-Vahl K. The therapeutic potential of cannabis and cannabinoids. Dtsch Arztebl Int. 2012;109(29–30):495–501.
- Novotna A, Mares J, Ratcliffe S, et al. A randomized, double-blind, placebo-controlled, parallel-group, enriched-design study of nabiximols* (Sativex(®)), as add-on therapy, in subjects with refractory spasticity caused by multiple sclerosis. Eur J Neurol. 2011;18(9):1122–1131.
- Rahn EJ, Hohmann AG. Cannabinoids as pharmacotherapies for neuropathic pain: from the bench to the bedside. Neurotherapeutics. 2009;6(4):713–737.
- Mechoulam R, Shvo Y. Hashish—I: the structure of Cannabidiol. Tetrahedron. 1963;19(12):2073–2078.
- Grotenhermen F, Müller-Vahl K. Medicinal uses of marijuana and cannabinoids. Crit Rev Plant Sci. 2016;35(5–6):378–405.
- Pertwee RG. Cannabinoid pharmacology: the first 66 years. Br J Pharmacol. 2006;147(S1):S163–S171.
- Hillig KW. Genetic evidence for speciation in Cannabis (Cannabaceae). Genet Resour Crop Evol. 2005;52(2):161–180.
- Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86(8):1646–1647.
- Simiyu DC, Jang JH, Lee OR. Understanding cannabis sativa L.: current status of propagation, use, legalization, and haploid-inducer-mediated genetic engineering. Plants (Basel, Switzerland). 2022. 11(9):1236.
- Jin D, Henry P, Shan J, et al. Identification of phenotypic characteristics in three chemotype categories in the genus cannabis. HortScience horts. 2021;56(4):481–490.
- ElSohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci. 2005;78(5):539–548.
- Gaoni Y, Mechoulam R. The isolation and structure of delta-1-tetrahydrocannabinol and other neutral cannabinoids from hashish. J Am Chem Soc. 1971;93(1):217–224.
- Citti C, Linciano P, Russo F, et al. A novel phytocannabinoid isolated from Cannabis sativa L. with an in vivo cannabimimetic activity higher than Δ9-tetrahydrocannabinol: Δ9-Tetrahydrocannabiphorol. Sci Rep. 2019;9(1):20335.
- Dufresnes C, Jan C, Bienert F, et al. Broad-scale genetic diversity of cannabis for forensic applications. PLOS ONE. 2017;12(1):e0170522.
- Hillig KW, Mahlberg PG. A chemotaxonomic analysis of cannabinoid variation in Cannabis (Cannabaceae). Am J Bot. 2004;91(6):966–975.
- Welling MT, Shapter T, Rose TJ, et al. A belated green revolution for cannabis: virtual genetic resources to fast-track cultivar development. Front Plant Sci. 2016;7:1113.
- Andre CM, Hausman J-F, Guerriero G. Cannabis sativa: the plant of the thousand and one molecules [Review]. Front Plant Sci. 2016;7:19.
- Pennypacker SD, Cunnane K, Cash MC, et al. Potency and THERAPEUTIC THC and CBD Ratios: U.S. cannabis markets overshoot [Original research]. Front Pharmacol. 2022;13:921493.
- Matheson J, Le Foll B. Cannabis legalization and acute harm from high potency cannabis products: a narrative review and recommendations for public health [Review]. Front Psychiatry. 2020;11:591979.
- ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995–2014): analysis of current data in the United States. Biol Psychiatry. 2016;79(7):613–619.
- Bergamaschi MM, Queiroz RHC, Chagas MHN, et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology. 2011;36(6):1219–1226.
- Pennypacker SD, Romero-Sandoval EA. CBD and THC: do they complement each other like Yin and Yang? Pharmacother J Human Pharmacol Drug Ther. 2020;40(11):1152–1165.
- Jones NA, Hill AJ, Smith I, et al. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J Pharmacol Exp Ther. 2010;332(2):569–577.
- Boggs DL, Nguyen JD, Morgenson D, et al. Clinical and preclinical evidence for functional interactions of cannabidiol and Δ9-tetrahydrocannabinol. Neuropsychopharmacology. 2018;43(1):142–154.
- Meiri E, Jhangiani H, Vredenburgh JJ, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23(3):533–543.
- Levin DN, Dulberg Z, Chan AW, et al. A randomized-controlled trial of nabilone for the prevention of acute postoperative nausea and vomiting in elective surgery. Can J Anaesth. 2017;64(4):385–395.
- Haney M, Gunderson EW, Rabkin J, et al. Dronabinol and marijuana in HIV-positive marijuana smokers. Caloric intake, mood, and sleep. J Acquir Immune Defic Syndr. 2007;45(5):545–554.
- Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the lennox-gastaut syndrome. N Engl J Med. 2018;378(20):1888–1897.
- Silvestro S, Mammana S, Cavalli E, et al. Use of cannabidiol in the treatment of epilepsy: efficacy and security in clinical trials. Molecules. 2019;24(8):1459.
- Kavia RB, De Ridder D, Constantinescu CS, et al. Randomized controlled trial of Sativex to treat detrusor overactivity in multiple sclerosis. Mult Scler. 2010;16(11):1349–1359.
- Legislatures NCoS. State medical cannabis laws [Internet]. Denver CO: National Conference of State Legislatures. June 2022 Available from: https://www.ncsl.org/research/health/state-medical-marijuana-laws.aspx.
- Yu B, Chen X, Chen X, et al. Marijuana legalization and historical trends in marijuana use among US residents aged 12–25: results from the 1979–2016 national survey on drug use and health. BMC Public Health. 2020;20(1):156.
- Booklet 3 - Drug market trends: opioids, cannabis [Internet]. United Nations Office on Drugs and Crime. June 2022. Available from: https://www.unodc.org/unodc/en/data-and-analysis/wdr-2021_booklet-3.html.
- Testai FD, Gorelick PB, Aparicio HJ, et al. Use of marijuana: effect on brain health: a scientific statement from the American heart association. Stroke. 2022;53(4):e176–e187.
- SAMHSA releases 2020 national survey on drug use and health. Rockville MD:SAMSHA. October 2021 Available from: https://www.samhsa.gov/newsroom/press-announcements/202110260320.
- Kocis PT, Vrana KE. Delta-9-Tetrahydrocannabinol and Cannabidiol Drug-Drug Interactions. Med Cannabis Cannabinoids. 2020;3(1):61–73.
- Turna J, Syan SK, Frey BN, et al. Cannabidiol as a novel candidate alcohol use disorder pharmacotherapy: a systematic review. Alcohol Clin Exp Res. 2019;43(4):550–563.
- Jin D, Dai K, Xie Z, et al. Secondary metabolites profiled in cannabis inflorescences, leaves, stem barks, and roots for medicinal purposes. Sci Rep. 2020;10(1):3309.
- Murillo-Rodriguez E. Molecular, biochemical and genetic aspects of cannabinoids. 2015.
- Hanuš LO, Meyer SM, Muñoz E, et al. Phytocannabinoids: a unified critical inventory. Nat Prod Rep. 2016;33(12):1357–1392.
- Lelario F, Pascale R, Bianco G, et al. Hemp chemotype definition by cannabinoids characterization Using LC-ESI(+)-LTQ-FTICR MS and infrared multiphoton dissociation. Separations. 2021;8(12):245.
- Wang M, Wang Y-H, Avula B, et al. Decarboxylation study of acidic cannabinoids: a novel approach using ultra-high-performance supercritical fluid chromatography/photodiode array-mass spectrometry. Cannabis Cannabinoid Res. 2016;1(1):262–271.
- Walsh KB, McKinney AE, Holmes AE. Minor cannabinoids: biosynthesis, molecular pharmacology and potential therapeutic uses. Front Pharmacol. 2021;12:777804.
- Abrahamov A, Abrahamov A, Mechoulam R. An efficient new cannabinoid antiemetic in pediatric oncology. Life Sci. 1995;56(23):2097–2102.
- Leas EC, Nobles AL, Shi Y, et al. Public interest in ∆(8)-Tetrahydrocannabinol (delta-8-THC) increased in US states that restricted ∆(9)-Tetrahydrocannabinol (delta-9-THC) use. Int J Drug Policy. 2022;101:103557.
- Bridgeman MB, Abazia DT. Medicinal cannabis: history, pharmacology, and implications for the acute care setting. P T. 2017;42(3):180–188.
- Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770–1804.
- Cox EJ, Maharao N, Patilea-Vrana G, et al. A marijuana-drug interaction primer: precipitants, pharmacology, and pharmacokinetics. Pharmacol Ther. 2019;201:25–38.
- Ohlsson A, Lindgren J-E, Andersson S, et al. Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom. 1986;13(2):77–83
- Desrosiers NA, Himes SK, Scheidweiler KB, et al. Phase I and II cannabinoid disposition in blood and plasma of occasional and frequent smokers following controlled smoked cannabis. Clin Chem. 2014;60(4):631–643.
- Schwope DM, Karschner EL, Gorelick DA, et al. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem. 2011;57(10):1406–1414.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42(4):327–360.
- Chayasirisobhon S. Mechanisms of action and pharmacokinetics of cannabis. Perm J. 2021;25(1):1–3.
- Lindgren JE, Ohlsson A, Agurell S, et al. Clinical effects and plasma levels of delta 9-tetrahydrocannabinol (delta 9-THC) in heavy and light users of cannabis. Psychopharmacology (Berl). 1981;74(3):208–212.
- Sharma P, Murthy P, Bharath MMS. Chemistry, metabolism, and toxicology of cannabis: clinical implications. Iran J Psychiatry. 2012;7(4):149–156.
- Perez-Reyes M. Marijuana smoking: factors that influence the bioavailability of tetrahydrocannabinol. NIDA Res Monogr. 1990;99:42–62.
- Nadulski T, Pragst F, Weinberg G, et al. Randomized, double-blind, placebo-controlled study about the effects of cannabidiol (CBD) on the pharmacokinetics of Delta9-tetrahydrocannabinol (THC) after oral application of THC verses standardized cannabis extract. Ther Drug Monit. 2005;27(6):799–810.
- Vandrey R, Herrmann ES, Mitchell JM, et al. Pharmacokinetic profile of oral cannabis in humans: blood and oral fluid disposition and relation to pharmacodynamic outcomes. J Anal Toxicol. 2017;41(2):83–99.
- Taylor L, Gidal B, Blakey G, et al. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053–1067.
- Tayo B, Taylor L, Sahebkar F, et al. A phase I, open-label, parallel-group, single-dose trial of the pharmacokinetics, safety, and tolerability of cannabidiol in subjects with mild to severe renal impairment. Clin Pharmacokinet. 2020;59(6):747–755.
- Anderson LL, Etchart MG, Bahceci D, et al. Cannabis constituents interact at the drug efflux pump BCRP to markedly increase plasma cannabidiolic acid concentrations. Sci Rep. 2021;11(1):14948.
- Karschner EL, Schwope DM, Schwilke EW, et al. Predictive model accuracy in estimating last Δ9-tetrahydrocannabinol (THC) intake from plasma and whole blood cannabinoid concentrations in chronic, daily cannabis smokers administered subchronic oral THC. Drug Alcohol Depend. 2012 Oct 1;125(3):313–319.
- Millar SA, Stone NL, Yates AS, et al. A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharmacol. 2018;9:1365.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol. 1992;16(5):276–282.
- Guy GW, Robson PJ. A phase I, double blind, three-way crossover study to assess the pharmacokinetic profile of Cannabis Based Medicine Extract (CBME) administered sublingually in variant cannabinoid ratios in normal healthy male volunteers (GWPK0215). J Cannabis Ther. 2004;3(4):121–152.
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008 Jan;153(2):199–215.
- Toennes SW, Ramaekers JG, Theunissen EL, et al. Comparison of cannabinoid pharmacokinetic properties in occasional and heavy users smoking a marijuana or placebo joint. J Anal Toxicol. 2008;32(7):470–477.
- Nasrin S, Watson CJW, Perez-Paramo YX, et al. Cannabinoid metabolites as inhibitors of major hepatic CYP450 enzymes, with implications for cannabis-drug interactions. Drug Metab Dispos. 2021a;49(12):1070–1080.
- Wall ME, Sadler BM, Brine D, et al. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin Pharmacol Ther. 1983;34(3):352–363.
- Harvey DJ, Mechoulam R. Metabolites of cannabidiol identified in human urine. Xenobiotica. 1990;20(3):303–320.
- Ujváry I, Hanuš L. Human metabolites of cannabidiol: a review on their formation, biological activity, and relevance in therapy. Cannabis Cannabinoid Res. 2016;1(1):90–101.
- Levitt DG. Heterogeneity of human adipose blood flow. BMC Clin Pharmacol. 2007;7:1.
- Thomas BF, Compton DR, Martin BR. Characterization of the lipophilicity of natural and synthetic analogs of delta 9-tetrahydrocannabinol and its relationship to pharmacological potency. J Pharmacol Exp Ther. 1990;255(2):624–630.
- Devinsky O, Cilio MR, Cross H, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014;55(6):791–802.
- Watanabe K, Yamamoto I, Oguri K, et al. Metabolic disposition of delta 8-tetrahydrocannabinol and its active metabolites, 11-hydroxy-delta 8-tetrahydrocannabinol and 11-oxo-delta 8-tetrahydrocannabinol, in mice. Drug Metab Dispos. 1981;9(3):261–264.
- Calapai F, Cardia L, Sorbara EE, et al. Cannabinoids, blood-brain barrier, and brain disposition. Pharmaceutics. 2020;12(3):265.
- Child RB, Tallon MJ. Cannabidiol (CBD) dosing: plasma pharmacokinetics and effects on accumulation in skeletal muscle, liver and adipose tissue. Nutrients. 2022;14(10):2101.
- Gronewold A, Skopp G. A preliminary investigation on the distribution of cannabinoids in man. Forensic Sci Int. 2011;210(1–3):e7–e11.
- Kudo K, Nagata T, Kimura K, et al. Sensitive determination of delta 9-tetrahydrocannabinol in human tissues by GC-MS. J Anal Toxicol. 1995;19(2):87–90.
- Widman M, Agurell S, Ehrnebo M, et al. Binding of (+)- and (minus)-delta-1-tetrahydrocannabinols and (minus)-7-hydroxy-delta-1-tetrahydrocannabinol to blood cells and plasma proteins in man. J Pharm Pharmacol. 1974;26(11):914–916.
- Skopp G, Pötsch L, Mauden M, et al. Partition coefficient, blood to plasma ratio, protein binding and short-term stability of 11-nor-Δ9-carboxy tetrahydrocannabinol glucuronide. Forensic Sci Int. 2002;126(1):17–23.
- Wahlqvist M, Nilsson IM, Sandberg F, et al. Binding of δ1-tetrahydrocannabinol to human plasma proteins. Biochem Pharmacol. 1970;19(9):2579–2582.
- Lucas CJ, Galettis P, Schneider J. The pharmacokinetics and the pharmacodynamics of cannabinoids. Br J Clin Pharmacol. 2018;84(11):2477–2482.
- Bland TM, Haining RL, Tracy TS, et al. CYP2C-catalyzed delta9-tetrahydrocannabinol metabolism: kinetics, pharmacogenetics and interaction with phenytoin. Biochem Pharmacol. 2005;70(7):1096–1103.
- Matsunaga T, Iwawaki Y, Watanabe K, et al. Metabolism of Δ9-tetrahydrocannabinol by cytochrome P450 isozymes purified from hepatic microsomes of monkeys. Life Sci. 1995;56(23):2089–2095.
- Watanabe K, Yamaori S, Funahashi T, et al. Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 2007;80(15):1415–1419.
- Lemberger L, Silberstein Stephen D, Axelrod J, et al. Marihuana: studies on the disposition and metabolism of delta-9-tetrahydrocannabinol in man. Science. 1970;170(3964):1320–1322.
- Mechoulam R, BenZvi Z, Agurell S, et al. Delta 6-tetrahydrocannabinol-7-oic acid, a urinary delta 6-THC metabolite: isolation and synthesis. Experientia. 1973;29(10):1193–1195.
- Huestis MA, Cone EJ. Differentiating new marijuana use from residual drug excretion in occasional marijuana users*. J Anal Toxicol. 1998;22(6):445–454.
- Grauwiler SB, Drewe J, Scholer A. Sensitivity and specificity of urinary cannabinoid detection with two immunoassays after controlled oral administration of cannabinoids to humans. Ther Drug Monit. 2008;30(4):530–535.
- Gasse A, Vennemann M, Köhler H, et al. Toxicogenetic analysis of Δ9-THC-metabolizing enzymes. Int J Legal Med. 2020;134(6):2095–2103.
- Mazur A, Lichti CF, Prather PL, et al. Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496–1504.
- Halldin MM, Widman M, Vb C, et al. Identification of in vitro metabolites of delta 1-tetrahydrocannabinol formed by human livers. Drug Metab Dispos. 1982;10(4):297–301.
- Huestis MA. Pharmacokinetics and metabolism of the plant cannabinoids, delta9-tetrahydrocannabinol, cannabidiol and cannabinol. Handb Exp Pharmacol. 2005;168:657–690.
- Harvey DJ, Martin BR, Paton WD. Identification and measurement of cannabinoids and their in vivo metabolites in liver by gas chromatography–mass spectrometry. Adv Biosci. 1978;22-23:45–62.
- Jiang R, Yamaori S, Okamoto Y, et al. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab Pharmacokinet. 2013;28(4):332–338.
- Beers JL, Fu D, Jackson KD. Cytochrome P450-catalyzed metabolism of cannabidiol to the active metabolite 7-hydroxy-cannabidiol. Drug Metab Dispos. 2021;49(10):882–891.
- Williams PL, Moffat AC. Identification in human urine of delta 9-tetrahydrocannabinol-11-oic acid glucuronide: a tetrahydrocannabinol metabolite. J Pharm Pharmacol. 1980;32(7):445–448.
- Vandevenne M, Vandenbussche H, Verstraete A. Detection time of drugs of abuse in urine. Acta Clin Belg. 2000;55(6):323–333.
- Heuberger JA, Guan Z, Oyetayo OO, et al. Population pharmacokinetic model of THC integrates oral, intravenous, and pulmonary dosing and characterizes short- and long-term pharmacokinetics. Clin Pharmacokinet. 2015;54(2):209–219.
- Johansson E, Halldin MM, Agurell S, et al. Terminal elimination plasma half-life of Δ1-tetrahydrocannabinol (Δ1-THC) in heavy users of marijuana. Eur J Clin Pharmacol. 1989;37(3):273–277.
- Smith-Kielland A, Skuterud B, Mørland J. Urinary excretion of 11-nor-9-carboxy-delta9-tetrahydrocannabinol and cannabinoids in frequent and infrequent drug users. J Anal Toxicol. 1999;23(5):323–332.
- Karschner EL, Schwilke EW, Lowe RH, et al. Implications of plasma Delta9-tetrahydrocannabinol, 11-hydroxy-THC, and 11-nor-9-carboxy-THC concentrations in chronic cannabis smokers. J Anal Toxicol. 2009;33(8):469–477.
- Taylor L, Crockett J, Tayo B, et al. A phase 1, open-label, parallel-group, single-dose trial of the pharmacokinetics and safety of cannabidiol (CBD) in subjects with mild to severe hepatic impairment. J Clin Pharmacol. 2019;59(8):1110–1119.
- Perkins D, Butler J, Ong K, et al. A phase 1, randomised, placebo-controlled, dose escalation study to investigate the safety, tolerability and pharmacokinetics of cannabidiol in fed healthy volunteers. Eur J Drug Metab Pharmacokinet. 2020;45(5):575–586.
- Consroe P, Kennedy K, Schram K. Assay of plasma cannabidiol by capillary gas chromatography/ion trap mass spectroscopy following high-dose repeated daily oral administration in humans. Pharmacol Biochem Behav. 1991;40(3):517–522.
- Di Marzo V. New approaches and challenges to targeting the endocannabinoid system. Nat Rev Drug Discov. 2018;17(9):623–639.
- Kendall DA, Yudowski GA. Cannabinoid receptors in the central nervous system: their signaling and roles in disease [Review]. Front Cell Neurosci. 2017;10. DOI:10.3389/fncel.2016.00294
- Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins, Leukotrienes and Essential Fatty Acids (PLEFA). 2002;66(2):101–121.
- Kumar RN, Chambers WA, Pertwee RG. Pharmacological actions and therapeutic uses of cannabis and cannabinoids. Anaesthesia. 2001;56(11):1059–1068.
- Matsuda LA, Lolait SJ, Brownstein MJ, et al. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564.
- Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65.
- Cristino L, Bisogno T, Di Marzo V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol. 2020;16(1):9–29.
- Galiègue S, Mary S, Marchand J, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232(1):54–61.
- Schatz AR, Lee M, Condie RB, et al. Cannabinoid receptors CB1 and CB2: a characterization of expression and adenylate cyclase modulation within the immune system. Toxicol Appl Pharmacol. 1997;142(2):278–287.
- Simard M, Rakotoarivelo V, Di Marzo V, et al. Expression and functions of the CB2 receptor in human leukocytes [Perspective]. Front Pharmacol. 2022;13:826400.
- Herkenham M, Lynn AB, Little MD, et al. Cannabinoid receptor localization in brain. Proc Natl Acad Sci U S A. 1990;87(5):1932–1936.
- Tsou K, Brown S, Sañudo-Peña MC, et al. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83(2):393–411.
- Bayewitch M, Rhee MH, Avidor-Reiss T, et al. (-)-Delta9-tetrahydrocannabinol antagonizes the peripheral cannabinoid receptor-mediated inhibition of adenylyl cyclase. J Biol Chem. 1996;271(17):9902–9905.
- Felder CC, Joyce KE, Briley EM, et al. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48(3):443–450.
- Iwamura H, Suzuki H, Ueda Y, et al. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J Pharmacol Exp Ther. 2001;296(2):420–425.
- Pertwee RG, Howlett AC, Abood ME, et al. International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB₁ and CB₂. Pharmacol Rev. 2010;62(4):588.
- Rhee MH, Vogel Z, Barg J, et al. Cannabinol derivatives: binding to cannabinoid receptors and inhibition of adenylylcyclase. J Med Chem. 1997;40(20):3228–3233.
- Showalter VM, Compton DR, Martin BR, et al. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278(3):989–999.
- Nachnani R, Raup-Konsavage WM, Vrana KE. The pharmacological case for cannabigerol. J Pharmacol Exp Ther. 2021;376(2):204.
- Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613–623.
- Laprairie RB, Bagher AM, Kelly ME, et al. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. 2015;172(20):4790–4805.
- Anderson LL, Absalom NL, Abelev SV, et al. Coadministered cannabidiol and clobazam: preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia. 2019;60(11):2224–2234.
- Bakas T, van Nieuwenhuijzen PS, Devenish SO, et al. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABA(A) receptors. Pharmacol Res. 2017;119:358–370.
- Ryberg E, Larsson N, Sjögren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092–1101.
- Scavone JL, Sterling RC, Van Bockstaele EJ. Cannabinoid and opioid interactions: implications for opiate dependence and withdrawal. Neuroscience. 2013;248:637–654.
- Muller C, Morales P, Reggio PH. Cannabinoid Ligands Targeting TRP Channels. Front Mol Neurosci. 2018;11:487.
- Vučković S, Srebro D, Vujović KS, et al. Cannabinoids and pain: new insights from old molecules [Review]. Front Pharmacol. 2018;9. DOI:10.3389/fphar.2018.01259.
- Russo EB, Burnett A, Hall B, et al. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res. 2005;30(8):1037–1043.
- O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152(5):576–582.
- Zagzoog A, Mohamed KA, Kim HJJ, et al. In vitro and in vivo pharmacological activity of minor cannabinoids isolated from Cannabis sativa. Sci Rep. 2020 Nov 23;10(1):20405.
- Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–141.
- Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396.
- In vitro drug interaction studies-cytochrome P450 enzyme- and transporter-mediated drug interactions guidance for industry [Internet]. Rockville MD:USFood & Drug Administration January 2022 Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions.
- Yamaori S, Kushihara M, Yamamoto I, et al. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem Pharmacol. 2010;79(11):1691–1698.
- Bansal S, Maharao N, Paine MF, et al. Predicting the potential for cannabinoids to precipitate pharmacokinetic drug interactions via reversible inhibition or inactivation of major cytochromes P450. Drug Metab Dispos. 2020;48(10):1008–1017.
- Bansal S, Paine MF, Unadkat JD. Comprehensive predictions of cytochrome P450 (CYP)-mediated in vivo cannabinoid-drug interactions based on reversible and time-dependent CYP inhibition in human liver microsomes. Drug Metab Dispos. 2022;50(4):DMD-AR-2021–000734.
- Yamaori S, Maeda C, Yamamoto I, et al. Differential inhibition of human cytochrome P450 2A6 and 2B6 by major phytocannabinoids. Forensic Toxicol. 2011a;29(2):117–124.
- Yamaori S, Ebisawa J, Okushima Y, et al. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011b;88(15–16):730–736.
- Yamaori S, Koeda K, Kushihara M, et al. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab Pharmacokinet. 2012;27(3):294–300.
- Yamaori S, Okamoto Y, Yamamoto I, et al. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos. 2011c;39(11):2049–2056.
- Doohan PT, Oldfield LD, Arnold JC, et al. Cannabinoid interactions with cytochrome P450 drug metabolism: a full-spectrum characterization. Aaps j. 2021;23(4):91.
- Geffrey AL, Pollack SF, Bruno PL, et al. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246–1251.
- Tolbert D, Bekersky I, Chu HM, et al. Drug-metabolism mechanism: knowledge-based population pharmacokinetic approach for characterizing clobazam drug-drug interactions. J Clin Pharmacol. 2016;56(3):365–374.
- Gaston TE, Bebin EM, Cutter GR, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58(9):1586–1592.
- Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90(14):e1204–e1211.
- Morrison G, Crockett J, Blakey G, et al., A phase 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019;8(8):1009–1031.
- Ridout KK, Young-Wolff KC, Ridout SJ. A change in blood carbamazepine levels associated with cannabis use: implications for clinical practice. J Clin Psychiatry. 2021;82(3). DOI:10.4088/JCP.20cr13777
- Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46(1):86–95.
- Qian Y, Gurley BJ, Markowitz JS. The Potential for Pharmacokinetic Interactions Between Cannabis Products and Conventional Medications. J Clin Psychopharmacol. 2019a;39(5):462–471.
- Thai C, Tayo B, Critchley D. A phase 1 open-label, fixed-sequence pharmacokinetic drug interaction trial to investigate the effect of cannabidiol on the CYP1A2 probe caffeine in healthy subjects. Clin Pharmacol Drug Dev. 2021;10(11):1279–1289.
- Roth MD, Marques-Magallanes JA, Yuan M, et al. Induction and regulation of the carcinogen-metabolizing enzymeCYP1A1 by marijuana smoke andδ 9 -tetrahydrocannabinol. Am J Respir Cell Mol Biol. 2001;24(3):339–344.
- Zhang W, Xu G, McLeod HL. Comprehensive evaluation of carboxylesterase-2 expression in normal human tissues using tissue array analysis. Appl Immunohistochem Mol Morphol. 2002;10(4):374–380.
- Taketani M, Shii M, Ohura K, et al. Carboxylesterase in the liver and small intestine of experimental animals and human. Life Sci. 2007;81(11):924–932.
- Hatfield MJ, Tsurkan L, Garrett M, et al. Organ-specific carboxylesterase profiling identifies the small intestine and kidney as major contributors of activation of the anticancer prodrug CPT-11. Biochem Pharmacol. 2011;81(1):24–31.
- Imai T, Taketani M, Shii M, et al. Substrate specificity of carboxylesterase isozymes and their contribution to hydrolase activity in human liver and small intestine. Drug Metab Dispos. 2006 Oct;34(10):1734–1741.
- Imai T, Yoshigae Y, Hosokawa M, et al. Evidence for the involvement of a pulmonary first-pass effect via carboxylesterase in the disposition of a propranolol ester derivative after intravenous administration. J Pharmacol Exp Ther. 2003 Dec;307(3):1234–1242.
- Wang D, Zou L, Jin Q, et al. Human carboxylesterases: a comprehensive review. Acta Pharm Sin B. 2018;8(5):699–712.
- Laizure SC, Herring V, Hu Z, et al. The role of human carboxylesterases in drug metabolism: have we overlooked their importance? Pharmacotherapy. 2013;33(2):210–222.
- Qian Y, Wang X, Markowitz JS. In vitro inhibition of carboxylesterase 1 by major cannabinoids and selected metabolites. Drug Metab Dispos. 2019b;47(5):465–472.
- Qian Y, Markowitz JS. Prediction of carboxylesterase 1 (CES1)-mediated in vivo drug interaction between methylphenidate and cannabinoids using static and physiologically based pharmacokinetic models. Drug Metab Dispos. 2022;50(7):968.
- Nasrin S, Watson CJW, Bardhi K, et al., Inhibition of UDP-glucuronosyltransferase enzymes by major cannabinoids and their metabolites. Drug Metab Dispos. 2021b;49(12):1081–1089.
- Meech R, Hu DG, McKinnon RA, et al. The UDP-Glycosyltransferase (UGT) superfamily: new members, new functions, and novel paradigms. Physiol Rev. 2019;99(2):1153–1222.
- Bushey RT, Lazarus P. Identification and functional characterization of a novel UDP-glucuronosyltransferase 2A1 splice variant: potential importance in tobacco-related cancer susceptibility. J Pharmacol Exp Ther. 2012;343(3):712–724.
- MacKenzie PI, Rogers A, Elliot DJ, et al. The novel UDP glycosyltransferase 3A2: cloning, catalytic properties, and tissue distribution. Mol Pharmacol. 2011;79(3):472–478.
- Williams JA, Hyland R, Jones BC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–1208.
- Al Saabi A, Allorge D, Sauvage FL, et al. Involvement of UDP-glucuronosyltransferases UGT1A9 and UGT2B7 in ethanol glucuronidation, and interactions with common drugs of abuse. Drug Metab Dispos. 2013;41(3):568–574.
- Rowland A, Miners JO, Mackenzie PI. The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol. 2013;45(6):1121–1132.
- Patsalos PN, Szaflarski JP, Gidal B, et al. Clinical implications of trials investigating drug-drug interactions between cannabidiol and enzyme inducers or inhibitors or common antiseizure drugs. Epilepsia. 2020;61(9):1854–1868.
- Seo KA, Bae SK, Choi YK, et al. Metabolism of 1’- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab Dispos. 2010;38(11):2007–2013.
- Abrams DI, Couey P, Shade SB, et al. Cannabinoid–opioid interaction in chronic pain. Clin Pharmacol Ther. 2011;90(6):844–851.
- Takeda S, Kitajima Y, Ishii Y, et al. Inhibition of UDP-glucuronosyltransferase 2b7-catalyzed morphine glucuronidation by ketoconazole: dual mechanisms involving a novel noncompetitive mode. Drug Metab Dispos. 2006;34(8):1277–1282.
- Anderson L, Doohan P, Oldfield L, et al. Citalopram and cannabidiol: in vitro and in vivo evidence of pharmacokinetic interactions relevant to the treatment of anxiety disorders in young people. J Clin Psychopharmacol. 2021;41(5):525–533.
- Devinsky O, Cross JH, Wright S. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;377(7):699–700.
- Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (London, England). 2018;391(10125):1085–1096.
- Grayson L, Vines B, Nichol K, et al. An interaction between warfarin and cannabidiol, a case report. Epilepsy Behav Case Rep. 2018;9:10–11.
- Damkier P, Lassen D, Christensen MMH, et al. Interaction between warfarin and cannabis. Basic Clin Pharmacol Toxicol. 2019;124(1):28–31.
- Yamreudeewong W, Wong HK, Brausch LM, et al. Probable interaction between warfarin and marijuana smoking. Ann Pharmacother. 2009;43(7):1347–1353.
- Hsu A, Painter NA. Probable interaction between warfarin and inhaled and oral administration of cannabis. J Pharm Pract. 2020;33(6):915–918.
- Brown GW, Bellnier TJ, Janda M, et al. Δ-9-tetrahydrocannabinol dose increase leads to warfarin drug interaction and elevated INR. J Am Pharm Assoc. 2021;61(1):e57–e60.
- Wilson M, Gogulski HY, Cuttler C, et al. Cannabis use moderates the relationship between pain and negative affect in adults with opioid use disorder. Addict Behav. 2018;77:225–231.
- Clem SN, Bigand TL, Wilson M. Cannabis use motivations among adults prescribed opioids for pain versus opioid addiction. Pain Manag Nurs. 2020 Feb;21(1):43–47.
- Thomas TF, Metaxas ES, Nguyen T, et al. Case report: medical cannabis-warfarin drug-drug interaction. J Cannabis Res. 2022;4(1):6.
- Engels FK, de Jong FA, Sparreboom A, et al. Medicinal cannabis does not influence the clinical pharmacokinetics of irinotecan and docetaxel. Oncologist. 2007;12(3):291–300.
- Riggs CE Jr., Egorin MJ, Fuks JZ, et al. Initial observations on the effects of delta 9-tetrahydrocannabinol on the plasma pharmacokinetics of cyclophosphamide and doxorubicin. J Clin Pharmacol. 1981;21(S1):90s–98s.
- Kosel BW, Aweeka FT, Benowitz NL, et al. The effects of cannabinoids on the pharmacokinetics of indinavir and nelfinavir. Aids. 2002;16(4):543–550.
- Basit A, Neradugomma NK, Wolford C, et al. Characterization of differential tissue abundance of major non-CYP enzymes in human. Mol Pharm. 2020;17(11):4114–4124.
- Margaillan G, Rouleau M, Fallon JK, et al. Quantitative profiling of human renal UDP-glucuronosyltransferases and glucuronidation activity: a comparison of normal and tumoral kidney tissues. Drug Metab Dispos. 2015;43(4):611.
- Yoshida K, Budha N, Jin JY. Impact of physiologically based pharmacokinetic models on regulatory reviews and product labels: frequent utilization in the field of oncology. Clin Pharmacol Ther. 2017;101(5):597–602.
- Huang SM, Abernethy DR, Wang Y, et al. The utility of modeling and simulation in drug development and regulatory review. J Pharm Sci. 2013;102(9):2912–2923.
- Grimstein M, Yang Y, Zhang X, et al. Physiologically based pharmacokinetic modeling in regulatory science: an update from the U.S. food and drug administration’s office of clinical pharmacology. J Pharm Sci. 2019;108(1):21–25
- Jamei M. Recent advances in development and application of Physiologically-Based Pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Current Pharmacol Rep. 2016;2(3):161–169.