
ABSTRACT
KATP channels are ligand-gated potassium channels that couple cellular energetics with membrane potential to regulate cell activity. Each channel is an eight subunit complex comprising four central pore-forming Kir6 inward rectifier potassium channel subunits surrounded by four regulatory subunits known as the sulfonylurea receptor, SUR, which confer homeostatic metabolic control of KATP gating. SUR is an ATP binding cassette (ABC) protein family homolog that lacks membrane transport activity but is essential for KATP expression and function. For more than four decades, understanding the structure-function relationship of Kir6 and SUR has remained a central objective of clinical significance. Here, we review progress in correlating the wealth of functional data in the literature with recent KATP cryoEM structures.
Introduction
Metabolism is a defining feature of life. Fitness and survival require an ability to tune physiological functions to changing metabolic environment. ATP-sensitive potassium (KATP) channels are ligand-gated potassium channels that regulate K+ efflux in response to changes in intracellular ATP and ADP concentrations. They are molecular sensors of cellular metabolism that endow those cells in which they are expressed with a homeostatic mechanism that modifies cellular activities controlled by membrane excitability in accord with the energetic state of the cell. The KATP channel current was originally identified four decades ago in cardiac myocytes [Citation1]. Soon after, pancreatic β-cells and then many other cell types were reported to possess K+ channels with similar properties [Citation2]. Extensive studies now show KATP channels are widely expressed among electrically excitable cells and regulate processes ranging from hormone secretion, vascular tone, learning and memory, to cardiac and neuronal protection against ischemic events [Citation3,Citation4]. Pharmacologically, direct regulation of KATP channel activity is a principal mechanism of action for anti-diabetic sulfonylurea drugs, and vasodilators diazoxide, minoxidil and pinacidil [Citation5].
Discovery that sulfonylureas promote insulin secretion by inhibiting the KATP channel in pancreatic β-cells [Citation6,Citation7] facilitated cloning of the KATP genes in the mid-90s [Citation2,Citation8,Citation9]. This revealed that KATP channels include as subunits both an inward rectifier potassium channel (Kir) family homolog, and an ATP Binding Cassette (ABC) transporter family protein. The K+ conduction unit is formed by one of two Kir6 gene products (Kir6.1 or Kir6.2). While Kir channels are named for their reduced activity at positive membrane potentials [Citation10], due to block by intracellular Mg2+ and polyamines [Citation11], Kir6 isoforms exhibit only mild inward rectification and thus KATP channel activity in cells has little voltage-dependence [Citation10]. The expression and gating of Kir6 in the KATP channel complex requires co-expression of the sulfonylurea receptor (SUR), named for its binding sulfonylureas [Citation12]. Evolved from the ABC transporter family, SUR retains active nucleotide binding functional domains, but does not itself possess transporting activity. Its sole known function is to regulate Kir6 activity. Single-particle EM structures discussed below show that the SUR central cavity, rather than accept transmembrane cargo, is a regulated binding site for the Kir6 N-terminal peptide.
Mammals contain a pair of chromosomally linked SUR and Kir6 genes (), which give rise to KATP channel isoforms with distinct activation characteristics, tissue specific expression, and cellular functions. A first pair, located on human chromosome 11 (and mouse Chr 7), comprises ABCC8 encoding SUR1 and KCNJ11 encoding Kir6.2. The Kir6.2/SUR1 isoform is the predominant isoform expressed by insulin-secreting β-cells of pancreatic islets, and by neurons in brain, and has essential roles in hormone secretion and learning and memory. A second pair of KATP genes, located on human chromosome 12 (and mouse Chr 6), includes ABCC9 encoding SUR2 and KCNJ8 encoding Kir6.1. The ABCC9 gene generates two major splice variant proteins, SUR2A and SUR2B. SUR2A and Kir6.2 form the predominant KATP channels found in ventricle myocytes, where they open in response to metabolic stress to shorten cardiac action potentials [Citation4,Citation13]. The SUR2B and Kir6.1 combination is the predominant subtype in vascular and non-vascular smooth muscle, and regulates blood pressure and lymph transport, as well as intestine contractions [Citation3,Citation14]. Mutations in KATP genes that cause gain- or loss-of-function characteristics in specific KATP channel isoforms are linked to a series of human diseases (see recent reviews in [Citation14–17]).
Figure 1. Molecular structure of KATP channels. (a) genes encoding SUR1 and Kir6.2. (b) genes encoding SUR2 (A/B) and Kir6.1. (c) topology of SUR and Kir6 proteins. Note the arginine-based ER retention motif is RKR in SUR1, Kir6.1, and Kir6.2, but RKQ in SUR2A/SUR2B. (d) CryoEM reconstruction of the Kir6.2/SUR1 KATP channel viewed from the side (left) and from the bottom (intracellularly) (right). (e) structural model of the Kir6.2/SUR1 KATP channel from the cryoEM reconstruction shown in (d), viewed from the side (left; only two SUR1 subunits are shown for clarity) and from the top (extracellularly) (right). Panels (d) and (e) are adapted from in Martin et al. eLife, 2017 [Citation28].
![Figure 1. Molecular structure of KATP channels. (a) genes encoding SUR1 and Kir6.2. (b) genes encoding SUR2 (A/B) and Kir6.1. (c) topology of SUR and Kir6 proteins. Note the arginine-based ER retention motif is RKR in SUR1, Kir6.1, and Kir6.2, but RKQ in SUR2A/SUR2B. (d) CryoEM reconstruction of the Kir6.2/SUR1 KATP channel viewed from the side (left) and from the bottom (intracellularly) (right). (e) structural model of the Kir6.2/SUR1 KATP channel from the cryoEM reconstruction shown in (d), viewed from the side (left; only two SUR1 subunits are shown for clarity) and from the top (extracellularly) (right). Panels (d) and (e) are adapted from Figure 2 in Martin et al. eLife, 2017 [Citation28].](/cms/asset/7bef8c34-14ed-49da-8b37-e0fed738c8bf/kchl_a_2327708_f0001_oc.jpg)
The biomedical importance of KATP channels as well as their unusual architecture has attracted considerable interest in understanding how two structurally dissimilar constituent proteins work in unison as a metabolic sensor. Studies occupying the past few decades have generated a wealth of inferred information on the channel’s structure-function relationship principally by correlating mutation perturbations and functional consequences on expression and activity. Over the past several years, high resolution cryoEM structures of KATP channels have unveiled the composition of structural elements and linkages that form, operate, and regulate the channel complex (). An expanding series of structural isoforms in various apo and liganded and mutation-bearing states has begun to reveal the dynamic conformational mechanisms by which KATP channels are gated physiologically by ligand binding, modulated by drugs, and perturbed by mutations [Citation18–21].
Here, we focus on the structural and functional role of SUR and Kir6 in KATP channel regulation, with particular attention to channel gating by intracellular ATP and ADP, and by membrane phospholipids, in particular PI(4,5)P2. Discussion necessarily centers on the SUR1/Kir6.2 channel, which is best studied to date, but includes insight from unique structural features of the SUR2B/Kir6.1 channel, and SUR2A in isolation, that address KATP functional diversity. Before assimilating the multiple binding sites and domain interactions through which SUR1 and Kir6.2 arbitrate channel activity, we briefly describe the macromolecular features of KATP channel structures, including subunit interfaces and conformational diversity, and review the antagonistic tension between several metabolic ligands that ultimately determines KATP activity.
Molecular structure of KATP channels
Biochemical and biophysical studies determined that the KATP channel is a hetero-octameric complex of four Kir6.x and four SURx subunits [Citation22–24]. Unassembled or partially assembled channel subunits are retained in the endoplasmic reticulum (ER) owing to an arginine-based ER retention motif present in both Kir6.x and SURx (-RKR- in SUR1, Kir6.1, and Kir6.2; -RKQ- in SUR2A/B; see ) [Citation25]. KATP high resolution structures derived from cryoEM single particle imaging confirmed the 4:4 stoichiometry, showing a Kir6 tetramer core surrounded by four SUR proteins (), including the SUR1/Kir6.2 [Citation26–28] and SUR2B/Kir6.1 subtypes [Citation29] (no SUR2A/Kir6.2 full channel structure has yet been reported). Both Kir6 and SUR are integral membrane proteins. Each Kir6 contains two transmembrane domain (TMD) helices (M1 and M2), which anchor a large cytosolic domain (CTD) comprising both N- and C-terminal chains (), characteristic of Kir channels [Citation10]. SUR bears two 6-helix TMDs (TMD1 and TMD2), each of which is followed by a cytoplasmic nucleotide binding domain (NBD), NBD1 and NBD2 respectively; these together comprise the ABC core module [Citation30]. SUR further includes an N-terminal 5-helix TMD, called TMD0, which is connected to the ABC core module via a cytoplasmic linker, L0 () [Citation12,Citation30].
Within the KATP complex, each Kir6 makes primary direct contact with one SUR through TMD interactions. The primary anchor between the two proteins includes the outer helix (M1) of Kir6 and the first TM helix in TMD0 of SUR (), accounting for early findings that SUR1 TMD0 alone assembles with Kir6.2 [Citation31,Citation32]. The Kir6 TMD and SUR TMD0 are consistently the best resolved domains in all published structures. Against this stable TMD interface, other SUR and Kir6 domains exhibit dynamic positioning. Most dramatically, the ABC modules of SUR1 in pancreatic KATP structures [Citation26], and of SUR2B in vascular KATP structures [Citation29], are observed to swing, as if hinged, into distinct rotational orientations relative to the Kir6/SUR-TMD0 tetrameric core. In top-down views, the distinct complexes resemble alternatively a propeller or a quatrefoil. In contrast, the cytoplasmic domains of the four Kir6 subunits associate to form a cohesive central unit, called the Kir6-CTD. In cryoEM structures, the Kir6-CTD displays a torsional flexibility relative to the Kir6 TMDs, wherein the CTD rotates about the central KATP axis to occupy particular orientations that correlate with distinct functional states of the channel [Citation33,Citation34]. In addition, a dynamic interaction is observed between the cytoplasmic domains of Kir6.2 and SUR1, in which the N- and C-terminal domains of Kir6.2 intertwine with cytoplasmic loops of TMD0 of SUR1 and with the L0 linker proximal to TMD0. This interface serves as a regulatory nexus between the two channel subunits and undergoes remodeling upon ligand binding and gating. Below, we discuss how structural observations to date inform the interplay between SUR and Kir6 that regulates ligand binding and gating of KATP channels.
Gating regulation of KATP channels
Most central to the unique function of KATP channels is their ability to couple cell metabolism with membrane excitability via an antagonistic control of gating by intracellular ATP and ADP. In electrophysiological recordings, ATP inhibits channel activity whether in the presence or absence of Mg2+, while Mg2+-complexed ADP stimulates channel activity by antagonizing the inhibitory effect of ATP. When intracellular ATP/ADP ratios rise, the ATP inhibitory effect dominates; conversely, when ATP/ADP ratios fall, the MgADP stimulatory effect dominates, thus linking cellular metabolism with KATP activity. Aside from nucleotide regulation, like all Kir channels, KATP channel activity is stimulated by membrane phosphoinositides [Citation10,Citation35]. Among the different phosphoinositides, PI(4,5)P2 (referred to as PIP2 hereinafter) is the most abundant in the plasma membrane and its effect on KATP channels has been extensively studied [Citation10,Citation35–38]. Depletion or scavenging endogenous PIP2 decreases channel activity, while increasing PIP2 in the membrane increases KATP channel open probability and antagonizes the inhibitory effect of ATP [Citation39,Citation40].
The discovery that truncating the C-terminus of Kir6.2 (Kir6.2ΔC, which removes the ER retention motif -RKR-) allows Kir6.2 channel expression in the plasma membrane in the absence of SUR1 facilitated dissection of the respective role of Kir6.2 and SUR1 in channel response to nucleotides and PIP2 [Citation25,Citation41]. Comparison of Kir6.2ΔC channels with or without SUR1 combined with mutation-function correlation studies revealed that ATP inhibition, MgADP stimulation and PIP2 gating are effected through distinct binding sites. ATP inhibits Kir6.2 channel gating by occupying a site composed principally of Kir6.2 residues from both N- and C-terminal cytoplasmic domains [Citation42–44]. PIP2 stimulates Kir6.2 gating by binding to a conserved PIP2 binding site in Kir-family channels that also involves residues from both N- and C-terminal domains [Citation37–40,Citation45]. The ATP binding site was predicted to be near the plasma membrane inner leaflet and immediately adjacent to the predicted PIP2 binding site based on homology modeling using crystal structures of other Kir channels [Citation45,Citation46]. Moreover, many mutations that affect ATP inhibition also affect PIP2 sensitivity, suggesting the two ligand binding sites may overlap [Citation37,Citation38]. Thus, ATP-inhibition of the KATP channel appears likely to derive from evolved antagonism of prior Kir-channel PIP2-activation mechanisms. Significantly, although Kir6.2 itself can be gated by ATP and PIP2, its sensitivity to both ligands is markedly increased by co-assembly with SUR1 [Citation41], suggesting SUR1 may participate in ATP and PIP2 binding or hypersensitizes channels to these ligands allosterically. In contrast, the stimulatory regulation of Kir6 channel gating in response to MgADP is translated from the SUR structural platform of paired ABC-domain NBDs, which preferentially bind Mg2+-complexed adenine nucleotides. Mg-nucleotide binding, in particular MgADP binding at NBD2, alters the interface between the two NBDs, resulting in a close interaction (termed NBD dimerization) associated with functional activation [Citation47]. A framework that emerges from the wealth of structure-function correlation data is that the Kir6 subunit serves as a basic module for PIP2 stimulation and ATP inhibition, and SUR modulates Kir6 sensitivity to both ligands via physical association as well as the action of MgATP/MgADP on the NBDs of SUR.
ATP inhibition mechanism
Inhibitory ATP binding site
CryoEM structures of KATP channels resolved in the presence of ATP (or ADP, which can also bind the inhibitory site but with much lower affinity [Citation48]) showed that ATP binds in the pocket formed by the N-terminal and C-terminal domains of adjacent Kir6.2 subunits () [Citation26,Citation28,Citation34]. The structures confirmed the ATP binding pocket proposed previously [Citation42] and clarified residues directly involved in ATP/ADP binding. These structures additionally revealed that the SUR1-L0 linker also participates in binding () [Citation28,Citation33,Citation49]. The proximal region of L0 immediately C-terminal to TMD0 was previously implicated in ATP inhibition [Citation50]. Stabilizing the interaction between SUR1-L0 and Kir6.2 N-terminal interfacial helix greatly increased ATP sensitivity or caused channel closure without ATP, whether through an engineered charge pairing between SUR1 amino acid (aa) position 203 in L0 (E203 of WT SUR1) and Kir6.2 aa position 52 (Q52 in WT Kir6.2), or alternatively engineering cysteine at these positions and crosslinking [Citation50]. Conversely, ATP inhibition is attenuated by mutation of SUR1-K205 in the L0 linker to alanine [Citation50]. The cryoEM structures showed that the side chain of SUR1-K205 is in position to form electrostatic interactions with bound ATP directly [Citation33,Citation49]. A primary role of this SUR1 residue in ATP binding is further validated using a FRET based assay involving genetically encoded fluorescent non-canonical amino acid ANAP and a fluorescent ATP analog [Citation51]. Thus, structural, functional and direct chemical evidence together buttress the hypothesis that SUR1 enhances the ATP sensitivity of Kir6.2 by directly contributing to ATP binding with SUR1-L0. Conceptually, SUR’s enhancement of ATP inhibition presents a point of gating control that, leveraged by regulated withdrawal, could initiate channel activation. Several additional interactions between SUR and Kir6 have been implicated in further mediating dynamic regulation of ATP inhibition and gating as discussed below.
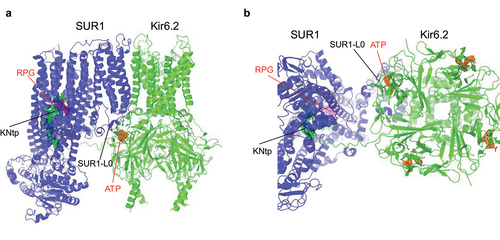
Figure 2. ATP- and Repaglinide (RPG)-bound SUR1/Kir6.2 structure (PDB ID 7TYS; EMD-26193). (a) side and (b) bottom (cytoplasmic) views showing the ATP (cryoEM map shown in orange, 3 σ contour) binding site coordinated by Kir6.2 residues from the N-terminus of one subunit and the C-terminus of an adjacent subunit, as well as L0 of the SUR1 subunit directly in association with the Kir6.2 subunit whose N-terminus contributes to ATP binding. Repaglinide (RPG; cryoEM map shown in purple, 1 σ contour) is bound in a transmembrane pocket in SUR1. The N-terminal peptide of Kir6.2 (KNtp; cryoEM density in green mesh, 1 σ contour) is inserted into the cavity of the two halves of the SUR1-ABC core with the very N-terminus adjacent to the bound RPG.
The Kir6 N-terminus
Multiple early studies implicated the distal N-terminal domain of Kir6.2 as a critical handle through which SUR1 maintains control of channel sensitivity to ATP inhibition. In particular, truncation of Kir6.2 N-terminal amino acids 2–30 (Kir6.2ΔN) increases channel open probability and reduces ATP sensitivity in SUR1/Kir6.2ΔN channels, but not in channels composed of Kir6.2 alone (Kir6.2ΔC) [Citation52–55]. Applying free synthetic Kir6.2 N-terminal peptide (aa 1–30) to WT SUR1/Kir6.2 channels mimicked the effects of Kir6.2ΔN [Citation52], suggesting the synthetic peptide competes with the N-terminal peptide domain in Kir6.2 for a binding site in SUR1 to modulate channel activity.
The Kir6 N-terminus linkage between SUR and the Kir6-CTD is an essential conduit for pharmacological inhibition of KATP gating by sulfonylureas such as glibenclamide and glinides such as repaglinide, which specifically bind to SUR. Progressive deletions of the Kir6.2 N-terminus reduce binding affinity of glibenclamide and repaglinide, and attenuate drug-dependent inhibition of the channel [Citation55,Citation56]. The observation that binding affinity of sulfonylureas to SUR1 is also reduced by Kir6.2 N-terminal deletion, but not eliminated, suggested that drug and Kir6.2 N-terminus bind cooperatively within SUR1, and thus pharmacological inhibition occurs primarily by increasing the avidity of Kir6.2 N-terminus binding to SUR1. CryoEM studies support this interpretation and specify the binding interactions involved.
CryoEM channel structures have resolved the N-terminal ~30 aa (KNtp) of Kir6.2 as an extended projection that inserts deeply into the central cleft between the two halves of the ABC core module of the SUR1 subunit that is in direct physical contact with the Kir6.2 () [Citation33,Citation57]. Notably, strong cryoEM density of KNtp in the SUR central cleft is correlated with the presence of pharmacological inhibitors, such as glibenclamide and repaglinide, which also bind within the SUR1 ABC core in a pocket next to the very N-terminus of Kir6.2 (). A weaker KNtp cryoEM density in the SUR1-ABC core cleft was seen in channels bound to ATP only, and the density was even weaker in the absence of any inhibitory ligands [Citation33,Citation57]. The results provide a striking structural correlate to prior biochemical and functional studies that implicated a role of Kir6.2 distal N-terminus in drug binding and inhibitory gating [Citation55,Citation56,Citation58]. Structure-guided crosslinking experiments provide direct evidence that the KNtp-SUR1 interface controls channel gating. Crosslinking of a cysteine engineered at amino acid position 2 in the Kir6.2 KNtp to an adjacent endogenous SUR1 cysteine (C1142) led to spontaneous loss of channel activity [Citation57], demonstrating that trapping KNtp in the SUR1-ABC core exerts ATP-independent allosteric control of Kir6.2 activity. Accordingly, KATP cryoEM structures of an SUR1-Kir6.2 fusion protein having a 39 amino acid linker (SUR1-39aa-Kir6.2) may have been found to lack KNtp density in the SUR1-ABC core, even with bound inhibitors, because such a linker tethers and constrains the KNtp from entering the SUR1 ABC core cavity [Citation34,Citation49]. Of note, functional studies have shown fusion channels have higher Po and reduced ATP sensitivity [Citation24], similar to SUR1/Kir6.2ΔN channels, consistent with the importance of the KNtp/SUR1 interface in channel gating. CryoEM structures of the vascular KATP channel prepared with glibenclamide and ATP also show the Kir6.1 N-terminus wedged in the TM cleft between the two halves of the SUR2B-ABC core next to the bound glibenclamide [Citation29], suggesting this inter-subunit regulatory mechanism is conserved among KATP isoforms.
PIP2 stimulation mechanism
PIP2 binding site
PIP2 opens KATP channels, and is expected to be present in open channel structures. However, initial open channel KATP structures reported by two separate groups have lacked clear PIP2 cryoEM density. These include the structure of a human channel consisting of SUR1 and a mutant Kir6.2 harboring a pore mutation C166S known to stabilize channel opening as well as a G334D mutation that prevents ATP binding at the inhibition site (denoted by Kir6.2C166S, G334D), and the structure of a rodent SUR1-Kir6.2 fusion protein channel containing a Kir6.2 mutation H175K (Kir6.2H175K), which was incorporated to enhance PIP2 binding. Recently, the cryoEM structure of a PIP2-bound open KATP channel was reported in a preprint by Driggers et al. [Citation21]. Using a Kir6.2 variant harboring the neonatal diabetes-causing mutation Q52R (Kir6.2Q52R), which has been shown to increase channel open probability and decrease channel sensitivity to ATP inhibition in a SUR1-dependent manner [Citation59], and by pre-incubating membranes expressing SUR1/Kir6.2Q52R channels with natural long-chain PIP2, which provides more hydrophobic anchoring compared to synthetic short-chain PIP2, the authors observed, surprisingly, that not one but two adjacent well-resolved PIP2 molecules occupy intramembrane positions between the Kir6.2 TMDs and SUR1-TMD0 ().
Figure 3. PIP2-bound open SUR1/Kir6.2Q52R channel structure. (a) CryoEM map features of two PIP2 molecules colored in magenta and cyan (0.08 V, 4 σ contour), respectively. (b) structural model of the PIP2 binding pocket (red boxed region in (a)) viewed from the side. Residues from both Kir6.2 (adjacent subunit denoted “b”) and SUR1 (blue outline) surrounding bound PIP2 molecules are labeled. (c) comparison of the SUR1-Kir6.2 cytoplasm-plasma membrane interface between open PIP2 (cyan and magenta sticks)-bound SUR1/Kir6.2Q52R (SUR1 in teal and Kir6.2 in yellow) KATP channel and closed ATP-(orange phosphate and gray carbon sticks) and repaglinide (out of view)-bound SUR1/Kir6.2 (SUR1 in pink and Kir6.2 in gray) KATP channel (PDB ID 7TYS) viewed from the extracellular side. Reorientation of side chains of the Kir6.2 inner helix gate residue F168 and M1 residue F60 in the two conformations is shown. A 6.4° clockwise rotation of the Kir6.2-CTD is indicated by the magenta curved arrow (with D323 Cα in each structure marked as spheres). Reorientation of the side chain of W51 at the bottom of TM1 of SUR1-TMD0 is evident. SUR1-L0, marked by the ATP-binding residue K205 and an adjacent residue E203, moves away from the ATP binding pocket (magenta arrow) in the open conformation. Panels (a) and (b) are taken from , and panel (c) from of bioRxiv preprint by Driggers et al.. (reference [Citation21]).
![Figure 3. PIP2-bound open SUR1/Kir6.2Q52R channel structure. (a) CryoEM map features of two PIP2 molecules colored in magenta and cyan (0.08 V, 4 σ contour), respectively. (b) structural model of the PIP2 binding pocket (red boxed region in (a)) viewed from the side. Residues from both Kir6.2 (adjacent subunit denoted “b”) and SUR1 (blue outline) surrounding bound PIP2 molecules are labeled. (c) comparison of the SUR1-Kir6.2 cytoplasm-plasma membrane interface between open PIP2 (cyan and magenta sticks)-bound SUR1/Kir6.2Q52R (SUR1 in teal and Kir6.2 in yellow) KATP channel and closed ATP-(orange phosphate and gray carbon sticks) and repaglinide (out of view)-bound SUR1/Kir6.2 (SUR1 in pink and Kir6.2 in gray) KATP channel (PDB ID 7TYS) viewed from the extracellular side. Reorientation of side chains of the Kir6.2 inner helix gate residue F168 and M1 residue F60 in the two conformations is shown. A 6.4° clockwise rotation of the Kir6.2-CTD is indicated by the magenta curved arrow (with D323 Cα in each structure marked as spheres). Reorientation of the side chain of W51 at the bottom of TM1 of SUR1-TMD0 is evident. SUR1-L0, marked by the ATP-binding residue K205 and an adjacent residue E203, moves away from the ATP binding pocket (magenta arrow) in the open conformation. Panels (a) and (b) are taken from Figure 2, and panel (c) from Figure 4 of bioRxiv preprint by Driggers et al.. (reference [Citation21]).](/cms/asset/f65f1a68-2fc7-4352-bc95-b8534461759b/kchl_a_2327708_f0003_oc.jpg)
The first PIP2 binding site occupies the predicted Kir6-specific pocket based on homologous structures for Kir2 and Kir3 bound to PIP2 [Citation45,Citation46,Citation60]. The lipid chains interact closely with Kir6.2 TMD1 and TMD2, and the head group phosphates are closely coordinated by Kir6.2 residues mostly conserved in Kir2 and Kir3 channels. The second, adjacent PIP2 binding site is formed by the transmembrane interface between the Kir6.2 TMDs and SUR1-TMD0, and is unique to the KATP channel. Perturbation of either binding site by mutating key residues coordinating PIP2 binding based on the structure markedly reduced channel open probability in electrophysiological studies under ambient membrane PIP2 conditions [Citation21], indicating both PIP2 binding sites are functionally important for regulating KATP channel activity in native membranes. The direct involvement of SUR1 in PIP2 binding accounts for the previously observed increase in PIP2 sensitivity conferred by SUR1 on Kir6.2 [Citation36,Citation39,Citation40].
The PIP2-bound open SUR1/Kir6.2Q52R structure also revealed a cation-π bonding between Kir6.2-Q52R and SUR1-W51. Disrupting this interaction by mutating SUR1-W51 to cysteine compromised the ability of Kir6.2-Q52R to increase channel open probability and reduce channel sensitivity to ATP inhibition [Citation21]. These observations indicate that cation-π interaction between Kir6.2-Q52R and SUR1-W51 stabilizes the channel in an open conformation, providing a molecular explanation for how Kir6.2-Q52R causes gain-of-function channel defect in a SUR1-dependent manner [Citation59].
Structural basis of PIP2 and ATP antagonism
PIP2 and ATP functionally compete to open and close KATP channels, respectively [Citation36,Citation61]. Comparison of the PIP2-bound open structure and the ATP-bound closed structure reveals that PIP2 binding disrupts ATP binding at two levels, first by directly competing for shared binding residues and second by causing a conformational change that disfavors ATP binding [Citation21] (). Specifically, Kir6.2-K39, which stabilizes ATP binding, is also involved in PIP2 binding at the novel site (). Additionally, the conformational change associated with channel opening by PIP2 results in SUR1-L0, including ATP coordinating residue SUR1-K205, moving away from the ATP binding site and an enlargement of the ATP binding pocket such that ATP can no longer bind tightly (see control of Kir6.2 close-open transition below). Occupation of PIP2 binding sites thereby destabilizes inhibitory ATP binding and increases channel open state probability. While SUR1-TMD0 directly supports PIP2 binding, favoring channel activation, SUR1-L0 also directly supports ATP binding toward inhibition. The direct involvement of SUR1 in regulating the binding of both ATP and PIP2 mirrors that of Kir6.2, whose antagonistic binding of ATP and PIP2 hinges on directly interacting sites. Thus, SUR1 appears to function as an amplifier of the intrinsic Kir6.2 ATP versus PIP2 gating switch, leveraged by a direct participation in binding both ATP and PIP2 that increases the sensitivity of Kir6.2 to inhibition as well as activation.
Control of Kir6.2 close-open transition
The structural and functional evidence cited above indicates SUR1 regulates Kir6.2 Po and ATP sensitivity by directly contributing to PIP2 and ATP binding, and by allosteric control via regulation of Kir6.2-CTD dynamics. How then are these influences translated to gating of the Kir6.2 channel pore? By comparing KATP channel structures having closed (including those bound to inhibitory ATP/ADP and/or pharmacological inhibitors [Citation26–28,Citation33,Citation34,Citation49,Citation62]) and open channel pores (including SUR1-NBD dimerized open human SUR1/Kir6.2C166S, G334D channel, SUR1-NBD dimerized pre-open rodent SUR1-Kir6.2H175K fusion channel, and rodent PIP2 bound open SUR1/Kir6.2Q52R channel [Citation21,Citation63,Citation64]), an alignment of the transmembrane domains reveals that the open channel state, regardless of SUR1-NBD dimerization or PIP2 binding status, is accompanied by a clockwise rotation of the Kir6.2-CTD to a new stable position, viewed from the extracellular side (; see also ) [Citation33,Citation63,Citation64]. It has been proposed that SUR’s ability to envelop the KNtp within its ABC cleft is a likely mechanism to control Kir6.2-CTD conformation [Citation33,Citation34]. The resolved structures clarify that insertion of KNtp into the SUR1-ABC core cavity physically constrains the Kir6.2-CTD such that it is unable to adopt the rotated conformation associated with channel opening. Conversely, when KNtp is not bound within the SUR1-ABC core, rotation of Kir6.2-CTD to an open-associated orientation is unconstrained. Accordingly, pharmacological inhibitors such as glibenclamide act to stabilize a closed channel precisely by increasing the avidity of the SUR1-KNtp interaction, and thus hinder the Kir6-CTD rotation. Consistent with this model, engineering a longer KNtp by insertion of additional amino acids both uncouples SUR1 control of Kir6.2-CTD and attenuates the ability of glibenclamide to close the channel [Citation34].
Figure 4. Molecular structure of KATP channels. (a) view of the inhibitory ATP binding site on Kir6.2 (light green) (PDB ID 6BAA) with ATP bound, in a closed pore conformation, with SUR1 (teal) subunits shown. (b) the same view of the pre-open Kir6.2 pore (dark green) (PDB ID 7W4O; pre-open SUR1-Kir6.2H175K fusion) with the inhibitory ATP pocket empty. The ATP-binding pocket is enlarged due to outward movement of SUR1 (blue) L0, and Kir6.2 CTD rotation. Note two other open structures: SUR1/Kir6.2C166S, G334D (PDB ID 7S5X) and PIP2-bound SUR1/Kir6.2Q52R (preprint in bioRxiv, reference [Citation21]) are very similar in Kir6.2-CTD rotation and pore opening, the movement of SUR1-L0 away from Kir6.2, and the enlargement of the ATP binding pocket.
![Figure 4. Molecular structure of KATP channels. (a) view of the inhibitory ATP binding site on Kir6.2 (light green) (PDB ID 6BAA) with ATP bound, in a closed pore conformation, with SUR1 (teal) subunits shown. (b) the same view of the pre-open Kir6.2 pore (dark green) (PDB ID 7W4O; pre-open SUR1-Kir6.2H175K fusion) with the inhibitory ATP pocket empty. The ATP-binding pocket is enlarged due to outward movement of SUR1 (blue) L0, and Kir6.2 CTD rotation. Note two other open structures: SUR1/Kir6.2C166S, G334D (PDB ID 7S5X) and PIP2-bound SUR1/Kir6.2Q52R (preprint in bioRxiv, reference [Citation21]) are very similar in Kir6.2-CTD rotation and pore opening, the movement of SUR1-L0 away from Kir6.2, and the enlargement of the ATP binding pocket.](/cms/asset/57dca35e-3fcd-4cf6-91c6-7b5079fec736/kchl_a_2327708_f0004_oc.jpg)
Mobility of the Kir6-CTD is further constrained by occupation of the ATP inhibitory site, in which SUR’s L0 linker participates as described above. In ATP-bound closed channel structures of the pancreatic Kir6.2/SUR1 isoform, ATP at the inhibitory site simultaneously bonds with residues of the Kir6.2-CTD (including R50 at the N-terminal domain, and K185 at the C-terminal domain of an adjacent Kir6.2 subunit) and also with SUR1-K205 [Citation28,Citation33,Citation49] (also see recent reviews [Citation18,Citation19]), which pulls the N-terminal portion of SUR-L0 in close proximity. Thus, ATP effectively glues Kir6.2-CTD to SUR1-L0, hindering Kir6.2-CTD rotation and stabilizing the Kir6.2 core in a closed conformation (). In the absence of ATP, Kir6.2-CTD rotation away from the closed conformation is unconstrained by SUR-L0 interaction. Instead, rotation of Kir6.2-CTD to the open position results in formation of interactions with SUR1-TMD0, in particular the cytoplasmic loops of TMD0 [Citation63,Citation64], which likely retard spontaneous counter-rotations back to the closed condition (). Interestingly, in rotating to the open conformation, the residues coordinating ATP binding become spread further apart [Citation63,Citation64]. Lengthening the bonds with ATP enlarges the binding pocket, decreases the strength of tethering to SUR1-L0, and favors ATP dissociation.
Mechanisms of channel stimulation by MgADP/MgATP
Stimulation of the KATP channel by MgADP/MgATP is mediated by the SUR subunit [Citation47]. SUR has two, but although both bind Mg-nucleotides, only NBD2 is capable of hydrolyzing MgATP. It is hypothesized that Mg-nucleotide binding at the two NBDs of SUR causes dimerization of the NBDs in a head-to-tail fashion [Citation2,Citation61,Citation65,Citation66], as it does in other ABC proteins [Citation30], and that NBD dimerization regulates the Kir6 subunit to decrease ATP inhibition and open the channel. Whether MgATP hydrolysis is involved in KATP channel stimulation has been investigated by several approaches. Although low level ATPase activity at NBD2 has been reported [Citation27,Citation66–68], analysis of single channel kinetics does not support the requirement of MgATP hydrolysis for MgADP-induced gating [Citation69]. Instead, preferential binding of MgADP at NBD2 as MgADP concentrations rise is thought to drive NBD dimerization [Citation70]. Supporting this idea, binding studies show NBD1 binds MgATP preferentially, whereas NBD2 binds MgADP with higher affinity [Citation71–73]. Moreover, cryoEM structures of KATP channels in which SUR NBDs are bound to Mg-nucleotides and dimerized show larger opening of the NBD2 MgADP binding site that would permit exchange with MgADP in solution [Citation26,Citation63,Citation64]. This feature distinguishes SUR from other ABC transporters, and favors an ability of SUR to function as a sensor of changing MgADP levels rather than an ATPase.
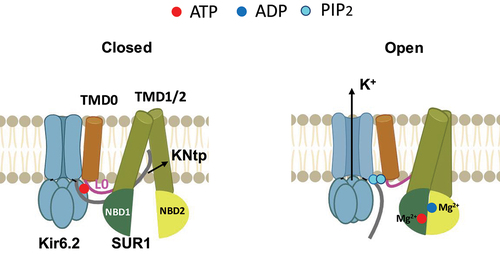
Compared to closed structures, SUR1 NBD-dimerized open structures (including open SUR1/Kir6.2C166S, G334D channel and pre-open SUR1-Kir6.2H175K fusion channel) have rearrangements at the subunit interface involving SUR1 TMD0-L0 and Kir6.2-CTD [Citation63,Citation64]. In particular, the L0 domain moves away from the ATP binding pocket, which becomes enlarged as the Kir6.2-CTD is rotated to the open position (see discussion in the previous section), and the TMD0 of SUR1 bends away at TM1 that directly contacts M1 of Kir6.2. The distancing of SUR1-L0 away from the inhibitory ATP binding pocket has been proposed to account for the antagonistic effect of MgADP/MgATP on ATP inhibition that leads to channel stimulation [Citation63].
The SUR1-NBD dimerized open and pre-open structures discussed above do not have discernable bound PIP2, yet they are very similar to the PIP2-bound SUR1/Kir6.2Q52R open structure without SUR1-NBD dimerization with regard to the Kir6.2 conformation and Kir6.2-SUR1 interface (). In both the open and pre-open structures, the SUR1-TMD0 and Kir6.2-TM interface would be expected to accommodate binding of two PIP2 molecules. Thus, it seems that PIP2 binding and Mg-nucleotide induced SUR1 NBD dimerization converge to a common structural mechanism to antagonize ATP inhibition and stimulate channel activity, as illustrated in the cartoon in .
Figure 5. Nucleotide regulation of KATP channels. Cartoon illustrating key structural differences between KATP channels bound to inhibitory ATP in closed conformation (left) and KATP channels in open conformation, with SUR1 NBD1/2 bound to MgATP/MgADP and dimerized (right). The PIP2 binding pocket in the SUR1 NBD-dimerized open conformation would accommodate the binding of two PIP2 molecules at the SUR1-Kir6.2 interface.
Regulation of MgADP/MgATP response in KATP subtypes
MgADP responsiveness varies among KATP subtypes containing different SUR isoforms [Citation74]. Significantly, activation of the cardiac KATP channel comprising SUR2A and Kir6.2 requires a higher MgADP concentration than channels containing SUR1 or SUR2B and Kir6.2 [Citation68,Citation74,Citation75]. The increased MgADP threshold for cardiac KATP channel activation ensures KATP channels open only under more intense metabolic stress such as exercise; opening of KATP channels shortens action potentials at higher heart rates and protects the heart from calcium loading [Citation4,Citation76,Citation77]. The splice variants SUR2A and SUR2B differ in their C-terminal 42 amino acids (C42), which are encoded by the last exon of the SUR2 gene [Citation8]. Interestingly, the sequence homology of C42 is only ~ 30% between SUR2B and SUR2A, but ~ 70% between SUR2B and SUR1. Direct functional evidence supports the suggestion that C42 plays a primary role in modulating the sensitivity of the MgADP response of KATP channels [Citation75].
R-helix regulation
A recent study of SUR2A monomer structures (without co-assembly with Kir6.2) in the presence of the inhibitor repaglinide with different combinations of Mg-nucleotides identified an additional regulatory element that may account for the reduced MgADP sensitivity in SUR2A/Kir6.2 channels [Citation78]. SUR2A structures in the presence of repaglinide and MgATP reveal a helical segment interposed between the two NBDs of SUR2A, forming a putative dimerization regulatory element termed the R-helix (). The corresponding sequence (aa 924–942) is part of the NBD1-TMD2 linker. The R-helix is not observed between the NBDs when SUR2A structures were imaged in conditions that had NBD2 occupied by MgADP, suggesting MgADP binding at NBD2 mobilizes the R-helix. Functional studies of complete SUR2A/Kir6.2 channels showed that mutating certain residues in the R-helix that interact with NBD1 or NBD2 enhanced the MgADP response. The authors propose that by wedging between the two NBDs, the R-helix acts as an NBD separator to prevent dimerization and channel activation, and that binding of MgADP to NBD2 at high ADP/ATP ratios then relieves R-helix inhibition to allow channel activation.
Figure 6. Structural elements implicated in MgADP regulation of KATP channels. (a) cartoon representation of the structures of either SUR1 (orange) (PDB ID 6JB1), SUR2A (purple) (PDB ID 7Y1J), or SUR2B (blue) (PDB ID 7Y1L) with the regulatory helix (R-Helix) shown as spheres (green). ATP (red spheres) and RPG (yellow spheres) are also shown. (b) overall structure of Kir6.1 (navy) plus one single SUR2B subunit (blue) in the quatrefoil like conformation (PDB ID 7MJO) showing the NBD1-TMD2 (N1T2) linker cryoEM density (green) in between SUR2B NBD2 and Kir6.1-CTD. Red boxed region is the approximate location of the ED domain.
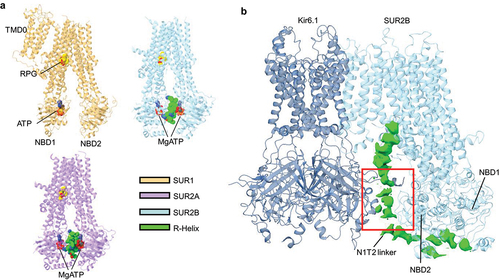
R-helix dynamics may partly account for differences in MgADP sensitivity among KATP isoforms. The peptide sequence corresponding to the R-helix is not conserved in SUR1, which could explain why no R-helix like cryoEM density was previously observed in published KATP structures () [Citation34,Citation49,Citation57,Citation62]. Regarding the SUR2B isoform, a structural analysis similar to SUR2A found that the R-helix was observed within the SUR2B NBD cleft under some conditions () [Citation78]. Interestingly, the SUR2B-NBD2 containing the alternatively spliced C42 appeared to be significantly more dynamic compared to SUR2A-NBD2, which may result in less stable inhibition by the R-helix. While the SUR structures reported in the study lack the Kir6.2 subunit, the structural variations in the different SUR isoforms observed, when coupled with functional evidence, offer a refined model for the differential MgADP sensitivity seen in different KATP channel subtypes.
Role of ED domain in the NBD1-TMD2 linker
The NBD1-TMD2 linker of SUR2A/2B contains a stretch of 15 negatively charged amino acids known as the ED domain [Citation79]. The ED domain has been implicated in regulating channel response to MgADP and the potassium channel opener pinacidil [Citation79]. The ED domain has not been resolved in pancreatic channel structures. However, in structures of SUR2B/Kir6.1 vascular KATP channels bound to glibenclamide and ATP, the NBD1-TMD2 cryoEM density that includes the ED domain is resolved at the subunit interface between the cytoplasmic domain of Kir6.1 and the NBD2 of SUR2B () [Citation33]. MD simulations of this interface structure show dynamic MgADP-dependent tripartite interactions between the ED domain linker, SUR2B, and Kir6.1. The structures captured implicate a progression of intermediate states between MgADP-free inactivated conformations and MgADP-bound activated conformations, wherein the ED-rich linker participates as mobile autoinhibitory domain, suggesting a conformational pathway toward KATP channel regulation [Citation29]. Vascular KATP channels composed of SUR2B and Kir6.1 are not spontaneously active and only open in the presence of MgATP/MgADP [Citation80]. The autoinhibitory mechanism would prevent MgADP-independent spontaneous activity. Whether the ED domain plays a similar structural role in SUR2A/Kir6.2 channels awaits further investigation.
Concluding remarks
The advent of cryoEM protein structure determination has dramatically accelerated progress in understanding how KATP channel activity is controlled by the relative intracellular concentrations of ADP and ATP, especially for the pancreatic/neuronal SUR1/Kir6.2 isoform. The core four-subunit Kir6.2 channel is activated by binding of the agonist PIP2, an ancestral feature shared with Kir-family channels. Uniquely, however, Kir6.2 channels have evolved to directly bind ATP as an inverse agonist in tension with PIP2 to retard channel activation by preventing Kir6-CTD from rotating to the open position. The SUR1 subunits directly amplify, strengthen and thus control the PIP2-ATP switch mechanism intrinsic to Kir6.2. SUR1 TMD0 and L0 linker domains contribute binding residues to both PIP2 and ATP ligand pockets, in the alternative open and closed states, which increases the alternative avidities for both ligands to ensure that KATP channels respond to these ligands with high efficiency. SUR1 further enforces ATP inhibition by binding Kir6.2 N-terminus in the ABC core of SUR1, which prevents Kir6.2-CTD from rotating to the open position.
When intracellular ADP concentrations rise under increased metabolic demand, MgADP binds to SUR1 to promote NBD dimerization. This excludes Kir6.2 N-terminus from SUR1’s ABC core and simultaneously pulls SUR1-L0 domain away from the ATP binding pocket to promote ATP dissociation at the inhibitory site, allowing Kir6.2-CTD rotation to the open position, which is stabilized by PIP2 binding. SUR1 conformational dynamics, controlled by MgADP binding that simultaneously weaken one interaction while favoring the other allows for variable control of channel activation. Thus, SUR1 and Kir6.2 integrate interactions with MgADP, tandem PIP2 binding, ATP, and each other in support of cellular homeostasis. Different SUR isoforms have additional regulatory elements and mechanisms, such as the R-helix and the ED domain to shift MgADP sensitivity. While drugs such as sulfonylureas and glinides stabilize the interface of the KNtp and SUR-ABC core to mimic ATP-bound closed conformation and inhibit channel activity [Citation20,Citation57], KATP openers such as NN-414 and pinacidil stabilize SUR in the NBD dimerized conformation to stimulate channel activity [Citation63,Citation81]. Numerous KATP gene mutations cause disease by altering channel sensitivity to ATP or MgADP [Citation14,Citation16,Citation17]. Improved understanding of structural mechanisms of KATP channel regulation will guide the development of isoform-specific KATP channel drugs to improve the treatment of human disease.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983 Sep 8-14;305(5930):147–16. doi: 10.1038/305147a0
- Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annu Rev Physiol. 1998;60(1):667–87. doi: 10.1146/annurev.physiol.60.1.667
- Shyng SL. K(ATP) channel function: more than meets the eye. Function (Oxf). 2022;3(1):zqab070. doi: 10.1093/function/zqab070
- Foster MN, Coetzee WA. KATP Channels in the Cardiovascular System. Physiol Rev. 2016 Jan;96(1):177–252. doi: 10.1152/physrev.00003.2015
- Gribble FM, Reimann F. Pharmacological modulation of K(ATP) channels. Biochem Soc Trans. 2002 Apr;30(2):333–339. doi: 10.1042/bst0300333
- Gaines KL, Hamilton S, Boyd AE 3rd. Characterization of the sulfonylurea receptor on beta cell membranes. J Biol Chem. 1988 Feb 25;263(6):2589–2592. doi: 10.1016/S0021-9258(18)69106-9
- Schmid-Antomarchi H, De Weille J, Fosset M, et al. The receptor for antidiabetic sulfonylureas controls the activity of the ATP-modulated K+ channel in insulin-secreting cells. J Biol Chem. 1987 Nov 25;262(33):15840–4. doi: 10.1016/S0021-9258(18)47664-8
- Inagaki N, Gonoi T, Iv JPC, et al. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996 May;16(5):1011–1017.
- Inagaki N, Gonoi T, Clement J, et al. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995 Nov 17;270(5239):1166–1170. doi: 10.1126/science.270.5239.1166
- Hibino H, Inanobe A, Furutani K, et al. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev. 2010 Jan;90(1):291–366.
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994 Nov 24;372(6504):366–9. doi: 10.1038/372366a0
- Aguilar-Bryan L, Nichols CG, Wechsler SW, et al. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995 Apr 21;268(5209):423–426. doi: 10.1126/science.7716547
- Zhang H, Flagg TP, Nichols CG. Cardiac sarcolemmal K(ATP) channels: Latest twists in a questing tale! J Mol Cell Cardiol. 2010 Jan;48(1):71–75. doi: 10.1016/j.yjmcc.2009.07.002
- McClenaghan C, Nichols CG. Kir6.1 and SUR2B in Cantú syndrome. Am J Physiol Cell Physiol. 2022 Jul 25;323(3):C920–C935. doi: 10.1152/ajpcell.00154.2022
- Tinker A, Aziz Q, Li Y, et al. ATP-Sensitive potassium channels and their physiological and pathophysiological roles. Compr Physiol. 2018 Sep 14;8(4):1463–1511.
- Pipatpolkai T, Usher S, Stansfeld PJ, et al. New insights into KATP channel gene mutations and neonatal diabetes mellitus. Nat Rev Endocrinol. 2020 Jul;16(7):378–393. doi: 10.1038/s41574-020-0351-y
- ElSheikh A, Shyng SL. K(ATP) channel mutations in congenital hyperinsulinism: progress and challenges towards mechanism-based therapies. Front Endocrinol. 2023;14:1161117. doi: 10.3389/fendo.2023.1161117
- Driggers CM, Shyng SL. Mechanistic insights on KATP channel regulation from cryo-EM structures. J Gen Physiol. 2023 Jan 2;155(1). doi: 10.1085/jgp.202113046
- Martin GM, Patton BL, Shyng SL. K(ATP) channels in focus: progress toward a structural understanding of ligand regulation. Curr Opin Struct Biol. 2023 Apr;79:102541. doi: 10.1016/j.sbi.2023.102541
- Wu JX, Ding D, Wang M, et al. Structural insights into the inhibitory mechanism of insulin secretagogues on the pancreatic ATP-Sensitive potassium channel. Biochemistry. 2020 Jan 14;59(1):18–25. doi: 10.1021/acs.biochem.9b00692
- Driggers CM, Kuo YY, Zhu P, et al. Structure of an open K (ATP) channel reveals tandem PIP (2) binding sites mediating the Kir6.2 and SUR1 regulatory interface. bioRxiv. 2023 Aug 1. 10.1101/2023.08.01.551546
- Clement J, Kunjilwar K, Gonzalez G, et al. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997 May;18(5):827–838.
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997 Jun 9;409(2):232–236. doi: 10.1016/S0014-5793(97)00488-2
- Shyng S, Nichols CG. Octameric stoichiometry of the KATP channel complex. J Gen Physiol. 1997 Dec;110(6):655–64. doi: 10.1085/jgp.110.6.655
- Zerangue N, Schwappach B, Jan YN, et al. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 1999 Mar;22(3):537–548.
- Lee KPK, Chen J, MacKinnon R. Molecular structure of human KATP in complex with ATP and ADP. Elife. 2017 Dec 29;6. doi: 10.7554/eLife.32481
- Li N, Wu JX, Ding D, et al. Structure of a pancreatic ATP-Sensitive potassium channel. Cell. 2017 Jan 12;168(1–2):101–110 e10. doi: 10.1016/j.cell.2016.12.028
- Martin GM, Yoshioka C, Rex EA, et al. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife. 2017 Jan 16;6. doi: 10.7554/eLife.24149
- Sung MW, Yang Z, Driggers CM. et al. Vascular KATP channel structural dynamics reveal regulatory mechanism by Mg-nucleotides. Proc Natl Acad Sci U S A. 2021 Nov 2;118(44). doi: 10.1073/pnas.2109441118
- Thomas C, Aller SG, Beis K, et al. Structural and functional diversity calls for a new classification of ABC transporters. FEBS Lett. 2020 Dec;594(23):3767–3775.
- Babenko AP, Bryan J. Sur domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem. 2003 Oct 24;278(43):41577–80. doi: 10.1074/jbc.C300363200
- Chan KW, Zhang H, Logothetis DE. N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits [research support, U.S. Gov’t, P.H.S.]. EMBO J. 2003 Aug 1;22(15):3833–3843. doi: 10.1093/emboj/cdg376
- Sung MW, Driggers CM, Mostofian B, et al. Ligand-mediated Structural Dynamics of a Mammalian Pancreatic KATP Channel. J Mol Biol. 2022 Aug 11;434(19):167789. doi: 10.1016/j.jmb.2022.167789
- Wu JX, Ding D, Wang M, et al. Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell. 2018 Jun;9(6):553–567.
- Rohacs T, Lopes CM, Jin T, et al. Specificity of activation by phosphoinositides determines lipid regulation of kir channels. Proc Natl Acad Sci U S A. 2003 Jan 21;100(2):745–750. doi: 10.1073/pnas.0236364100
- Enkvetchakul D, Loussouarn G, Makhina E, et al. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys J. 2000 May;78(5):2334–2348.
- Cukras CA, Jeliazkova I, Nichols CG. The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels [research support, U.S. Gov’t, P.H.S.]. J Gen Physiol. 2002 Sep;120(3):437–46. doi: 10.1085/jgp.20028621
- Shyng SL, Cukras CA, Harwood J, et al. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J Gen Physiol. 2000 Nov;116(5):599–608.
- Baukrowitz T, Schulte U, Oliver D, et al. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998 Nov 6;282(5391):1141–4. doi: 10.1126/science.282.5391.1141
- Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998 Nov 6;282(5391):1138–41. doi: 10.1126/science.282.5391.1138
- Tucker SJ, Gribble FM, Zhao C, et al. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997 May 8;387(6629):179–83. doi: 10.1038/387179a0
- Antcliff JF, Haider S, Proks P, et al. Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J. 2005 Jan 26;24(2):229–39. doi: 10.1038/sj.emboj.7600487
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci U S A. 1998 Nov 10;95(23):13953–8. doi: 10.1073/pnas.95.23.13953
- Tucker SJ, Gribble FM, Proks P, et al. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998 Jun 15;17(12):3290–3296. doi: 10.1093/emboj/17.12.3290
- Whorton MR, MacKinnon R. Crystal structure of the mammalian GIRK2 K+ channel and gating regulation by G proteins, PIP2, and sodium. Cell. 2011 Sep 30;147(1):199–208. doi: 10.1016/j.cell.2011.07.046
- Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. 2011 Aug 28;477(7365):495–8. doi: 10.1038/nature10370
- Gribble FM, Tucker SJ, Haug T, et al. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc Natl Acad Sci U S A. 1998 Jun 9;95(12):7185–7190. doi: 10.1073/pnas.95.12.7185
- Ribalet B, John SA, Weiss JN. Molecular basis for Kir6.2 channel inhibition by adenine nucleotides. Biophys J. 2003 Jan;84(1):266–76. doi: 10.1016/S0006-3495(03)74847-4
- Ding D, Wang M, Wu JX, et al. The structural basis for the binding of repaglinide to the Pancreatic KATP Channel. Cell Rep. 2019 May 7;27(6):1848–1857 e4. doi: 10.1016/j.celrep.2019.04.050
- Pratt EB, Zhou Q, Gay JW, et al. Engineered interaction between SUR1 and Kir6.2 that enhances ATP sensitivity in KATP channels. J Gen Physiol. 2012 Aug;140(2):175–87.
- Usher SG, Ashcroft FM, Puljung MC. Nucleotide inhibition of the pancreatic ATP-sensitive K+ channel explored with patch-clamp fluorometry. Elife. 2020 Jan 7;9.
- Babenko AP, Bryan J. SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. Defining intersubunit gating interactions. J Biol Chem. 2002 Nov 15;277(46):43997–4004. doi: 10.1074/jbc.M208085200
- Babenko AP, Gonzalez G, Bryan J. The N-terminus of KIR6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem Biophys Res Commun. 1999 Feb 16;255(2):231–8. doi: 10.1006/bbrc.1999.0172
- Koster JC, Sha Q, Shyng S, et al. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J Physiol. 1999 Feb 15;515(Pt 1):19–30. doi: 10.1111/j.1469-7793.1999.019ad.x
- Reimann F, Tucker SJ, Proks P, et al. Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol. 1999 Jul 15;518(Pt 2):325–336. doi: 10.1111/j.1469-7793.1999.0325p.x
- Kuhner P, Prager R, Stephan D, et al. Importance of the Kir6.2 N-terminus for the interaction of glibenclamide and repaglinide with the pancreatic K(ATP) channel [research support, non-U.S. Gov’t]. Naunyn-Schmiedeberg’s Arch Pharmacol. 2012 Mar;385(3):299–311.
- Martin GM, Sung MW, Yang Z, et al. Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. Elife. 2019 Jul 25;8. doi: 10.7554/eLife.46417
- Koster JC, Sha Q, Nichols CG. Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol. 1999 Aug;114(2):203–213. doi: 10.1085/jgp.114.2.203
- Tammaro P, Proks P, Ashcroft FM. Functional effects of naturally occurring KCNJ11 mutations causing neonatal diabetes on cloned cardiac KATP channels. J Physiol. 2006 Feb 15;571(Pt 1):3–14. doi: 10.1113/jphysiol.2005.099168
- Niu Y, Tao X, Touhara KK, et al. Cryo-EM analysis of PIP(2) regulation in mammalian GIRK channels. Elife. 2020 Aug 26;9. doi: 10.7554/eLife.60552
- Nichols CG. KATP channels as molecular sensors of cellular metabolism [review]. Nature. 2006 Mar 23;440(7083):470–6. doi: 10.1038/nature04711
- Martin GM, Kandasamy B, DiMaio F, et al. Anti-diabetic drug binding site in a mammalian KATP channel revealed by cryo-EM. Elife. 2017 Oct 24;6. doi: 10.7554/eLife.31054
- Wang M, Wu JX, Ding D, et al. Structural insights into the mechanism of pancreatic KATP channel regulation by nucleotides. Nat Commun. 2022 May 19;13(1):2770. doi: 10.1038/s41467-022-30430-4
- Zhao C, MacKinnon R. Molecular structure of an open human KATP channel. Proc Natl Acad Sci U S A. 2021 Nov 30;118(48). doi: 10.1073/pnas.2112267118
- Aittoniemi J, Fotinou C, Craig TJ, et al. Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator. Philos Trans R Soc Lond B Biol Sci. 2009 Jan 27;364(1514):257–67. doi: 10.1098/rstb.2008.0142
- Zingman LV, Alekseev AE, Bienengraeber M, et al. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001 Aug 2;31(2):233–45. doi: 10.1016/S0896-6273(01)00356-7
- de Wet H, Mikhailov MV, Fotinou C, et al. Studies of the ATPase activity of the ABC protein SUR1. FEBS J. 2007 Jul;274(14):3532–3544.
- Masia R, Enkvetchakul D, Nichols CG. Differential nucleotide regulation of KATP channels by SUR1 and SUR2A. J Mol Cell Cardiol. 2005 Sep;39(3):491–501. doi: 10.1016/j.yjmcc.2005.03.009
- Choi KH, Tantama M, Licht S. Testing for violations of microscopic reversibility in ATP-sensitive potassium channel gating. J Phys Chem B. 2008 Aug 21;112(33):10314–21. doi: 10.1021/jp712088v
- Sikimic J, McMillen TS, Bleile C, et al. ATP-binding without hydrolysis switches sulfonylurea receptor 1 (SUR1) to outward-facing conformations that activate KATP channels. J Biol Chem. 2018 Dec 26;294(10):3707–3719. doi: 10.1074/jbc.RA118.005236
- Ueda K, Inagaki N, Seino S. MgADP antagonism to Mg2±independent ATP binding of the sulfonylurea receptor SUR1. J Biol Chem. 1997 Sep 12;272(37):22983–22986. doi: 10.1074/jbc.272.37.22983
- Ueda K, Komine J, Matsuo M, et al. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc Natl Acad Sci U S A. 1999 Feb 16;96(4):1268–72. doi: 10.1073/pnas.96.4.1268
- Matsuo M, Tanabe K, Kioka N, et al. Different binding properties and affinities for ATP and ADP among sulfonylurea receptor subtypes, SUR1, SUR2A, and SUR2B. J Biol Chem. 2000 Sep 15;275(37):28757–63. doi: 10.1074/jbc.M004818200
- Fujita A, Kurachi Y. Molecular aspects of ATP-sensitive K+ channels in the cardiovascular system and K+ channel openers. Pharmacol Ther. 2000 Jan;85(1):39–53. doi: 10.1016/S0163-7258(99)00050-9
- Matsushita K, Kinoshita K, Matsuoka T, et al. Intramolecular interaction of SUR2 subtypes for intracellular ADP-Induced differential control of K(ATP) channels. Circ Res. 2002 Mar 22;90(5):554–561. doi: 10.1161/01.RES.0000012666.42782.30
- Nichols CG. Adenosine triphosphate-sensitive potassium currents in heart disease and cardioprotection. Card Electrophysiol Clin. 2016 Jun;8(2):323–335. doi: 10.1016/j.ccep.2016.01.005
- Kane GC, Liu XK, Yamada S, et al. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005 Jun;38(6):937–943.
- Ding D, Hou T, Wei M, et al. The inhibition mechanism of the SUR2A-containing K(ATP) channel by a regulatory helix. Nat Commun. 2023 Jun 17;14(1):3608. doi: 10.1038/s41467-023-39379-4
- Karger AB, Park S, Reyes S, et al. Role for SUR2A ED domain in allosteric coupling within the K(ATP) channel complex. J Gen Physiol. 2008 Mar;131(3):185–196.
- Yamada M, Isomoto S, Matsumoto S, et al. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J Physiol. 1997 Mar 15;499(Pt 3):715–720. doi: 10.1113/jphysiol.1997.sp021963
- Ding D, Wu JX, Duan X, et al. Structural identification of vasodilator binding sites on the SUR2 subunit. Nat Commun. 2022 May 13;13(1):2675. doi: 10.1038/s41467-022-30428-y