
ABSTRACT
The development of an injectable drug-device combination (DDC) product for biologics is an intricate and evolving process that requires substantial investments of time and money. Consequently, the commercial dosage form(s) or presentation(s) are often not ready when pivotal trials commence, and it is common to have drug product changes (manufacturing process or presentation) during clinical development. A scientifically sound and robust bridging strategy is required in order to introduce these changes into the clinic safely. There is currently no single developmental paradigm, but a risk-based hierarchical approach has been well accepted. The rigor required of a bridging package depends on the level of risk associated with the changes. Clinical pharmacokinetic/pharmacodynamic comparability or outcome studies are only required when important changes occur at a late stage. Moreover, an injectable DDC needs to be user-centric, and usability assessment in real-world clinical settings may be required to support the approval of a DDC. In this review, we discuss the common issues during the manufacturing process and presentation development of an injectable DDC and practical considerations in establishing a clinical strategy to address these issues, including key elements of clinical studies. We also analyze the current practice in the industry and review relevant and status of regulatory guidance in the DDC field.
| Abbreviations | ||
| ADA | = | anti-drug antibody |
| AI | = | auto-injector |
| AUC | = | area under the curve |
| BE | = | bioequivalence |
| BLA | = | biologics license application |
| Cmax | = | maximum concentration |
| DDC | = | drug-device combination |
| GMR | = | geometric mean ratio |
| HF | = | human factor |
| IM | = | intramuscular |
| IV | = | intravenous |
| LVD | = | large volume wearable injector device |
| LYOIV | = | lyophilized power in vial |
| MS | = | multiple sclerosis |
| PD | = | pharmacodynamics |
| PFS | = | prefilled syringe |
| PK | = | pharmacokinetics |
| RA | = | rheumatoid arthritis |
| SC | = | subcutaneous |
| SIV | = | solution in vial |
| VS | = | vial and syringe |
Introduction
Over the past thirty years, biotechnology products, including monoclonal antibodies (mAbs), have become the focus of targeted therapeutics development by the pharmaceutical industry, and they have proven to be promising treatment options for a variety of difficult-to-treat diseases. As of 2013, over 900 biologics targeting more than 100 indications were in clinical development or waiting for regulatory actions.Citation1 Biologics have accounted for approximately one third of new medicine approvals in the past decade,Citation1 and in 2015 alone, Food and Drug Administration marketing approvals for biologics, including 10 mAbs, reached an all-time high.Citation2,3 Owing to better understanding of diseases at the molecular and genetic levels, the pipeline of biologic drug candidates has been growing steadily, and in the near future, some of these are poised to make substantial impacts in a broad range of indications, especially in under- or un-represented disease areas.Citation1,4
Biologics are well known for their manufacturing challenges due to the complex nature of these molecules. From genetic development to cell banks, to drug substance production, and finally to drug product production, the creation and implementation of a manufacturing process for a biologic is an intricate and sensitive path. Slight changes in a single parameter within the process may affect the biological activity, and consequently the clinical efficacy and safety profile, which could necessitate additional clinical studies when these changes occur later in development.Citation5–8
The structural complexity of biologics and their inherent susceptibility to degradation also bring unique challenges both to the formulation of these macromolecules and to their effective delivery to the target sites of activity. Unlike their small molecule counterparts, biologics are almost exclusively administered by parenteral routes (e.g., intravenous (IV), subcutaneous (SC) or intramuscular (IM)). Although novel delivery methods for biologics (e.g., transdermal, pulmonary delivery) are being aggressively pursued with significant progress made over the past few years, to date, parenteral delivery remains the standard for biologic drugs.Citation9 A dose of several milligrams per kilogram body weight for these protein drugs is often required, presenting significant challenges in formulation and delivery. IV infusion makes a higher dose possible, but it has disadvantages, including inconvenience for patients and the high cost for patient care.Citation10 SC or IM administration requires minimal skills and is less invasive, thus enabling patients to self-administer in a home setting. But the convenience of SC/IM administration comes with a cost. Because it is limited by the administration volume (<2 mL per injection), a SC/IM formulation often needs to achieve high drug concentration with acceptable viscosity and stability, which can be technically difficult and require substantial resources. Despite the challenges, these extravascular delivery routes, especially SC, have become an attractive option for many protein therapeutics for chronic indications because of the advantage of self-administration and the associated benefits.
The increasing trend of developing biologics for SC/IM administration has led to intensive efforts to develop various types of injectable drug-device combination (DDC) products to improve convenience and compliance through modern technologies. Efficacy and safety of a therapy are necessary, but not sufficient, for its therapeutic success. It has been increasingly recognized that disease management for patients suffering from a chronic disease is a critical determinant in patient adherence to long-term therapies, which plays a crucial role in achieving optimal treatment benefits and outcomes. It has been reported that adherence rate to chronic therapy in developed countries averages only ∼ 50%, and is much lower in developing countries.Citation11 The non-adherence contributes to the gap between the clinical efficacy demonstrated in well-controlled clinical trials and effectiveness in real-world settings. Closing the gap is the ultimate goal of DDC product development in which patient factors must be considered in the design of the device to promote consistent and persistent use of these products in chronic disease management.
A DDC product, as the name implies, refers to two or more single-entity products combined for use. These products may be combined either chemically, physically, via co-packaging or provided separately and specifically labeled for use together. Commonly seen formats for injectable DDC products include the traditional vial and syringe (VS), as well as the more advanced and popular presentations, such as prefilled syringe (PFS), prefilled pen and auto-injector (AI).
With many possible twists and turns on the development path for an injectable DDC, questions often arise regarding whether or not clinical studies are needed when some changes are introduced. If clinical studies are needed, what kind of studies would suffice for adopting the changes? What is needed for a safe and efficient transition from the original process or product to the new one?
This review provides an in-depth look at the practical issues that are often encountered during the manufacturing process and presentation development of an injectable DDC. Considerations in establishing a scientifically sound and efficient clinical strategy to address these issues, as well as key elements of clinical studies, are discussed. Current trends in the biopharmaceutical industry and relevant regulatory guidance are also presented.
Development paradigm of injectable drug-device combination products for biologics
The constituent parts of an injectable DDC are the drug substance in its formulation, the primary container closure, and the device in which they are packaged, used or integrated together. The device may measure the dose or deliver the drug to the target site of action. The development of a DDC can be delineated into manufacturing process development and device development (). The manufacturing process development refers to the development of drug substance, formulation and primary container. Device development, as its name implies, refers to the design and development of the delivery device.
Figure 1. Overview of Injectable DDC Product Development Process and Typical Timelines in Relation to the Clinical Development Timeline.
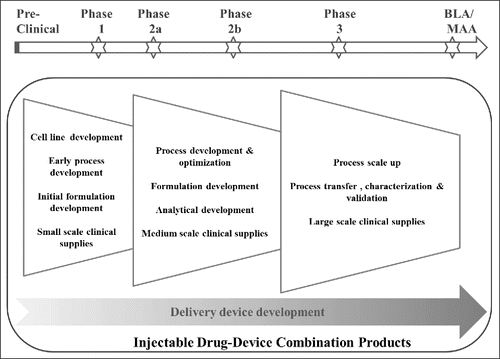
Manufacturing process development of a biologic is normally launched immediately after identification of a drug candidate with desirable biological and pharmacological activity for the disease target. Unlike small molecules that are chemically synthesized, biologics are mostly derived from living organisms and made by genetically engineering living cells (i.e., cell lines). A typical manufacturing process involves 5 sequential phases: 1) genetic engineering of a cell and establishing a cell line, 2) cell culture and fermentation, 3) protein isolation and purification, 4) formulation, and 5) fill and finish. Process development activities are interrelated, and the development workstream from cell line to final formulation and fill should be designed and structured according to the clinical development timeline in order to meet the demand and regulatory requirements for drug supplies at each stage ().
Since biologics are complex molecules, the manufacturing processes of these macromolecules are complicated. A high level of precision is required in the commercial manufacturing process to ensure a safe, active and consistent product is made from every batch. Numerous factors along the production line can influence the characteristics of the protein produced by the cells; therefore, each of the manufacturing process development phases is equally crucial in the success of the final drug product, and all aspects of the complicated process, including source and type of raw materials, environmental conditions, containers, equipment and procedures, need to be closely monitored, controlled and documented.
Within manufacturing process development, formulation represents a major area requiring extensive development efforts. Injectable formulation for a biologic usually evolves as the drug advances through clinical development, and typically the final formulation is not mature and ready for use until the late stages of clinical development, i.e., Phase 2b or Phase 3 studies. For multiple reasons, IV administration is often implemented in early clinical development when dose-ranging clinical studies (Phase 1 or Phase 2a) are conducted. In the early stages of development, the targeted therapeutic dose, which will drive the target concentration and formulation, is unknown, and an IV formulation is a good option for small-scale clinical trials because it is relatively easy to prepare. Moreover, for the first-in-human studies, a wide range of doses need to be explored in order to characterize the safety and activity of the drug. IV infusion allows flexibility while doses for subsequent pivotal trials are being determined. Last but not the least, IV administration ensures complete delivery of the drug to the body (100% bioavailable) and allows comprehensive understanding of the systemic pharmacokinetic (PK) profiles, which is crucial at this stage of clinical development and often one of the primary or secondary objectives of early trials. Depending on the design of the trial and availability of a preliminary SC/IM formulation, in many cases, SC/IM cohorts are included in the early trials to obtain information, such as SC/IM bioavailability and SC/IM safety and tolerability. The bioavailability by SC/IM administration is an important determinant for the feasibility of a DDC.
A major inherent limitation for SC/IM administration is the fluid characteristics (e.g., viscosity) and allowable volume for a convenient injection, which is typically less than 2 mL per injection. Consequently, an injectable formulation for a biologic often requires a relatively high protein concentration to achieve the therapeutic dose, which can create substantial development challenges, such as solubility limitations, physical instability due to aggregation, increased viscosity, and prolonged injection time. In addition, from a user's perspective, injecting the full amount of a viscous solution may be difficult, and cause discomfort and pain at the injection site. In recent years, a number of approaches have been taken to overcome these hurdles and improve the acceptability of SC/IM injections from both a technical point of view and from the user's perspective. One of the approaches is to use recombinant human hyaluronidase as a pharmaceutical adjuvant together with the biologic of interest to achieve higher injection volume. The recombinant human hyaluronidase can transiently degrade hyaluronic acid (aka. hyaluronan), a key structural component of tissues, in the interstitium and thereby increase the diffusion of proteins at the injection site,Citation9 allowing a larger volume to be injected.
Designing and selecting a compatible primary container closure is an integral part of the formulation development. At the Phase 1/2a study stage, a glass vial with rubber stopper presentation is widely used, which provides the convenience and flexibility for dilution in order to test a wide range of doses. This presentation may also meet the demands for a Phase 2b trial. However, for the pivotal Phase 3 trial and beyond, one must consider additional factors, e.g., dose requirements, patient factors and commercial preferences, when selecting and finalizing a primary container for the final drug product presentation. If a ready-to-use type of product is desired, e.g., PFS instead of a traditional VS format, it should be recognized that potential incompatibility of the biological molecules with various contact surfaces and interaction with leachables from the glass barrel or plunger stopper of the new container closure may present distinctive challenges. If any changes become necessary during or after the pivotal trials, a cascade of additional stability tests as well as formulation work will be triggered, and significant clinical bridging data may also be required.Citation12–16
Device development for injectable DDC products for biologics is a process that spans the entire period of manufacturing process development and clinical development, and sometime extends to the post-approval stage as part of life-cycle management. In the past 15 years or so, driven by an increased demand for patient-centric DDC products, device development has gained significant momentum and been recognized as an important part of the overall drug development plan.
Introducing a SC/IM injectable product through devices such as PFS or AI offers a multitude of advantages. It not only differentiates the product on the market through innovative and user-friendly designs, but also offers economical and patient benefits by allowing convenient self-injection and reducing systemic health care burdens.
In the early clinical development stage, device development activities usually focus on planning and feasibility assessment. The activities intensify as the drug candidate advances through the later clinical stages. The most common injectable DDC product presentations for extravascular administration are VS, PFS, pen injector and AI. In the VS combination, the vial is the primary container and a separate syringe is used to measure and deliver the therapy. In a PFS, pen injector or AI, the drug is pre-measured and filled in the integrated product device that also delivers the therapy. As discussed earlier, the VS combination product is often used as an early drug product presentation in Phase 1, Phase 2a and sometimes Phase 2b proof-of-concept studies while device development is still underway, in parallel with clinical development. Ideally, the to-be-marketed product, either continuing to be the VS, or a PFS or an AI, should be used in the pivotal trials to support its approval and labeling. In reality, more advanced devices like AIs are often introduced to the market after the initial product launch as an alternative delivery system due to process gaps, technical challenges or intended delay of investment to minimize product attrition risks. This delay is becoming less significant as these devices for use in injectable DDC products are gaining unprecedented popularity and biopharmaceutical companies are investing earlier than before on device development work.
Clinical comparability assessment following manufacturing process changes
The manufacturing process for biologics is sophisticated and complicated, and so is the development path leading to the final process. All phases involved in the process development are closely nested and should not proceed in isolation. From drug substance to formulation development, and to primary container closure, multiple iterations are often necessary to achieve the optimum balance of drug stability, process efficiency, patient tolerability/acceptance and other factors. Therefore, manufacturing process development for an injectable biologic is a convoluted process, and it is not uncommon that process changes to the drug product need to be made while Phase 2 or even Phase 3 clinical trials are already underway. Changes to the manufacturing process can even occur after a product is approved for marketing. These changes can be introduced by altering the cell line, scaling up from pilot production to full-scale manufacturing, improving manufacturing efficiency, improving product quality, transferring manufacturing facilities, modifying formulation or meeting regulatory commitments. Compared to small molecules, biologics are much more sensitive to manufacturing process changes, and these variations in the production process could result in alterations to the primary, secondary, tertiary or quaternary structure of the molecule, and have the potential for unpredictable effects on safety and efficacy of the product.Citation17–19,20 Consequently, comparison of biological products pre- and post-manufacturing changes is required to demonstrate that the products are sufficiently similar or comparable.
Risk-based comparability assessment
There is no single paradigm for comparability assessment that would be appropriate for all, and it is well accepted that comparability assessment should be a risk-based hierarchical approach with patient safety as the primary consideration. The degree of risk for any process change(s) is a composite of the nature of change(s) and the timing of change(s) relative to the clinical development timeline. The goal for a comparability assessment is to demonstrate that the product from the new process is “equivalent” to the pre-change product and to ensure that data collected from studies with the pre-change product are applicable for the post-change product; or if not, additional studies are conducted to ensure safe product transition during clinical development or after marketing has begun.
There are 3 components in the potential comparability data package: analytical characterization, nonclinical assessment and clinical assessment. Whenever manufacturing process changes occur, robust analytical characterization would be the minimally required data and the prerequisite for any in vivo assessment (i.e., nonclinical or clinical). Comparison of the product before and after process changes by analytical characterization should show that the product has highly similar quality attributes and potency so that the process maintains or improves its capabilities and that the changes have no effects on the product's safety and efficacy. The analytical technology and methods used in such assessment and the associated pros-and-cons have been discussed elsewhereCitation21–23 and is outside of the scope of this review. It should be recognized that, even with the most advanced technology and methods, changes to the complex protein molecules after process modification may not be fully characterized.Citation21 Therefore, the limitations of analytics together with the risk associated with the changes and the timing of the changes dictate the rigor of the in vivo data package (e.g., amount of preclinical and clinical data) and the degree of assurance and stringency of the data that are necessary to demonstrate comparability ().
Figure 2. Nature and Timing of Biologics Manufacturing Process Changes Determines the Overall Potential Risk on Patient Safety and the Rigor of Comparability Assessment.
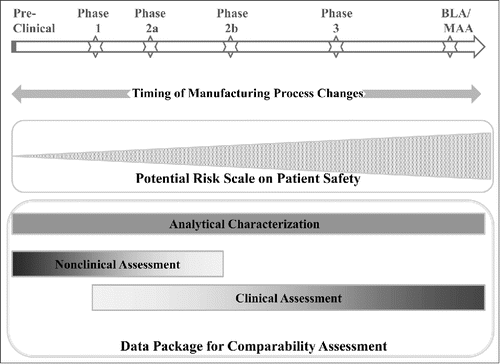
In vivo comparability assessment can be conducted in animal models or directly in humans. Animal studies are mostly used when changes occur very early during development. However, animal studies have their own inherent limitations and have not been widely conducted to support process changes. In principle, clinical studies are only required when major changes, such as changes of cell line, manufacturing site, formulation or manufacturing scale, occur at a late stage of clinical development (e.g., during or after completion of pivotal trials).
During early clinical development, i.e., Phase 1 and Phase 2a stage, it is unlikely that manufacturing processes or formulation studies will be completed or finalized by the time these clinical trials commence, and prototype formulation are often developed initially for use in these studies. By the time clinical development reaches Phase 3, the manufacturing process is generally mature and the drug product is either in its final presentation or very close to the one that will ultimately be used in the market. Therefore, most manufacturing process changes generally occur during Phase 1 and Phase 2, and most companies manage to complete these changes prior to initiating registrational clinical studies in order to obtain the majority of the safety and efficacy data with the final formulation, and thereby minimize risks. However, it is sometimes necessary to make changes during pivotal trials or after the completion of these trials. This is when definitive and extensive clinical comparability assessment is most likely required.
The type and scope of clinical bridging studies supporting comparability and transition between pre- and post-change products also depends on the nature and timing of changes relative to the clinical development stage. A PK/PD focused comparability approach has been widely taken to support process changes prior to the conduct of large clinical efficacy and safety trials. This type of study aims at providing PK or PD comparison between products and directly addresses the question regarding dose selection for the pivotal efficacy and safety trials based on the Phase 1 and 2 results. The rigor of a PK/PD comparability study can vary from a small and non-bioequivalence (non-BE) study in healthy subjects to a large BE study in patients, and the acceptability of the approach is determined case-by-case depending on the degree of changes and the associated risks. Most dedicated PK/PD comparability studies are conducted as single dose studies, with PK or PD as the primary objective, and safety/tolerability/immunogenicity as secondary objectives.
As the molecule moves into the later stage of clinical development, important process changes, as aforementioned, may trigger an efficacy and safety trial in the target patient population with or without a stand-alone PK/PD comparability study. In the latter case (i.e., without a stand-alone PK/PD study), PK or PD data can be collected from the efficacy and safety trial. Besides PK/PD data, anti-drug antibody (ADA) data, another important parameter for comparability assessment, can also be collected. Compared to data from a single dose PK/PD study, ADA data from a repeat-dose efficacy and safety trial would be more relevant for comparability assessment.
We conducted a review of mAbs approved by the Food and Drug Administration (FDA) from January 2006 to March 2017 in order to understand the trends in the pharmaceutical /biotechnology industry and the thinking of the FDA in comparability assessment to support biologics manufacturing process changes. The information was obtained from FDA reviews of the original biologics license applications (BLAs) in the FDA database (Drugs@FDA, www.FDA.gov). There were 42 mAbs granted first approvals in this period (); data for any supplemental approvals of these mAbs were not considered. As specific information related to manufacturing process changes are mostly proprietary and cannot be made public in the FDA reviews, the nature and timing of the process changes may not be specified in the review documents. In some cases, information was taken from publications and clinicaltrials.gov to supplement our analyses. These mAbs are categorized into 4 groups as follows, based on whether or not, and when, process changes were made during clinical development and whether or not dedicated animal or human PK or PK/PD comparability studies or efficacy/safety trials were conducted to support the product transition.
Table 1. List of FDA Approved Monoclonal Antibodies from January 2006 to March 2017 and the Clinical Comparability Assessment Studies to Support the Manufacturing Process Changes during Development Footnotea, b.
Group 1: No changes are cited in the BLA reviews and no dedicated animal or human PK or PK/PD comparability studies were conducted.
Group 2: Changes are cited in the BLA reviews, but no dedicated animal or human PK or PK/PD comparability studies were conducted.
Group 3: Changes are cited in the BLA review and animal PK or PK/PD comparability, but no dedicated human studies were conducted.
Group 4: Changes are cited in the BLA review and clinical trials were conducted, either dedicated human PK or PK/PD comparability studies or PK/PD as part of efficacy/safety trials.
Among these cases, there are 14 in Group 1 (33.3%), 11 in Group 2 (26.2%), 3 in Group 3 (7.1%) and 14 in Group 4 (33.3%). Therefore, most mAbs in this analysis (67%; 28 of 42) went through some degree of manufacturing changes during development; and for half of them (14/28), clinical studies were conducted to support the process changes. These products were predominantly for IV (n=28) or SC (n=14) administration (including 1 product with both IV and SC options (tocilizumab)), except for one administered via intravitreal injection. Fifteen IV products (∼54%) vs. all SC products (100%) involved manufacturing process changes. This is likely because formulation development is generally more straightforward for IV than for SC administration, reducing the chance of significant changes. In terms of the timing of changes, 14 mAbs had changes only prior to the initiation of pivotal trials, 2 had changes only after, and 12 had changes both prior to and after initiation of pivotal trials (including one likely prior to and during the pivotal trials).
For the Group 2 cases (n=11), most (73%; 8 of 11) had process changes only prior to the initiation of pivotal trials, and the other three had changes both prior to and after pivotal trials were started. All changes were deemed minor, not warranting clinical bridging studies. Although no dedicated PK or PK/PD studies were conducted, comparability assessment by cross-study PK comparison or population PK analysis was performed in 3 of the cases to supplement analytical data. In one case (mepolizumab), FDA limited the use of PK data from earlier trials in the label because there were significant differences in the drug products between those earlier trials and the pivotal trials.
In Group 3 (n=3), two mAbs had process changes only prior to, and one had changes both prior to and after, the initiation of pivotal trials. The nature of changes was related to formulation change or cell line change in two cases, and not specified for the third case. The proceeding or adoptions of these changes were adequately justified by the Chemistry, Manufacturing and Controls analytical data package together with nonclinical PK comparability studies. Meanwhile, in two of the cases, supplementary cross-study data comparison or population PK analysis was performed to further support comparability assessment.
Among the Group 4 cases (n=14), four had process changes only prior to the initiation of pivotal trials and ten had changes after, with eight of those also having changes prior to the initiation of pivotal trials. Where specified, the process changes involved change of cell line or formulation, or site transfer. Comparability was evaluated by dedicated PK or PK/PD comparability studies (n=9), dedicated bridging efficacy/safety trials or as part of other efficacy/safety trials (n=4), or both PK/PD and efficacy/safety trials (n=1). Supplementary data comparison across studies or population PK analysis was provided in three of the cases.
From these examples, it can be seen that the timing and nature of changes cannot be taken in isolation to determine the comparability package. Even when the drug products in pivotal trials are different from the to-be-marketed drug products, as presented in some cases, in vivo assessment may not be needed if the changes are minor and do not pose unknowns risks to patient safety. However, when changes occur early, in vivo assessment may still be necessary to justify the use of the data from the previous version of drug product for critical decision-making, such as dose selection or labeling. The key point is whether or not the comparability strategy is scientifically justified based on knowns and unknowns associated with the process change, which must be evaluated on a case-by-case basis.
Considerations in the design of human pk or pk/pd comparability studies
As part of the comparability assessment to support manufacturing process changes, human PK or PK/PD comparability studies are essential. Major questions related to the design of these studies include choice of study subjects, selection of dose and regimen, sample size and primary endpoints.
The choice of the population for such studies, healthy subjects vs. patients, takes into account the same considerations as for other Phase 1 studies for biologics, and is primarily driven by the drug's mechanism of action. While in most cases, heathy subjects are justified, for some drugs, such as those for oncology indications, patients may be more appropriate. When PD evaluation is part of a comparability study, the PD effects of interest and their assessment and relevance in the study subjects of choice must be considered.
Single dose parallel design is generally considered acceptable for these studies due to slow elimination, long elimination half-lives of biologics (days to weeks) and their propensity of eliciting ADA. However, these studies offer limited information in the comparability assessment of anti-drug antibody formation, which, if deemed necessary, is best evaluated in repeat-dose studies in the target patient population. Regarding dose selection, it can be in the steep part of the dose-response curve, as recommended in the European Medicines Agency (EMA) guidance document, in order to detect relevant differences, or in accordance with the target dose for pivotal trials and marketing.Citation24 If a final therapeutic dose is not determined at the time of a comparability study, multiple dose levels may be needed, especially if PK cannot be extrapolated from one dose to another for reasons such as nonlinear PK or different formulations at different doses.
Sample size is often the most debated question that many face when designing comparability studies for biologics. As the key objective of these studies is to demonstrate comparability, not equivalency, it is not necessary to power the studies as a standard bioequivalence (BE) study, a requirement for small molecule generic drugs. In practice, however, BE studies are sometimes conducted to mitigate risks and the level of acceptable risk often drives the sample size. Predefined criteria to declare comparability should be set in place in the data analysis plan. The criteria should be objective and justified on a case-by-case basis with considerations for factors such as PK variability and exposure-response relationships for efficacy and safety parameters. While the 80-125% boundary for geometric mean ratio (GMR) for PK parameters (i.e., a conventional BE criteria) is routinely used in many comparability studies for biologics, it is not a regulatory requirement.
Since these comparability studies aim to demonstrate PK comparability, the primary objective should be comparison of PK between pre-change and post-change products, rather than characterization of PK profiles per se, so only the relevant PK parameters are necessary. If PD markers are available, evaluation of PD is often preferably integrated into the comparability studies.
There are situations in which a dedicated clinical PK or PK/PD comparability study is not sufficient to support product changes, and additional clinical data are warranted to support efficacy and safety, including immunogenicity. In such situations, PK or PD evaluations can be part of the endpoints for these trials.
Among the previously reviewed cases in Group 4, ten biologics had a total of 13 reported Phase 1 PK or PK/PD trials conducted, with 10 in healthy subjects and 3 in patients (all three were for oncology indications). The key study features are summarized in . Except for one conducted as a crossover repeat-dose study, all studies used a single-dose parallel design. Sample size varies dramatically, being as low as 7 per arm to as high as 175 per arm. Of note, the 90% confidence interval for a PK parameter comparison between test and reference products (GMR) within the standard equivalence limits of 80-125% criterion is used for comparability evaluation in every case except two (alirocumab and dupilumab). For alirocumab and dupilumab, with a small sample size (n<20 per arm), the GMRs in the comparability studies were close to 1.0 and the 90% CI fell slightly outside the 0.80-1.25 boundary. The test and reference products in these studies were deemed comparable by the FDA [Source: Drugs@FDA database, www.FDA.gov].
Table 2. Study Features of Human PK Comparability Trials to Support Manufacturing Process Changes in the FDA Database from January 2006 to March 2017.
In summary, the study design for PK comparability trials can vary substantially. While demonstrating strict BE may not be required, all elements of a comparability study should aim at maximizing the potential for detecting relevant differences and be justified based on the product characteristics.
Clinical strategy to support presentation changes for injectable ddc products
The device in a DDC product is an integral part of a biological treatment, and an important factor in the overall safety and efficacy of the drug product. It needs to ensure that the active drug is delivered safely and accurately at the intended dose level. Adding another dimension to a biological product, the device itself requires its own design, development and manufacturing.
Device types for DDC products
There are numerous DDC products for injectable biologics on the market or in development, such as VS, pen injectors, PFS, AI, and devices based on recent innovative technologies, such as large volume wearable injector device (LVD).
A pen injector is a cartridge-based device that was introduced to the market for insulin in the 1980s and has since become widely used for peptide therapies, such as insulin and human growth hormone. It is designed as either a disposable or reusable injector (for either single-dose or repeat-dose), to meet the requirements of the therapy, such as frequent manual injections or variable doses. The volume per injection is normally within 0.5 mL, and each cartridge may hold enough quantity of drug to accommodate multiple injections for days or weeks depending on dosing regimen and product stability.
Other types of DDC products are mainly used for large molecule biotechnology products, such as mAbs. The VS presentation is the basic DDC format, and often the first commercially available for many of these products. In the VS format, the drugs are presented in the vial either in a lyophilized form for reconstitution before injection or as a ready-for-injection liquid formulation, and injected in a traditional way using a syringe pre-packaged in the VS presentation.
The PFS, as the name implies, is a container closure syringe system with a pre-attached needle and is prefilled with liquid-based formulation (single-chamber) or lyophilized drugs (multiple-chamber). Over the past decade, PFS has been a fast-growing choice for biologics intended for fixed-dose SC/IM administration because it offers great advantages over the traditional VS format, such as convenience to both healthcare providers and patients, reduction of dosing errors, prevention of contamination, thereby improving safety, cost-saving by eliminating drug overfill required for VS. The field of PFS development continues to grow rapidly, driven by the incessant search for greater capability, better safety and more convenience.
The development of PFS is also of broad interest due to its potential to combine with an autoinjector (AI), the widely accepted gold standard self-injection device. For many drug developers, PFS has been developed as the precursor for an AI DDC product, in which a PFS is prepackaged within the AI. An AI simplifies the use of PFS by automating the manual tasks of needle insertion and injection, which enables caregivers or patients to complete the injection with greater ease and improved needle safety. Besides the advantages offered by PFS within an AI product, using an AI may also help to reduce emotional hurdles surrounding needle use, as well as injection-associated pain and discomfort, and thereby facilitate long-term adherence to a therapy. Moreover, AI can accommodate specific features to enable self-injection for patients with manual dexterity issues, such as elderly patients and those with disabling diseases (e.g., rheumatoid arthritis (RA) or multiple sclerosis (MS)).
Other devices based on emerging technologies, such as LVD, aim at overcoming the volume limitation for SC/IM injections and enable delivery of a larger volume of biologic therapies than can be delivered with handheld devices (VS or PFS/AI). For the latter, the injection volume is usually limited to 1 mL and may not exceed 2 mL, and the injection can be completed in less than a minute (often to be within 20 seconds by design for AI). However, a LVD, for example, is a wearable and disposable device that can be attached to the body to deliver a volume of greater than 2 mL in a designated period of time, and is then removed and disposed upon injection completion. This body-worn feature of LVD is ideal for many patients due to the convenience of completing injections at flexible locations at any time.
Clinical studies to support DDC product presentation changes
Similar to manufacturing process development, DDC device development is also an iterative and evolving process in order to achieve the optimum balance of drug stability, safe use and patient compliance. Development of an advanced DDC presentation requires substantial financial resources and manpower. Therefore, DDC products in the VS format are often by default the initial product presentation used in clinical trials with a lyophilized or a liquid formulation, and the development of other formats (e.g., PFS or AI), if deemed desirable, usually lags the clinical trials, making clinical bridging a necessary step before introduced to a large population (e.g., large pivotal trials) or the market. The type of clinical studies that are required for bridging, in essence, are similar to those conducted in support of manufacturing process development.
DDC device development, in large part, is driven by the need of simple and safe self-administration for patients that enhances user acceptability, improves patient experience and facilitates adherence, ultimately helping to maximize therapeutic outcomes. Success of any DDC product is contingent upon its ability to provide patients or intended users a safe and reliable method to take the medication, as well as to promote long-term consistent and persistent use of the product. From a regulatory perspective, demonstration of simple and safe handling by patients, as well as accurate and reliable drug delivery through usability assessment, is a key requirement to support marketing approvals of a DDC product. Usability of devices are normally addressed by human factor (HF) studies in the representative target population(s), and these studies focus on assessing the adequacy of the device-user interface in order to eliminate or mitigate potential user related hazards.Citation25 Through DDC product development, HF studies are conducted to support device design and confirm if the device executes its intended use. These studies are performed in the anticipated patient demographic and gather actual user feedback for validation in a simulated environment; therefore, they are not considered clinical studies. The data collected in the HF studies are objective and subjective observational data, related not to clinical endpoints, but rather to the user's interaction with the device when injections are performed in a simulated-use setting (e.g., injection into an injection pad) with or without the active drug substance. For most DDC products, these HF studies are sufficient to assess the adequacy of the device-user interface design and to support labeling if the product is approved.
However, recently there have been increasing cases when usability data are collected in clinical studies.Citation26–37 The data collection may be incorporated into a major clinical trial (Phase 2, Phase 3 or open-label extension) with clinical outcomes as the primary endpoints; alternatively, stand-alone studies (the so-called “actual use” studies) can be conducted with usability as the primary endpoints.
Another type of study that is commonly used to support DDC device development involves collection of data on patient experience and feedback. Patients' long-term compliance with chronic treatment is vital for the therapeutic outcome. For this reason, it is important to understand a patient's ability to use the DDC product, and his/her acceptance and experience with the product. User experience and feedback are usually collected in the form of trainings and questionnaires, and can be built as a stand-alone study or incorporated into other studies, including HF studies, actual use studies or major clinical trials.Citation26,38–41 This type of evaluation usually has not been viewed as a regulatory requirement for approval.
Clinical bridging studies
In the development of DDC products, the criteria used to assess the need for clinical bridging studies are the same as that for manufacturing changes, i.e., studies are only required when major changes occur at a late stage of clinical development. The process of designing and developing DDC products is complex and involves substantial resource investment, which explains why product presentation changes often occur rather late. In the majority of cases (), the desired commercial DDC product was not ready at the initiation of Phase 3 pivotal trials or only became available close to the time of initial submission for marketing approval or even after.
Table 3. List of FDA Approved Monoclonal Antibodies DDC Products for SC Administration from January 2006 to March 2017: Clinical Studies in Support of Marketing Approvals of DDC Products.
Unlike manufacturing process changes, the changes of concern in DDC products are related to either changes in drug product contact (i.e., primary container closure) or changes in technical characteristics from the original presentation to the new presentation that have the potential to change the mode of administration, and consequently affect PK or PD, efficacy and safety profiles in a clinically relevant manner.
The most commonly seen presentation switching during biologics development is either between VS and PFS or AI, or between PFS and AI. Switching from a VS presentation to either a PFS or an AI likely involves change in drug product contact (i.e., primary container closure), which is a major change requiring a clinical bridging study or studies. In contrast, switching from a PFS to an AI, or vice versa, the drug product contact is likely intact as it is often the case that the same PFS is entirely housed within the AI. Therefore, from a primary container closure point of view, clinical bridging may not be needed when a DDC presentation changes between PFS and AI. However, the mode of administration via different DDC presentations can be affected in all these switches, depending on how the key administration specifications, such as injection force, needle gauge, needle insertion depth, injection time, or accuracy of dose delivery, differ between the original and the new presentations. In the case of switching from VS to PFS or to AI, it is likely for changes to one or more of these key specifications to occur. But it is easier to avoid or minimize such changes when switching between PFS and AI.
If the key administration specifications for the original and new presentations are sufficiently similar to justify that the mode of administration is unchanged, demonstration of analytical comparability of DDC product at release and at the end of shelf-life would be sufficient, and clinical comparability may not be required in support of the marketing approval of the DDC product of interest. Otherwise, clinical bridging studies (PK/PD comparability or outcome studies) may be warranted. In many cases, demonstrating PK or PK/PD comparability would be sufficient. If, however, the design of the new presentation raises concerns or uncertainties in terms of altering existing safety or efficacy profiles of the biologics, or if the PK or PK/PD comparability study does not demonstrate comparability, additional clinical studies for safety or efficacy will be necessary.
In support of presentation switches, clinical PK or PK/PD comparability studies commonly use BE criteria (80-125% for 90% confidence interval) for key PK parameters (e.g., AUC and Cmax) to define comparability, and the size of such studies are generally in the range of 100-300 subjects for a 2-arm parallel design. It still remains an open question in the field whether or not it is necessary to use the BE boundary to define comparability for biologics. As many of these large molecules have rather wide therapeutic windows and precise dosing is not critical, scientifically it is not necessary for a comparability assessment to be a BE exercise. In fact, trial sponsors often use BE to be conservative and to increase the likelihood of data acceptance by regulatory authorities, but the number of subjects or patients is substantially higher for a BE study than what would be required for a study using a less conservative approach (e.g., maximum imprecision). Ethical as well as resource factors must be taken into account when a comparability strategy is being considered.
Among the mAbs with initial approvals from January 2006 to March 2017, there were 13 products with one or two DDC presentations other than VS combination (e.g., PFS and AI) approved for marketing (). Based on product prescribing information and the information available in the FDA BLA reviews and on the clinicaltrials.gov website, we identified five products (alirocumab, brodalumab, daclizumab, dupilumab and tocilizumab) that used the same presentations in the pivotal Phase 3 trials as the commercial DDC products; therefore, no clinical bridging studies were conducted. The other 8 products had approved commercial DDC presentations that were different from the one(s) used in the pivotal trials. Among these 8 cases, 3 products (certolizumab, denosumab and ustekinumab) used either solution-in-vial (SIV) or lyophilized powder-in-vial (LYOIV) formulation administered by VS presentation in the pivotal Phase 3 trials, but had both VS and PFS approved. The bridging from SIV-VS or LYOIV-VS to PFS for certolizumab and denosumab was based on PK comparability in healthy subjects, and no information was found regarding efficacy or safety data to support bridging. For ustekinumab, no information, including PK comparability, was found.
Four other products (evolocumab, golimumab, ranibizumab and secukinumab) also used either SIV-VS or LYOIV-VS in the pivotal Phase 3 trials, but have PFS and AI approved without VS in the label. For evolocumab, golimumab and secukinumab, the bridging from SIV-VS or LYOIV-VS to PFS and AI was supported by both dedicated PK or PK/PD comparability and additional efficacy and safety data. The efficacy and safety data were collected either within pivotal Phase 3 (switching in the middle of a study) or in non-pivotal Phase 3 or open-label extension studies. No clinical bridging between PFS and AI was conducted for these three mAb drugs. For ranibizumab, no information was found.
For ixekizumab, PFS was ready at the time of the pivotal trials and was used throughout the Phase 3 program. Both PFS and AI were approved in the initial approval, and there was PK bridging data in patients in a non-pivotal Phase 3 study supporting the approval of AI.
For all cases where there were cited PK or PK/PD comparability studies, the sample sizes are in the range of approximately 50-175 per arm, and PK comparability was declared per BE criteria (). For 3 of the 4 cases where both PFS and AI were approved (evolocumab, golimumab, secukinumab, ixekizumab), available information in the data source indicated that the approved presentations contained the same solution formulation and the same primary syringe for all except for evolocumab, for which no such information was found.
Usability assessment in real-world clinical settings
Usability assessment is primarily carried out with HF studies, as discussed previously, and this usually satisfies the regulatory requirements. However, recently there seems to be an upswing in the trend of conducting clinical trials for such assessment.Citation26–37 Unlike HF studies, which are well defined regulatory-required studies, usability assessment in real-world clinical settings in terms of trial conduct and the required outcomes have not been specifically described in regulatory guidance documents. A search of the ClinicalTrials.gov website returned 15 studies for injectable DDC products (specifically, mAbs) involving either PFS or AI devices, with usability as the primary or secondary endpoints (). A review of these studies shows that various measurements with usability-related endpoints have been used, either in major clinical trials or in stand-alone clinical actual use studies, and the measurements collectively assess successful and safe self-administration (device failure, use failures, PK/exposure monitoring), occasionally with endpoints addressing patient preference and acceptability. “Major” clinical trials refer to Phase 2, Phase 3 or open-label extension studies with clinical outcomes as main objectives. Actual use studies are those with usability assessment and device-use related safety (e.g., pain, injection site reactions) as main objectives conducted in the target patient population. Five of the 15 studies are major clinical trials, 9 studies are actual use studies in target patient populations, and 1 study is a usability study in healthy subjects.
Table 4. Clinical Studies with Usability-related Outcome Measurements for mAb DDC productsFootnotea.
Although there are similar issues that can be addressed by both a HF study and usability assessment in a major clinical trial or an actual use study, there are some issues that can only be addressed in real-world clinical settings. A critical aspect of a DDC product is the accurate delivery of the intended dose by the device, which can be affected by various user-related factors, such as user ability and probability for user errors.Citation42–45 In some disease populations, administration of injectable therapies using any device may prove difficult and it is imperative to understand and evaluate user-related factors in usability assessments. It has been reported that patients may make errors in drawing and injecting the correct dose of insulin due to reduced manual dexterity or impaired eyesight. In a study by Kesson and Bailie it was demonstrated that there could be considerable error in insulin dosage. They reported that older diabetic patients (mean: 58 years old; range: 41-77 years old) were both highly inaccurate and less consistent in drawing up their prescribed doses of insulin with a percentage error of approximately 19.3% in the accuracy of the insulin doses drawn, as compared to 5.8% in a younger group of patients (mean: 23 years old; range: 13-36 years old).Citation46 Another study reported a smaller but still significant inaccuracy, approximately 12.2% (mean) in a diabetic population of syringe users aged >55 years.Citation47 Therefore, usability assessments done in a real-world clinical setting can provide the most relevant data regarding patients' ability to use the product and the potential clinical impact.
Regulatory requirements on injectable ddc products for biologics
Over the past decade, there has been a steady increase in the number of combination products. Among these, many fall into the category of injectable DDC products. This rapid growth of injectable DDC products, as well as their inherent complexity, unavoidably presents unique regulatory challenges.
There are specific and well-developed regulatory requirements regulating manufacturing processes and quality aspects of each of the constituent parts of injectable DDC products, but there is a great need for a more streamlined and integrated regulatory framework for the combination products. Responding to the need, FDA finalized its regulation (21 CFR part 4) in 2013;Citation48 and provided a draft guidance “Current Good Manufacturing Practice Requirements for Combination Products” in 2015 and the final guidance in January 2017.Citation49 In the European Union, in addition to the existing guidelines, EMA indicated its intention to develop a new guidance document on quality requirements of medicinal products containing a device component for delivery or use of the medicinal product, and released a new concept paper on this guidance in February 2017.Citation50 It is clear that continuous efforts are underway within regulatory agencies to address manufacturing and quality issues related to DDC products, including injectable mAbs.
In comparison, there are more gaps in regulatory requirements for clinical investigations in support of injectable DDC product development and approval. The FDA first developed and released the comparability guidance “Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology-derived Products” in 1996 to respond to the expansion in biotechnology product development. In this guidance, the general considerations for clinical comparability studies were outlined.Citation51 Similar guidance documents were released by the EMA and other health authorities. With collective efforts across health authorities in the world and in partnership with biopharmaceutical industry, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Q5E guidelineCitation52 was published in 2005, which was later adopted by the FDA; and to date it is still being accepted and referenced globally.Citation53 For the most part, this guidance is very useful for establishing a comparability strategy to address manufacturing process changes, but not as much for device transition during DDC product development. The most relevant regulatory guidance related to device switching for injectable mAb products and the required clinical data package is the FDA's guidance document specifically for RA drug development, which is understandable as the RA population is among the first to use injectable mAb and other protein DDC products and has a greater variety of DDC products available compared to other patient populations.Citation54 There is a specific section in this FDA document dedicated to “Drug-Device Combination Product Considerations”. While the general principle, i.e., “the extent of clinical data needed depends on the nature of the change and the development stage”, is applicable to all products for RA as well as for other diseases, it is questionable whether the minimally required data package outlined in this document is appropriate or necessary in all situations for all products, even within RA. The example FDA provides in the document is a transition from a PFS to an AI, and FDA states that the clinical data package “involves the following, at a minimum: (1) human factor studies to evaluate potential use-related risks of the modified combination product; (2) a pharmacokinetic bridging study that demonstrates similar delivery of the drug product to the same biospace across a range of body weights; and (3) real-life patient handling experience to assess device performance as discussed above. Depending on the extent of the proposed changes, additional clinical data may be needed to support efficacy and safety, including immunogenicity”. HF studies, as discussed previously, are not clinical studies, but are normally required for all DDC products. PK bridging studies, however, may not be needed in cases where it is fully justified that the original device and the new device are identical in primary container closure and mode of administration. Real-world usability assessment has become common for injectable DDC products, especially those for patients with disability and dexterity issues. There are, however, many questions to be addressed regarding usability assessment in clinical trials. Establishing the reliability or usability of injection devices in the real-world clinical setting using the real product in the target patient population is clearly an objective, but these trials have been conducted using a number of different approaches and endpoints (). Although clinical trial requirements should be product specific, standard requirements for these studies with respect to design, conduct and expected outcomes are also needed. The biopharmaceutical industry has reached a crucial point that calls for guidance and recommendations from regulatory authorities regarding injectable DDC products, which will foster consistency in practice, and promote quality and efficiency in DDC product development.
The future success of injectable DDC product development for mAbs will rely heavily on a streamlined and transparent science-based regulatory system. The currently available regulatory framework for development and marketing approval of these products needs to be further clarified and expanded. There is an urgent need for greater partnership between the regulatory bodies and the industry to make this happen and bring meaningful progress to the field.
Conclusion
The development of injectable DDC products for biologics is now a strategic imperative across the biopharmaceutical industry with important implications for product value propositions. There is also a high demand for DDC products in many therapeutic areas, driven by the desire for more convenience for patients and health care providers, less economic burden and better treatment compliance in long-term disease management.
Both manufacturing process development and device development for injectable DDC products are continuously evolving and iterative processes. When significant changes are made at a relatively late stage during clinical development of a DDC product, clinical bridging studies may be required. The rigor of the clinical data package required to support the transition between pre-change and post-change products depends on the nature and timing of the changes. The review of FDA-approved injectable DDC products for biologics, specifically mAbs, in recent years has revealed the current trends and common practices for designing and conducting clinical comparability studies as well as other supportive studies.
There have been great initiatives within health authorities in the United States and Europe for more streamlined and transparent science-based regulatory framework to guide DDC product development, but gaps still remain. The future success of injectable DDC product development for biologics undoubtedly depends on strong collaboration between regulatory agencies and the biopharmaceutical industry with aligned expectations and defined goals.
Disclosure
Zhaoyang Li and Rachael Easton are employees of Sanofi. The authors report no conflict of interest.
Funding
This work was supported by Sanofi.
References
- Pharmaceutical Research and Manufacturers of America. Medicines in Development: Biologics; 2013 [last accessed 2017 October 06]. http://phrma.org/sites/default/files/pdf/biologicsoverview2013.pdf
- Food and Drug Administration. CDER New Drug Review: 2015 Update [last accessed 2017 October 06]. http://www.fda.gov/downloads/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDER/UCM477020.pdf
- Cai HH. Therapeutic monoclonal antibodies approved by FDA in 2015. MOJ Immunology. 2016;3(2):00087 [last accessed 2017 Oct 06]. http://medcraveonline.com/MOJI/MOJI-03-00087.pdf. doi:10.15406/moji.2016.03.00087.
- Shih HH, Miller P, Harnish DC. Chapter 1. An Overview of discovery and development process for biologics. In: Wang W and Singh M, editors. Biological Drug Products: Development and Strategies, First Edition. John Wiley & Sons; 2014. p.3–30.
- Kozlowski S, Swann P. Current and future issues in the manufacturing and development of monoclonal antibodies. Adv Drug Deliv Rev. 2006;58(5-6):707–22. doi:10.1016/j.addr.2006.05.002. PMID:16828921
- Jefferis R. Aggregation, immune complexes and immunogenicity. MAbs. 2011;3(6):503–4. doi:10.4161/mabs.3.6.17611. PMID:22123066
- Roberts CJ. Protein aggregation and its impact on product quality. Curr Opin Biotechnol. 2014;30: 211–7. doi:10.1016/j.copbio.2014.08.001. PMID:25173826
- Hmiel LK, Brorson KA, Boyne MT 2nd. Post-translational structural modifications of immunoglobulin G and their effect on biological activity. Anal Bioanal Chem. 2015;407(1):79–94. doi:10.1007/s00216-014-8108-x. PMID:25200070
- Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opinion on Drug Delivery. 2007;4 (4):427–440. doi:10.1517/17425247.4.4.427. PMID:17683255
- Goswami S, Wang W, Arakawa T and Ohtake S. Developments and Challenges for mAb-Based Therapeutics. Antibodies. 2013;2 (3):452–500. doi:10.3390/antib2030452.
- World Health Organization Report: Adherence to long-term therapies: evidence for action. 2003 [last accessed 2017 July 27]. www.who.int/chp/knowledge/publications/adherence_report/en/
- Badkar A, Wolf A, Bohack L, Kolhe P. Development of biotechnology products in pre-filled syringes: technical considerations and approaches. AAPS Pharm Sci Tech. 2011;12 (2):564–72. doi:10.1208/s12249-011-9617-y.
- Majumdar S, Ford BM, Mar KD, Sullivan VJ, Ulrich RG, D'souza AJ. Evaluation of the effect of syringe surfaces on protein formulations. J Pharm Sci. 2011;100(7):2563–73. doi:10.1002/jps.22515. PMID:21319164
- Bai S, Landsman P, Spencer A, DeCollibus D, Vega F, Temel DB, Houde D, Henderson O, Brader ML. Evaluation of incremental siliconization levels on soluble aggregates, submicron and subvisible particles in a prefilled syringe product. J Pharm Sci. 2016;105(1):50–63. doi:10.1016/j.xphs.2015.10.012. PMID:26852839
- Jezek J, Darton NJ, Derham BK, Royle N, Simpson I. Biopharmaceutical formulations for pre-filled delivery devices. Expert Opin Drug Deliv. 2013;10(6):811–28. doi:10.1517/17425247.2013.780023. PMID:23506207
- Jenke DR. Extractables and leachables considerations for prefilled syringes. Expert Opin Drug Deliv. 2014;11(10):1591–600. doi:10.1517/17425247.2014.928281. PMID:24955780
- Schellekens H. Biosimilar therapeutics—what do we need to consider? NDT Plus. 2009;2(suppl 1):i27–i36. doi:10.1093/ndtplus/sfn177. PMID:19461855
- Lee JF, Litten JB, Grampp G. Comparability and biosimilarity: considerations for the healthcare provider. Curr Med Res Opin. 2012;28(6):1053–1058. doi:10.1185/03007995.2012.686902. PMID:22519391
- Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–419. doi:10.1093/annonc/mdm345. PMID:17872902
- Choy E, Jacobs IA. Biosimilar safety considerations in clinical practice. Semin Oncol. 2014;41 Suppl 1:S3–14. doi:10.1053/j.seminoncol.2013.12.001. PMID:24560025
- Webber JTK. Demonstrating comparability for well-characterized biotechnology products. BioProcess Int 2008:32–43 [last accessed 2017 Oct 06]. http://www.bioprocessintl.com/analytical/product-characterization/demonstrating-comparability-for-well-characterized-biotechnology-products-182256/
- Lubiniecki AS, Marcia Federici M. Comparability is not just analytical equivalence. Biologicals. 2006;34(1):45–7. doi:10.1016/j.biologicals.2005.08.016. PMID:16330221
- Federici M1, Lubiniecki A, Manikwar P, Volkin DB. Analytical lessons learned from selected therapeutic protein drug comparability studies. Biologicals. 2013 May;41(3):131–47. doi:10.1016/j.biologicals.2012.10.001.
- European Medicines Agency. Guideline on Comparability of Biotechnology-Derived Medicinal Products after a Change in the Manufacturing Process: Non-Clinical and Clinical Issues. November 2007. EMEA/CHMP/BMWP /101695/2006. [last accessed 2017 Oct 06] http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003935.pdf
- Food and Drug Administration. Draft Guidance: Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development. February 2016 [last accessed 2017 July 07]. https://www.fda.gov/downloads/regulatoryinformation/guidances/ucm484345.pdf
- Kivitz A1, Cohen S, Dowd JE, Edwards W, Thakker S, Wellborne FR, Renz CL, Segurado OG. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28(10):1619–29. doi:10.1016/j.clinthera.2006.10.006. PMID:17157117
- Phillips JT, Fox E, Grainger W, Tuccillo D, Liu S, Deykin A. An open-label, multicenter study to evaluate the safe and effective use of the single-use autoinjector with an Avonex® prefilled syringe in multiple sclerosis subjects. BMC Neurol. 2011;11:126. doi:10.1186/1471-2377-11-126. PMID:21999176
- Wray S, Armstrong R, Herrman C, Calkwood J, Cascione M, Watsky E, Hayward B, Mercer B, Dangond F. Results from the single-use autoinjector for self-administration of subcutaneous interferon beta-1a in patients with relapsing multiple sclerosis (MOSAIC) study. Expert Opin Drug Deliv. 2011;8(12):1543–53. doi:10.1517/17425247.2011.628656. PMID:22032264
- Paul C, Stalder JF, Thaçi D, Vincendon P, Brault Y, Kielar D, Tebbs V. Patient satisfaction with injection devices: a randomized controlled study comparing two different etanercept delivery systems in moderate to severe psoriasis. J Eur Acad Dermatol Venereol. 2012;26(4):448–55. doi:10.1111/j.1468-3083.2011.04093.x. PMID:21557778
- Xie L, Zhou S, Wei W, et al. Does pen help? A real-world outcomes study of switching from vial to disposable pen among insulin glargine-treated patients with type 2 diabetes mellitus. Diabetes Technol Ther. 2013;15:230–236. doi:10.1089/dia.2012.0253. doi:10.1089/dia.2012.0253. PMID:23336845
- Pivot X, Gligorov J, Müller V, Barrett-Lee P, Verma S, Knoop A, Curigliano G, Semiglazov V, López-Vivanco G, Jenkins V, et al. Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2-positive early breast cancer (PrefHer): an open-label randomised study. Lancet Oncol. 2013;14(10):962–70. doi:10.1016/S1470-2045(13)70383-8. PMID:23965225
- Freundlich B, Kivitz A, Jaffe JS. Nearly pain-free self-administration of subcutaneous methotrexate with an autoinjector: results of a phase 2 clinical trial in patients with rheumatoid arthritis who have functional limitations. J Clin Rheumatol. 2014;20(5):256–60. doi:10.1097/RHU.0000000000000117. PMID:25036562
- Seddighzadeh A, Hung S, Selmaj K, Cui Y, Liu S, Sperling B, Calabresi PA. Single-use autoinjector for peginterferon-β1a treatment of relapsing-remitting multiple sclerosis: safety, tolerability and patient evaluation data from the Phase IIIb ATTAIN study. Expert Opin Drug Deliv. 2014;11(11):1713–20. doi:10.1517/17425247.2014.944159. PMID:25073663
- Hupperts R, Becker V, Friedrich J, Gobbi C, Salgado AV, Sperling B, You X. Multiple sclerosis patients treated with intramuscular IFN-β-1a autoinjector in a real-world setting: prospective evaluation of treatment persistence, adherence, quality of life and satisfaction. Expert Opin Drug Deliv. 2015;12(1):15–25. doi:10.1517/17425247.2015.989209. PMID:25430947
- Paul C, Lacour JP, Tedremets L, Kreutzer K, Jazayeri S, Adams S, Guindon C, You R, Papavassilis C, JUNCTURE study group. Efficacy, safety and usability of secukinumab administration by autoinjector/pen in psoriasis: a randomized, controlled trial (JUNCTURE). J Eur Acad Dermatol Venereol. 2015;29(6):1082–90. doi:10.1111/jdv.12751. PMID:25243910
- Blauvelt A, Prinz JC, Gottlieb AB, Kingo K, Sofen H, Ruer-Mulard M, Singh V, Pathan R, Papavassilis C, Cooper S, FEATURE Study Group. Secukinumab administration by pre-filled syringe: efficacy, safety and usability results from a randomized controlled trial in psoriasis (FEATURE). Br J Dermatol. 2015;172(2):484–93. doi:10.1111/bjd.13348. PMID:25132411
- Pivot X, Spano JP, Espie M, Cottu P, Jouannaud C, Pottier V, Moreau L, Extra JM, Lortholary A, Rivera P, et al. Patients' preference of trastuzumab administration (subcutaneous versus intravenous) in HER2-positive metastatic breast cancer: Results of the randomised MetaspHer study. Eur J Cancer. 2017;82:230–236. doi:10.1016/j.ejca.2017.05.009. PMID:28648618
- Borrás-Blasco J, Gracia-Pérez A, Rosique-Robles JD, Casterá MD, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10(3):301–7. doi:10.1517/14712590903530633. PMID:20059372
- Roth EM, Bujas-Bobanovic M, Louie MJ, Cariou B. Patient and physician perspectives on mode of administration of the PCSK9 monoclonal antibody alirocumab, an injectable medication to lower LDL-C levels. Clin Ther. 2015;37(9):1945–1954.e6. doi:10.1016/j.clinthera.2015.07.008. PMID:26278513
- Korytkowski M1, Bell D, Jacobsen C, Suwannasari R, FlexPen Study Team. A multicenter, randomized, open-label, comparative, two-period crossover trial of preference, efficacy, and safety profiles of a prefilled, disposable pen and conventional vial/syringe for insulin injection in patients with type 1 or 2 diabetes mellitus. Clin Ther. 2003;25(11):2836–48. doi:10.1016/S0149-2918(03)80337-5. PMID:14693308
- Stockl K1, Ory C, Vanderplas A, Nicklasson L, Lyness W, Cobden D, Chang E. An evaluation of patient preference for an alternative insulin delivery system compared to standard vial and syringe. Curr Med Res Opin. 2007;23(1):133–46. doi:10.1185/030079906X159524. PMID:17257475
- Sheikhzadeh A, Yoon J, Formosa D, Domanska B, Morgan D, Schiff M. The effect of a new syringe design on the ability of rheumatoid arthritis patients to inject a biological medication. Appl Ergon. 2012;43(2):368–75. doi:10.1016/j.apergo.2011.05.014. PMID:21696704
- Schiff M, Jaffe J, Freundlich B, Madsen P. New autoinjector technology for the delivery of subcutaneous methotrexate in the treatment of rheumatoid arthritis. Expert Rev Med Devices. 2014;11(5):447–55. doi:10.1586/17434440.2014.929492. PMID:24934630
- Deleu D, Alsharoqi I, Al Jumah MA, Al Tahan AR, Bohlega S, Dahdaleh M, Inshasi J, Khalifa A, Szólics M, Yamout BI. Will new injection devices for interferon β-1a s.c. affect treatment adherence in patients with multiple sclerosis? An expert opinion in the Middle East. Int J Neurosci. 2011;121(4):171–5. doi:10.3109/00207454.2010.541572. PMID:21329472
- Devonshire VA, Verdun di Cantogno E. Review of subcutaneous interferon β-1a, delivered via the electronic self-injection device RebiSmart™, for the treatment of multiple sclerosis. Ther Deliv. 2011;2(11):1455–65. doi:10.4155/tde.11.116. PMID:22844681
- Kesson CM, Bailie GR. Do diabetic patients inject accurate doses of insulin? Diabetes Care. 1981;4:333. doi:10.2337/diacare.4.2.333a. PMID:7011752
- Puxty JA, Hunter DH, Burr WA. Accuracy of insulin injection in elderly patients. BMJ (Clin Res Ed). 1983;287:1762. doi:10.1136/bmj.287.6407.1762.
- Food and Drug Administration. 21 CFR Part 4 [Docket No. FDA–2009–N–0435]. Current Good Manufacturing Practice Requirements for Combination Products; January 2013 [last accessed 2017 Oct 06]. https://www.gpo.gov/fdsys/pkg/FR-2013-01-22/pdf/2013-01068.pdf
- Food and Drug Administration. Final Guidance: Current Good Manufacturing Practice Requirements for Combination Products; January 2017 [last accessed 2017 Oct 06]. https://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM429304.pdf
- European Medicines Agency. Concept Paper on Developing a Guideline on Quality Requirements of Medicinal Products Containing a Device Component for Delivery or Use of the Medicinal Product; November 2016 [last accessed 2017 Oct 06]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/02/WC500221747.pdf
- Food and Drug Administration. Draft Guidance: Demonstration of comparability of human biological products, including therapeutic biotechnology-derived products; April 1996 [last accessed 2017 Oct 06]. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human use. ICH Harmonised Tripartite Guideline Q5E. International Conference on Harmonization/ICH Expert Working Group. Comparability of biotechnological/biological products subject to changes in their manufacturing process; November 2004 [last accessed 2017 Oct 06]. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf
- Food and Drug Administration. Guidance: Q5E Comparability of biotechnological/biological products subject to changes in their manufacturing process; June 2005 [last accessed 2017 Oct 06]. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM073476.pdf
- Food and Drug Administration. Draft Guidance: Rheumatoid Arthritis: Developing Drug Products for Treatment; May 2013 [last accessed 2017 Oct 06]. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm354468.pdf