
ABSTRACT
The ESCRT machinery plays a pivotal role in membrane-remodeling events across multiple cellular processes including nuclear envelope repair and reformation, nuclear pore complex surveillance, endolysosomal trafficking, and neuronal pruning. Alterations in ESCRT-III functionality have been associated with neurodegenerative diseases including Frontotemporal Dementia (FTD), Amyotrophic Lateral Sclerosis (ALS), and Alzheimer’s Disease (AD). In addition, mutations in specific ESCRT-III proteins have been identified in FTD/ALS. Thus, understanding how disruptions in the fundamental functions of this pathway and its individual protein components in the human central nervous system (CNS) may offer valuable insights into mechanisms underlying neurodegenerative disease pathogenesis and identification of potential therapeutic targets. In this review, we discuss ESCRT components, dynamics, and functions, with a focus on the ESCRT-III pathway. In addition, we explore the implications of altered ESCRT-III function for neurodegeneration with a primary emphasis on nuclear surveillance and endolysosomal trafficking within the CNS.
Introduction
The endosomal sorting complex required for transport (ESCRT) is a multi-functional membrane-remodeling complex present in archaea and eukaryotes [Citation1–9]. The ESCRT pathway plays a pivotal role in multiple cellular processes including nuclear envelope repair, nuclear pore complex (NPC) quality control, plasma membrane repair, lysosome repair, intraluminal vesicle (ILV) and multivesicular body (MVB) formation, endolysosomal trafficking, neuronal pruning, and exosome biogenesis and release () among others as has been recently described [Citation4,Citation5,Citation10–30]. Notably, alterations in cell biological processes linked to ESCRT function, specifically nuclear surveillance and endolysosomal trafficking/autophagy, have been highlighted through studies in various model systems of neurodegenerative diseases including Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD) [Citation31–39]. Collectively, this work implicates ESCRT dysfunction as a significant contributor to ALS and FTD pathogenesis.
Figure 1. Overview of a subset ESCRT-III mediated cellular processes. A subset of known cellular processes that involved ESCRT-III membrane remodeling functions depicted in neurons.
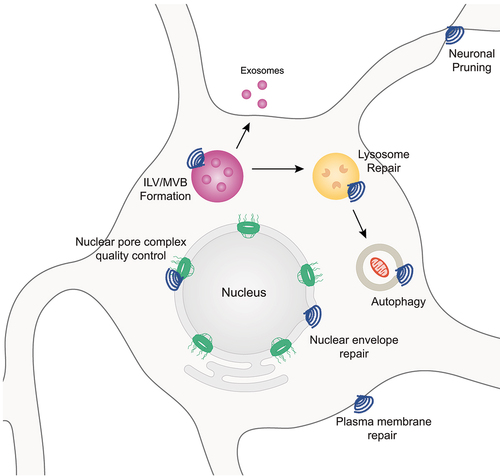
Neurodegenerative diseases such as ALS and FTD are characterized by the selective death and dysfunction of neuronal and glial cell populations within distinct CNS regions. Specifically, neurons and glial cells within the motor circuitry of the motor cortex and spinal cord affected in ALS and neuronal and glial cells within the frontal and temporal cortices impacted in FTD. Given the cell type and CNS region specificity of these neurodegenerative diseases, ALS leads to a deterioration of motor function and FTD impacts decision-making, personality, behavior, and language [Citation40–49]. Although largely considered distinct neurodegenerative diseases, ALS and FTD share a number of genetic and pathological underpinnings. For example, mutations in C9orf72 and CHMP2B are two examples of genetic mutations causative of both ALS and FTD [Citation50–56]. Interestingly, amongst patients with the C9orf72 mutation, a subset (~10–15%) are diagnosed with both ALS and FTD [Citation44,Citation57–64]. Pathologically, nuclear clearing and associated nuclear loss of function as well as subsequent cytoplasmic mislocalization and/or aggregation of the RNA binding protein TDP-43 has been observed in the majority of ALS cases and ~50% of FTD cases [Citation65–77]. Collectively, studies in various model systems of ALS and FTD have documented impairments in multiple cellular processes including protein degradation, nucleocytoplasmic transport, and RNA processing [Citation39,Citation57,Citation73,Citation78–89]. Intriguingly, the ESCRT-III pathway and its protein constituents play a pivotal role in both the direct and indirect regulation, coordination, and execution of cellular processes disrupted in ALS and FTD, highlighting a role for the ESCRT-III pathway in disease pathogenesis.
In this review, we will discuss the function of the ESCRT-III pathway in nuclear membrane sealing and repair, nuclear pore complex surveillance, and endolysosomal trafficking and degradation, given the documented importance and relevance of these cell biological processes to neurodegeneration, namely ALS and FTD. As the fundamental biology and dynamics of the ESCRT-III machinery has been recently and thoroughly reviewed elsewhere [Citation4,Citation5,Citation8,Citation21], the central focus of our review will be on the implications of disruptions in specific ESCRT-III functions for ALS and FTD pathogenesis. Where appropriate, we will discuss studies directly implicating ESCRT-III dysfunction in models of ALS and FTD. This review will emphasize the fundamental and pathologic involvement of the ESCRT-III nuclear surveillance pathway in the central nervous system (CNS) given that a number of recent studies have documented nuclear pore complex and nuclear envelope alterations in neurodegenerative diseases [Citation90–97]. Lastly, we will discuss the role of mutations in the ESCRT-III protein CHMP2B in FTD and ALS.
Overview of ESCRT proteins and dynamics
The ESCRT machinery is comprised of five core subcomplexes: ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III, and VPS4. These ESCRT complexes can physically interact but have defined functional roles within the cell. In total, over 30 individual proteins make up the mammalian (over 20 in yeast) ESCRT machinery [Citation2–8,Citation98–101]. Briefly, the mammalian ESCRT-0 complex is comprised of two proteins HRS and STAM and plays an essential role in multivesicular body (MVB) formation by binding multiple ubiquitin molecules and localizing ubiquitinated proteins to the endosome [Citation13,Citation102–104]. VPS23/TSG101, VPS28, VPS37A, VPS37B, VPS37C, VPS37D, MVBB, and UBAP1 make up the mammalian ESCRT-I complex and collectively function as a bridge between ESCRT-0 and ESCRT-II, handing off ubiquitinated proteins from ESCRT-0 to ESCRT-II. The ESCRT-II complex, made of VPS22, VPS25, and VPS36 in mammals, is essential for delivering ubiquitinated proteins to endosomes [Citation2,Citation4,Citation8,Citation105].
The ESCRT-III complex is perhaps the most versatile and critical of the ESCRT machinery as it functions in all ESCRT processes (). It is comprised of ~12 proteins in mammals including CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4A, CHMP4B, CHMP4C, CHMP5, CHMP6, CHMP7, and IST1/CHMP8. Unlike other ESCRT complexes, the ESCRT-III machinery is only transiently activated or assembled. Assembly begins when early acting factors such as ESCRT-I/-II, the ESCRT associated protein ALIX, or the ESCRT-II/-III hybrid protein CHMP7 bind adapter proteins, ubiquitinated cargo, and/or membranes to cluster cargo and vesicles and initiate membrane bending. In turn, this facilitates ESCRT-III subunit assembly and filamentation along bound membranes [Citation4,Citation7,Citation98,Citation106]. When ESCRT-III assembly is not required, aberrant polymerization of ESCRT-III filaments is prevented by autoinhibition via ‘closed’ conformations of ESCRT-III protein monomers [Citation107–111]. The closed-protein conformation is dependent upon the C terminus as removal of the C terminal region converts ESCRT-III proteins into an ‘open’ conformation and enables subsequent polymerization [Citation112]. This conformational change is essential for ECSRT-III protein assembly at target membranes and in turn ESCRT-III protein function. Under basal cellular conditions, the conversion from a ‘closed’ to ‘open’ state to remove autoinhibition is facilitated by protein – protein interactions. This activation can either occur via interactions between individual ESCRT-III subunits or resident proteins within target membranes [Citation98,Citation99,Citation113–117].
ESCRT-III proteins are recruited in a sequential manner and assemble into multi-protein polymers that undergo subunit exchange to facilitate membrane remodeling [Citation118–124]. Recent evidence suggests that membrane remodeling can occur in both an inside-out (toward the cytoplasm) and outside-in (away from the cytoplasm) manner. The binding of ESCRT proteins to membranes stabilizes membrane curvature thereby allowing the formation of membrane necks. Membrane scission for repair and remodeling is then catalyzed by recruitment and function of the AAA – ATPase VPS4 [Citation4–6,Citation20,Citation98,Citation105,Citation109,Citation120,Citation122,Citation123,Citation125–127]. In addition, VPS4 facilitates ESCRT-III polymer disassembly [Citation118,Citation121,Citation122,Citation128–133]. VPS4 recruitment is facilitated by direct interactions with a microtubule interacting and transport domain interacting motif (MIM) sequence in the C terminus of most ESCRT-III proteins [Citation98,Citation133,Citation134]. However, as is the case for CHMP7 due to an amino acid charge substitution, not all ESCRT-III proteins are capable of recruiting VPS4 through this MIM domain sequence [Citation135]. Once recruited, VPS4 catalyzes ATP hydrolysis to exchange ESCRT-III protein subunits from polymer filaments, initiating their ‘deactivation’ and recycling [Citation118,Citation121,Citation122,Citation128–133]. For a more in depth and comprehensive review of ESCRT-III protein domain structure and interactions, we refer readers to a recent publication by McCullough and colleagues [Citation4]. However, it is important to note, especially in considering mechanisms underlying ESCRT-III dysfunction in neurodegenerative disease, that the sequence of recruitment events and requirement of individual ESCRT-III proteins for function is not known for every membrane throughout the cell. Additionally, it is unknown whether the sequence of events and protein requirements is conserved across organisms and cell types. In fact, recent work has established fundamental differences in the requirement of specific nuclear envelope proteins required for CHMP7/ESCRT-III function in nuclear surveillance in human neurons compared to yeast and non-neuronal immortalized mammalian cell lines [Citation90,Citation93,Citation114–117].
Nuclear membrane sealing and repair
Maintenance of nuclear envelope integrity is essential for maintaining cellular compartmentalization and separation of genetic material from the cytosol of eukaryotic cells. Impaired nuclear envelope integrity can result in the uncontrolled exchange of cellular materials, DNA damage, and ultimately impaired cellular function [Citation136]. Pathologic alterations to nuclear envelope integrity or mutations in resident nuclear envelope proteins have been linked to multiple diseases including tauopathies, laminopathies, ataxia, and muscular dystrophy [Citation96,Citation137–143]. In addition, during cell migration or in the case of cancer cell metastasis, cells navigate through confined spaces leading to nuclear constriction and rupture [Citation22]. Thus, mechanical stress to the nucleus or via forces exerted through the Linker of the Nucleoskeleton and Cytoskeleton (LINC) complex via actin, microtubules, and intermediate filaments within the cytoplasm can compromise nuclear integrity [Citation144–148].
In order to prevent catastrophic loss of nuclear integrity and uncontrolled nuclear ruptures, the ESCRT-III pathway safeguards the nuclear periphery by sealing holes within nuclear membranes in coordination with resident nuclear envelope proteins [Citation22,Citation25,Citation26,Citation29,Citation119,Citation149–151] (). The ESCRT-II/ESCRT-III hybrid protein CHMP7 plays a fundamental role in initiating ESCRT-III function in nuclear envelope sealing (). CHMP7 passively diffuses into the nucleus where it is then appropriately positioned to interact with the inner nuclear membrane protein LEM2/LEMD2 [Citation30,Citation114–117,Citation152]. In yeast, direct binding to membrane lipids is required for CHMP7’s nuclear localization and interaction with LEMD2 [Citation153]. Once localized to nuclear ruptures, LEMD2 phase separation promotes ‘activation’ of CHMP7 and recruitment of ESCRT-III factors to facilitate nuclear envelope sealing [Citation116]. However, the requirement and contribution of additional ESCRT-III subunits and the cell-type specific requirements of ESCRT-III proteins in nuclear envelope resealing remains unknown. As much research to date has focused on the role of the CHMP7 in nuclear envelope sealing during ‘regulated’ formation of membrane holes during cell division, this is particularly important when considering the role of the ESCRT-III pathway in maintaining nuclear integrity in non-dividing cells such as neurons.
Figure 2. ESCRT-III functions at the nuclear envelope and NPC. (a) Nuclear envelope sealing: upon nuclear envelope rupture, barrier to autoregulation factor (BAF) is recruited to ‘plug’ larger holes prior to sealing. For smaller ruptures, CHMP7 localizes to the nuclear periphery where it is exposed to the inner nuclear membrane protein LEMD2. Protein – protein interactions between CHMP7 and LEMD2 facilitate activation of CHMP7 and subsequent recruitment and polymerization of ESCRT-III subunits to seal nuclear envelope holes. VPS4 recruitment mediates ESCRT-III polymer disassembly and subunit reuse. (b) NPC surveillance and quality control: upon NPC misassembly within the nuclear envelope, CHMP7 localizes to the nuclear periphery where it is positioned to interact with LEMD2. CHMP7 is activated through physical association with LEMD2. ESCRT-III subunits are recruited to generate and stabilize membrane buds. Polymerization of ESCRT-III subunits and recruitment of VPS4 facilitates scission of membrane buds containing NPCs for degradation. VPS4 mediates ESCRT-III polymer disassembly and subunit exchange for reuse.
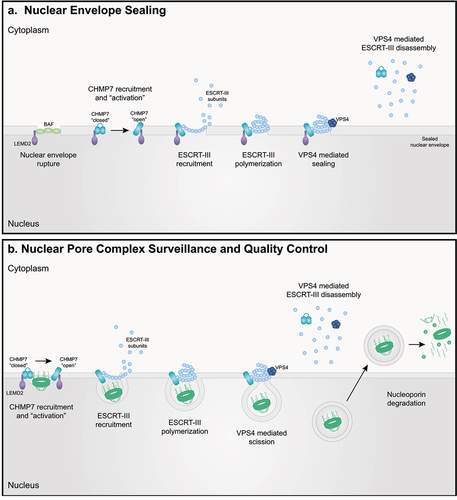
Substantial evidence suggests that CHMP7/LEMD2 are involved in sealing small (e.g. <100 nm) holes within the nuclear envelope [Citation30,Citation114–117,Citation152,Citation154]. However, repair of larger ruptures requires barrier to autoregulation factor (BAF/BANF1). Upon nuclear rupture, BAF rapidly translocates and binds to DNA, thereby accumulating at the rupture site and effectively ‘plugging’ the nuclear membrane hole. BAF then recruits LEM domain proteins to sites of nuclear rupture to facilitate repair [Citation149]. Interestingly, a recent study suggests that assembly of nuclear envelopes is dependent on CHMP7 in the absence of BAF mediated hole closure [Citation150]. This is consistent with a report demonstrating the formation of ‘grommets’ comprised of ESCRT-III proteins in order to maintain nucleocytoplasmic compartmentalization prior to the sealing of larger nuclear envelope holes during spindle body extrusion during mitosis in yeast [Citation119].
Uncontrolled ESCRT-III function and regulation itself can compromise nuclear integrity and initiate DNA damage [Citation154] highlighting an essential role and delicate balance for ESCRT-III function in facilitating nuclear envelope repair and maintaining nuclear integrity. In addition to the characteristic autoinhibitory conformations of ESCRT-III proteins [Citation107–112], regulatory factors such as CC2D1B and CDK1 prevent unnecessary CHMP7 – LEMD2 interactions and ESCRT-III function at the nuclear envelope [Citation135,Citation155]. Specifically, CC2D1B can bind to CHMP7 and impact the timing of ESCRT-III subunit recruitment and the quality of ESCRT-III polymers during nuclear envelope sealing in cell division [Citation155]. On the other hand, CDK1 can phosphorylate CHMP7 thereby inhibiting interactions with LEMD2 and preventing ESCRT-III polymer assembly during the M phase and exit of cell division [Citation135]. The interaction between CHMP7 and LEMD2 can additionally be influenced by membrane binding, lipid synthesis, and alternative splicing of LEMD2 [Citation152,Citation153,Citation156].
Nuclear membrane invaginations containing NPCs, mRNA, and other proteins have been documented in model systems of FTD and postmortem human FTD tissues [Citation92,Citation95,Citation96,Citation157–159] and Profilin 1 (PFN) centric models of ALS [Citation160]. In FTD, these invaginations appear to localize to areas containing perinuclear aggregates of the microtubule associated protein Tau (MAPT) and reduction of Tau aggregation/accumulation diminishes nuclear abnormalities [Citation95]. While the mechanisms underlying Tau or PFN1 mediated nuclear invagination remain unknown, it is plausible that destabilization of the microtubule or actin cytoskeleton respectively either due to mutations in cytoskeletal proteins or mechanical stress induced by protein aggregation, compromise the integrity and stability of the of the LINC complex leading to nuclear envelope disruption. While it is unclear whether these invaginations contain nuclear perforations, impaired nucleocytoplasmic compartmentalization can also be observed [Citation95,Citation158]. Interestingly, a recent study demonstrated that reduction of BAF promotes the pathologic accumulation of Tau and overexpression of LEMD2 or CHMP7 exerts protection against tau aggregation [Citation159]. However, the impact of BAF, LEMD2, and CHMP7 manipulation on nuclear invaginations and whether their impact on Tau aggregation is mechanistically linked to nuclear envelope integrity directly remains unknown at this time. Nonetheless, collectively, these studies suggest that impaired or altered function of CHMP7 and the ESCRT-III pathway in maintenance of nuclear envelope integrity may contribute to FTD pathogenesis.
Nuclear pore complex quality control
In addition to its role in maintaining nuclear envelope integrity, CHMP7 and the ESCRT-III pathway plays a crucial role in maintaining NPC homeostasis and managing NPC quality control in yeast and human cells [Citation30,Citation93,Citation161–163] (). NPCs are comprised of multiple copies of ~30 individual nucleoporin proteins totaling >1000 individual protein molecules per NPC in mammals. These large multi-protein structures function to regulate and coordinate essential cellular processes including nucleocytoplasmic transport and genome organization [Citation164–177]. NPCs assemble in a coordinated manner dependent on the sequential recruitment and insertion of nucleoporin proteins and nuclear pore subcomplexes into the nuclear envelope and partially assembled NPC [Citation169,Citation178–188]. The proper assembly and maintenance of NPC integrity over the lifetime of a cell is essential for continued function and ultimately cellular health.
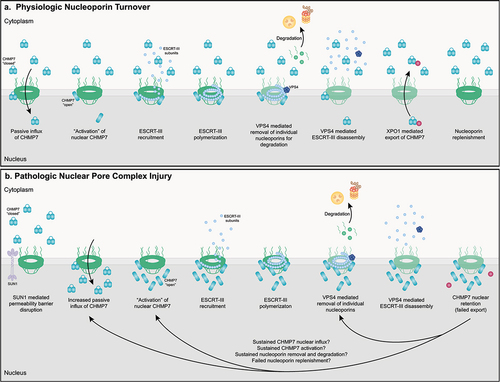
Recent studies support a role for CHMP7 and the ESCRT-III pathway in ensuring the proper assembly and insertion of NPCs in yeast [Citation30,Citation117]. During NPC surveillance, NPC intermediates, but not mature and properly assembled NPCs, are bound by the LEM family of inner nuclear membrane proteins. This positions them in close proximity to ESCRT-III subunits which are recruited to sites of misassembled or improperly inserted NPCs by LEM family inner nuclear membrane proteins. NPC intermediates are then cleared in a VPS4-dependent manner. Reduction of ESCRT-III subunits compromises NPC surveillance leading to the accumulation of NPC intermediates within storage of improperly assembled nuclear pore complexes (SINC) compartments which are retained in mother cells to safeguard nucleocytoplasmic compartmentalization in daughter cells [Citation30]. Interestingly, these SINCs resemble perinuclear vesicles generated during the nuclear egress of viruses and megaRNP particles [Citation189–191], the process of which has also been linked to ESCRT-III pathway function [Citation2–4,Citation8,Citation12]. Although the precise mechanism by which ESCRT-III clears aberrant NPC intermediates is not fully understood, NPC and nucleoporin degradation have been linked to both the lysosomal (autophagy) and proteasomal degradation pathways [Citation30,Citation115,Citation117,Citation192,Citation193]. ESCRT-III subunits and/or VPS4 may facilitate budding of NPC intermediates and/or directly facilitate stabilization of nuclear envelope invaginations during NPC assembly. However, ESCRT-III surveillance of NPC insertion and assembly was subsequently shown to be coupled to nuclear envelope sealing and linked to the function of the yeast orthologue of CHMP7, Chm7 [Citation117]. Although the mechanisms that lead to CHMP7 influx and ESCRT-III activation upon NPC misassembly remain unknown, one might speculate that improperly assembled NPCs compromise nucleocytoplasmic compartmentalization thereby culminating in CHMP7 nuclear influx and facilitating interactions with LEMD2. Indeed, a recent study has demonstrated that impaired NPC permeability barrier integrity promotes excessive nuclear influx of CHMP7 in ALS neurons [Citation90].
While ESCRT-III surveillance of NPC assembly and insertion has been described in yeast, it is unclear whether analogous mechanisms take place in dividing mammalian cells. Nonetheless, two studies have implicated the ESCRT-III pathway in the maintenance of NPCs throughout the lifetime of non-dividing mammalian cells [Citation93,Citation161,Citation163]. A subset of nucleoporins represent some of the longest lived proteins in the mammalian CNS [Citation194]. In order to sustain nucleocytoplasmic compartmentalization and cellular health, NPCs must be maintained throughout the lifetime of cells, in particular when considering infrequent protein turnover. In non-dividing muscle myoblasts, knockdown of CHMP2A and CHMP3 significantly increased the retention of ‘old’ Nup93 molecules within NPCs [Citation163]. In addition, increased nuclear localization of CHMP7 appears to lead to the abnormal reduction of specific nucleoporins from the NPC in induced pluripotent stem cell (iPSC) derived neurons (iPSNs) [Citation93]. Collectively, these studies implicate at least a subset of ESCRT-III proteins in the piecemeal turnover and degradation of individual nucleoporin proteins from existing NPCs. Although the molecular mechanisms that facilitate and regulate ESCRT-III mediated nucleoporin turnover in fully assembled NPCs remain largely unknown, this currently a topic of active investigation given the potential implications for age-related neurodegenerative diseases where ensuring properly executed nucleoporin molecule turnover is thought to be essential for maintaining NPC functionality.
As has been recently reviewed, nucleoporin and nucleocytoplasmic transport alterations are prevalent in neurodegenerative diseases such as ALS and FTD [Citation195–198]. Multiple studies have now documented diminished nuclear localization or expression of specific nucleoporins in sporadic and genetic forms of ALS and FTD [Citation91,Citation93–95,Citation160]. Recent work has established a role for CHMP7 and the ESCRT-III nuclear surveillance pathway in the pathologic initiation of NPC injury [Citation90,Citation93,Citation161,Citation199] (). Using an iPSN model of sporadic ALS (sALS) and C9orf72 ALS/FTD, we demonstrated that abnormal and excessive nuclear localization of CHMP7 precedes the pathologic reduction of specific nucleoporins from ALS nuclei and NPCs [Citation93,Citation94]. Importantly, in iPSNs, CHMP7 relocalization is not initiated by altered NPC composition resulting from nucleoporin reduction [Citation93]. However, it is currently unknown whether protein coding variants in nucleoporins, such as those recently identified in Nup50 [Citation200], can subtly alter the structure or function of the NPC and contribute to nuclear localization and/or function of CHMP7 and ESCRT-III nuclear surveillance. Interestingly, our group recently established that alterations in NPC permeability barrier integrity are dependent on the LINC complex protein SUN1 and facilitate increased nuclear influx of CHMP7, thereby increasing its nuclear localization in sALS iPSNs [Citation90]. Although the potential contribution of impaired nuclear export of CHMP7 to increased nuclear localization remains unknown, this study suggests that subtle alterations to NPC or nuclear envelope structure and integrity may compromise the integrity of the passive diffusion permeability barrier of the NPC central channel in the pathologic disruption of CHMP7/ESCRT-III nuclear surveillance. Notably, these studies suggest that the mechanisms underlying ESCRT-III surveillance of NPC assembly during cell division and maintenance throughout the lifetime of a cell may at least be in part distinct. In support of this, two publications now provide evidence that LEMD2 does not facilitate CHMP7/ESCRT-III NPC maintenance in iPSNs [Citation90,Citation93]. In contrast, as is typical for overall ESCRT functionality in multiple cellular processes [Citation2–4,Citation8,Citation105,Citation109,Citation201], pathological nucleoporin reduction requires the recruitment of VPS4 in a manner dependent on CHMP7 in iPSNs [Citation161]. A subsequent study confirmed these findings by demonstrating that increased expression of VPS4 leads to excessive degradation of nucleoporins in a Drosophila model of C9orf72 ALS/FTD [Citation199]. Interestingly, expression of a dominant negative VPS4 led to the intranuclear accumulation of POM121 following its reduction from NPCs in iPSNs [Citation161] suggesting that the ESCRT-III pathway may facilitate the nuclear internalization/budding of nucleoporins for physiologic and/or pathologic degradation. Together, these studies suggest that NPC injury observed in ALS and FTD may at least be in part due to pathologic overactivation of the ESCRT-III nuclear surveillance pathway. Future and current investigations into the molecular mechanisms underlying ESCRT-III mediated disruptions in NPC homeostasis is of high priority for our group and is essential for our understanding of pathomechanisms of neurodegenerative disease.
Figure 3. ESCRT-III mediated physiologic maintenance and pathologic disruption of NPCs in human neurons. (a) In normal human neurons, turnover of individual nucleoporin proteins to maintain NPC integrity and function is initiated by passive diffusion of CHMP7 to the nuclear space. CHMP7 is activated via currently undefined but LEMD2 independent protein – protein interactions. Activation of CHMP7 likely facilitates the recruitment and polymerization of ESCRT-III subunits. Individual nucleoporin proteins are removed from the NPC and degraded in a VPS4 dependent manner. VPS4 likely facilitates ESCRT-III polymer disassembly and subunit exchange and reuse. Exportin-1 (XPO1) actively exports CHMP7 from the nucleus, resulting in its ‘inactivation’ and maintaining low nuclear levels of CHMP7 under basal cellular conditions. Nucleoporins are synthesized and reinserted into the NPC. (b) In ALS neurons, the integrity of the passive diffusion permeability barrier is disrupted in a SUN1 dependent manner leading to a pathologic increase in the nuclear influx of CHMP7. CHMP7 is activated via currently undefined by LEMD2 independent protein – protein interactions. Activation of CHMP7 likely facilitates the recruitment and polymerization of ESCRT-III subunits. Individual nucleoporin proteins are removed from the NPC and degraded in a VPS4 dependent manner. VPS4 likely facilitates ESCRT-III polymer disassembly and subunit exchange and reuse. We hypothesize impaired interactions between CHMP7 and XPO1 or other Exportins abrogate its active nuclear export and facilitate nuclear accumulation of CHMP7 observed in ALS/FTD. In addition, we hypothesize that pathologic removal and degradation of nucleoporins is sustained via 1. sustained excessive nuclear influx of CHMP7 and/or 2. sustained LEMD2 independent activation of CHMP7 (e/g/persistent nuclear protein – protein interactions and/or 3. sustained VPS4 mediated nucleoporin removal from NPCs and/or 4. impaired nucleoporin reincorporation perhaps as a result of altered nuclear transport receptor and active nuclear import function. Collectively, sustained ‘overactivation’ of ESCRT-III mediated nucleoporin turnover gives rise to NPC disruptions in ALS/FTD human neurons.
Endolysosomal trafficking and protein degradation
Endolysosomal trafficking proceeds through a series of vesicular intermediates ultimately culminating in the degradation of cargo molecules within the lysosome [Citation202,Citation203]. Thus, this pathway plays a pivotal role in maintaining protein homeostasis within cells. A number of studies have identified a role for ESCRT proteins in endosomal sorting, MVB biogenesis, and endosomal and lysosomal membrane repair [Citation13,Citation15,Citation28,Citation37,Citation99–101,Citation204–206]. Briefly, endosomal sorting and MVB biogenesis proceeds first by the binding of ESCRT-0 complexes to endosomal membranes resulting in the clustering of ubiquitinated cargoes [Citation2,Citation3,Citation13,Citation105,Citation125,Citation201,Citation207]. ESCRT-I and ESCRT-II complexes then bind ubiquitinated cargoes and deform MVB membranes to form a cargo containing bud [Citation2,Citation3,Citation13,Citation37,Citation105,Citation125,Citation201,Citation207]. Studies also suggest that ESCRT-III proteins may play a role in the induction of membrane curvature when localized to endosomal and MVB membranes at high concentrations [Citation2,Citation3,Citation13,Citation105,Citation125,Citation208,Citation209]. Nonetheless, recruitment of ESCRT-III proteins is essential for formation of vesicles via scission of the neck of membrane buds [Citation8,Citation125,Citation210]. Moreover, in addition to their role in recruitment of VPS4 for polymer disassembly as discussed above, ESCRT-III subunits recruit deubiquitinating enzymes for protein deubiquitination during sorting of cargoes within endosomes into MVBs [Citation2,Citation3,Citation13,Citation105,Citation125,Citation211].
Ultimately, MVBs fuse with either autophagosomes or lysosomes to promote protein degradation through the autophagy pathway [Citation8,Citation212,Citation213]. Consistent with a role for ESCRT-III proteins in facilitating endolysosomal trafficking and degradation, reduction in CHMP4B or CHMP3 expression leads to accumulation of autophagosomes in rodent cortical neurons [Citation214] and HeLa cells [Citation215] respectively and knockdown of CHMP3 or CHMP5 impairs receptor degradation [Citation216,Citation217]. As will be discussed in more detail below, mutations in CHMP2B can also impact endolysosomal pathway function [Citation52,Citation218–220]. Although the molecular mechanisms by which ESCRT-III proteins regulate autophagy and lysosomal fusion remain unclear, ESCRT-III proteins also play a critical role in repair of lysosomal membranes upon damage, in turn, preventing cell death [Citation28,Citation221–223].
Neurodegenerative diseases are often characterized by the presence of protein aggregates at end-stage disease [Citation85,Citation224–230]. Thus, this pathology is suggestive of impaired protein homeostasis and degradation. A number of studies have implicated alterations in autophagy and lysosomal degradation as a contributing factor to neurodegenerative disease pathologies [Citation34,Citation36,Citation38,Citation39,Citation231–235]. Interestingly, a recent study has demonstrated that upregulation of ESCRT pathway proteins promotes the degradation of multiple proteins associated with neurodegeneration including Tau and Huntingtin [Citation236]. In addition, reduction in CHMP2A, CHMP2B, or CHMP6 can compromise endolysosomal function and promote Tau propagation and aggregation [Citation237] and Tau accumulation can subsequently impact the expression of Ist1/CHMP8 therefore impeding ESCRT-III function [Citation238]. These studies suggest that feedback loops between endolysosomal dysfunction and protein aggregation may perpetuate deficiencies in protein homeostasis in neurodegenerative disease.
Importantly, mutations in genes that function in the endolysosomal or autophagy pathway (e.g. C9orf72, VAPB, VCP, TBK1, UBQLN2, CHMP2B) are causative of neurodegenerative disease [Citation50,Citation51,Citation53,Citation56,Citation239–242] suggesting that impairments in endolysosomal function may be a primary contributor to disease pathophysiology. We refer readers to a recent review [Citation39] for a comprehensive discussion of implications of these genetic mutations on endolysosomal trafficking and protein degradation in ALS and FTD.
From a nuclear perspective, multiple studies have now demonstrated that NPCs and nucleoporins can be degraded via autophagy pathways [Citation192,Citation193,Citation243–246]. The data suggest that autophagy impairments may compromise nucleoporin turnover and may contribute to NPC disruptions in neurodegenerative disease. Although the mechanisms that connect ESCRT-III mediated NPC surveillance to autophagy and lysosomal degradation remain unknown, as discussed above, there are demonstrations that altered ESCRT-III nuclear surveillance may initiate aberrant nucleoporin degradation in ALS/FTD [Citation90,Citation93,Citation161,Citation199]. However, other reports suggest that NPCs and nucleoporins are degraded via the proteasome [Citation30,Citation199]. Thus, the mechanisms and pathways implicated in physiologic and pathologic degradation of nucleoporins and NPCs remain controversial. In the future, it will be necessary to determine whether distinct pathways facilitate cell type, organismal, and even nucleoporin-specific degradation.
Mutations in ESCRT-III proteins in neurodegenerative disease
Autosomal dominant mutations in the ESCRT-III protein CHMP2B have been implicated in rare genetic forms of FTD, ALS, and other motor neuron diseases [Citation53–56,Citation247]. The first identified mutation lies within the splice acceptor site for the final exon and results in two novel transcripts referred to as CHMP2BIntron5 (CHMP2BM178V) and CHMP2BΔ10 [Citation55,Citation216]. A second study identified an autosomal dominant point mutation at amino acid 165 whereby glutamine is replaced by a stop codon (CHMP2BQ165X) [Citation56]. Both mutations, and all three resulting transcripts result in the production of truncated CHMP2B proteins lacking a portion of the C terminus [Citation55,Citation56,Citation216]. Subsequent reports have identified a number of disease causative point mutations in patients with FTD (CHMP2BD148Y) and motor neuron disease (CHMP2BI29V, CHMP2BT104N, CHMP2BQ206H) [Citation53,Citation55,Citation56,Citation247]. Thus, the genetic link between CHMP2B and FTD/ALS suggests that ESCRT-III disruption may be a primary contributor to neurodegenerative disease pathogenesis.
CHMP2B FTD/ALS cases are unique in that they lack the characteristic cytoplasmic aggregation TDP-43 despite the presence ubiquitin and p62 positive inclusions at end-stage disease [Citation248]. In contrast, a recent study demonstrated that overexpression of wildtype CHMP2B or the CHMP2BIntron5 mutation resulted in accumulation of phosphorylated TDP-43 in N2a cells [Citation249]. Whether this discrepancy from human pathology is cell type specific or related to artifacts of overexpression remains unclear at this time. However, endocytosis and the ESCRT machinery have both been linked to TDP-43 turnover and clearance [Citation215,Citation250] albeit largely in model systems based on TDP-43 overexpression. We also note that the lack of cytoplasmic TDP-43 aggregation and phosphorylation may not accurately reflect and indicate the status of TDP-43 function, specifically as it relates to loss of transcription and splicing activity in the nucleus. In fact, even in ALS, cells that harbor TDP-43 aggregated at end-stage disease are rare [Citation65,Citation251,Citation252]. A substantially higher percentage of cells display a gradient-like distribution of non-aggregated TDP-43 throughout the nucleus and cytoplasm and a substantial proportion of cells even display ‘normal’ nuclear localization and expression of TDP-43 [Citation253]. Thus, it is possible that TDP-43 nuclear function may be disrupted and/or TDP-43 May mislocalize to the cytoplasm independent of cytoplasmic aggregation, a phenomenon that may not be detected by histological analyses. As a result, future studies are necessary to determine whether TDP-43 function is disrupted in models of CHMP2B FTD/ALS and whether this contributes to disease pathogenesis as has been recently observed in other forms of ALS/FTD [Citation63,Citation64,Citation70,Citation76,Citation254–256].
To date, most studies detailing the cellular consequences of CHMP2B mutations have focused on endolysosomal trafficking and autophagy functions of the ESCRT-III pathway. Histologically, multiple groups have observed pathologic accumulation of enlarged endosomes and autophagic structures in in vitro and in vivo model systems of CHMP2B FTD/ALS [Citation52,Citation55,Citation214,Citation216,Citation257]. In contrast to the normally diffuse and predominantly cytoplasmic distribution of wildtype CHMP2B, CHMP2BIntron5 and CHMP2BΔ10 proteins form cytoplasmic puncta that colocalize with the endosomal and lysosomal marker CD63 when overexpressed in PC12 cells [Citation55]. In addition, when overexpressed in primary neurons, cytoplasmic CHMP2BIntron5 puncta colocalize with markers of recycling, late, and early endosomes [Citation258] and CHMP2BIntron5 expression can lead to CHMP4B sequestration within Rab7 positive endosomal structures [Citation214]. Together, these observations are suggestive of widespread disruptions to endolysosomal trafficking and function. This is supported by observations of large endosome accumulations in patient fibroblasts, postmortem cortical tissue and primary rat neurons overexpressing CHMP2BIntron5 [Citation214,Citation216,Citation258] as well as autofluorescent accumulations in transgenic mice [Citation52]. When expressed in HEK293 cells, CHMP2BIntron5 more strongly associates with CHMP4B compared to its wildtype counterpart [Citation214]. This is reminiscent of enhanced ESCRT-III subunit association upon expression of a dominant negative VPS4 [Citation214] suggesting that ESCRT-III polymer disassembly may be impaired in the context of CHMP2B mutations.
Given that a number of disease associated mutations in CHMP2B result in C-terminal protein truncation [Citation55,Citation56,Citation216], it is possible that autoinhibition is prevented leading to constitutive CHMP2B polymerization. This hypothesis is supported by the punctate distribution of CHMP2B mutants as well as the failure to dissociate from CHMP4B [Citation55,Citation214]. Whether CHMP2B and its potential constitutive activation compromise nuclear surveillance functions of the ESCRT-III pathway in disease are currently unclear but under investigation at least within our group. However, a recent study demonstrated that CHMP2BIntron5 exhibits stronger binding to the microtubule-severing enzyme Spastin than its wildtype counterpart [Citation259]. In addition, Spastin is colocalized with CHMP2BIntron5 accumulations presumably impacting its solubility [Citation259]. Interestingly, Spastin is recruited by IST1/CHMP8 for mitotic spindle disassembly and nuclear envelope sealing [Citation29]. Thus, these data support a potential link between CHMP2B mutations in dysregulation of nuclear envelope and nuclear pore complex maintenance and surveillance functions of the ESCRT-III pathway.
Conclusions and perspectives
The ESCRT-III pathway is a multifaceted cellular pathway with critical roles in membrane dynamics, protein degradation, and maintenance of cellular integrity. Therefore, disruptions to ESCRT-III protein subunits and overall pathway function are likely to have widespread impacts on cellular health and survival. Recent studies have highlighted a role for ESCRT-III pathway alterations, particular NPC and nuclear envelope surveillance and endolysosomal trafficking as important contributors to neurodegenerative disease pathogenesis. However, the molecular mechanisms by which ESCRT-III dysfunction gives rise to pathophysiologic events in disease remain largely unknown. Specifically, from the perspective of NPC surveillance, little is known regarding the maintenance of NPCs throughout the lifetime of non-dividing neurons. Thus, as it relates to neurodegenerative disease pathogenesis, future studies are necessary to understand both the physiologic function and pathologic consequences of impaired ESCRT-III nuclear surveillance function in a cell type and organism-specific manner. Ultimately, understanding the mechanisms by which ESCRT-III dysfunction contributes to neurodegenerative disease, may yield novel therapeutic targets and strategies for disease. Modulation of specific ESCRT-III functions, proteins, or subunit interactions could hold promise for future therapies. In support of this idea, a recent study demonstrated that antisense oligonucleotide (ASO) mediated knockdown of CHMP7 was sufficient to repair NPCs, alleviate alterations to TDP-43 function and localization, and improve neuronal survival in genetic and sporadic ALS iPSNs [Citation93]. Thus, continuing to unravel the intricacies underlying ESCRT-III function and dysfunction specifically in human neurons has the potential to have a significant impact on our understanding of neurodegenerative disease and novel treatment strategies.
Author contributions
ANC wrote and edited the manuscript. OK carried out all manuscript revisions and assisted with final editing. ANC conceptualized and generated figures. Conceptualization and oversight were carried out by ANC. All authors reviewed and approved the final manuscript.
Acknowledgments
We thank America Chandia Cristi and Ssu-Ying Chen for feedback on this manuscript.
Disclosure statement
ANC has submitted patents on methods and drugs to modulate various ESCRT-III proteins in neurodegeneration.
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Hatano T, Palani S, Papatziamou D, et al. Asgard archaea shed light on the evolutionary origins of the eukaryotic ubiquitin-ESCRT machinery. Nat Commun. 2022;13(1):3398. doi: 10.1038/s41467-022-30656-2
- Hurley JH. The ESCRT complexes. Crit Rev Biochem Mol Biol. 2010;45(6):463–20. doi: 10.3109/10409238.2010.502516
- Hurley JH. Escrts are everywhere. Embo J. 2015;34(19):2398–2407. doi: 10.15252/embj.201592484
- McCullough J, Frost A, Sundquist WI. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu Rev Cell Dev Biol. 2018;34(1):85–109. doi: 10.1146/annurev-cellbio-100616-060600
- Olmos Y. The ESCRT machinery: remodeling, repairing, and sealing membranes. Membranes. 2022;12(6):633. doi: 10.3390/membranes12060633
- Remec Pavlin M, Hurley JH. The ESCRTs - converging on mechanism. J Cell Sci. 2020;133(18). doi: 10.1242/jcs.240333
- Schmidt O, Teis D. The ESCRT machinery. Curr Biol. 2012;22(4):R116–20. doi: 10.1016/j.cub.2012.01.028
- Vietri M, Radulovic M, Stenmark H. The many functions of ESCRTs. Nat Rev Mol Cell Biol. 2019;21(1):25–42. doi: 10.1038/s41580-019-0177-4
- Schöneberg J, Pavlin MR, Yan S, et al. ATP-dependent force generation and membrane scission by ESCRT-III and Vps4. Science. 2018;362(6421):1423–1428.
- Krylova SV, Feng D. The machinery of exosomes: biogenesis, release, and uptake. Int J Mol Sci. 2023;24(2). doi: 10.3390/ijms24021337
- Lusk CP, Ader NR. Chmpions of repair: emerging perspectives on sensing and repairing the nuclear envelope barrier. Curr Opin Cell Biol. 2020;64:25–33. doi: 10.1016/j.ceb.2020.01.011
- Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe. 2013;14(3):232–241. doi: 10.1016/j.chom.2013.08.012
- Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464(7290):864–869. doi: 10.1038/nature08849
- Zhou F, Wu Z, Zhao M, et al. Rab5-dependent autophagosome closure by ESCRT. J Cell Bio. 2019;218(6):1908–1927. doi: 10.1083/jcb.201811173
- Yang Y, Wang M, Zhang Y-Y, et al. The endosomal sorting complex required for transport repairs the membrane to delay cell death. Front Oncol. 2022;12:1007446. doi: 10.3389/fonc.2022.1007446
- Colombo M, Moita C, van Niel G, et al. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J Cell Sci. 2013;126(Pt 24):5553–5565. doi: 10.1242/jcs.128868
- Juan T, Fürthauer M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin Cell Dev Biol. 2018;74:66–77. doi: 10.1016/j.semcdb.2017.08.022
- Larios J, Mercier V, Roux A, et al. ALIX- and ESCRT-III–dependent sorting of tetraspanins to exosomes. Journal of Cell Biology. 2020;219(3): doi: 10.1083/jcb.201904113
- Borah S, Dhanasekaran K, Kumar S. The LEM-ESCRT toolkit: repair and maintenance of the nucleus. Front Cell Dev Biol. 2022;10:989217. doi: 10.3389/fcell.2022.989217
- Gatta AT, Carlton JG. The ESCRT-machinery: closing holes and expanding roles. Curr Opin Cell Biol. 2019;59:121–132. doi: 10.1016/j.ceb.2019.04.005
- Isono E. ESCRT is a great Sealer: non-endosomal function of the ESCRT Machinery in Membrane Repair and autophagy. Plant Cell Physiol. 2021;62(5):766–774. doi: 10.1093/pcp/pcab045
- Denais CM, Gilbert RM, Isermann P, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352(6283):353–358. doi: 10.1126/science.aad7297
- Jimenez AJ, Maiuri P, Lafaurie-Janvore J, et al. ESCRT machinery is required for plasma membrane repair. Science. 2014;343(6174):1247136.
- Loncle N, Agromayor M, Martin-Serrano J, et al. An ESCRT module is required for neuron pruning. Sci Rep. 2015;5(1):8461. doi: 10.1038/srep08461
- Olmos Y, Hodgson L, Mantell J, et al. ESCRT-III controls nuclear envelope reformation. Nature. 2015;522(7555):236–239. doi: 10.1038/nature14503
- Raab M, Gentili M, de Belly H, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science. 2016;352(6283):359–362.
- Scheffer LL, Sreetama SC, Sharma N, et al. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5(1):5646. doi: 10.1038/ncomms6646
- Skowyra ML, Schlesinger PH, Naismith TV, et al. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science. 2018;360(6384). doi: 10.1126/science.aar5078
- Vietri M, Schink KO, Campsteijn C, et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522(7555):231–235. doi: 10.1038/nature14408
- Webster BM, Colombi P, Jäger J, et al. Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell. 2014;159(2):388–401. DOI:10.1016/j.cell.2014.09.012
- Lee JA, Gao FB. Neuronal functions of ESCRTs. Exp Neurobiol. Exp Neurobiol. 2012;21(1):9–15. doi: 10.5607/en.2012.21.1.9
- Chandra S, Lusk CP. Emerging connections between nuclear pore complex homeostasis and ALS. Int J Mol Sci. 2022;23(3). doi: 10.3390/ijms23031329
- Kaul Z, Chakrabarti O. Endosomal sorting complexes required for ESCRTing cells toward death during neurogenesis, neurodevelopment and neurodegeneration. Traffic. 2018;19(7):485–495. doi: 10.1111/tra.12569
- Hu YB, Dammer EB, Ren R-J, et al. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener. 2015;4(1):18. doi: 10.1186/s40035-015-0041-1
- Lee JA, Gao FB. ESCRT, autophagy, and frontotemporal dementia. BMB Rep. 2008;41(12):827–832. doi: 10.5483/BMBRep.2008.41.12.827
- Lee JA, Gao FB. Roles of ESCRT in autophagy-associated neurodegeneration. Autophagy. 2008;4(2):230–232. doi: 10.4161/auto.5384
- Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458(7237):445–452. doi: 10.1038/nature07961
- Rusten TE, Simonsen A. ESCRT functions in autophagy and associated disease. Cell Cycle. 2008;7(9):1166–1172. doi: 10.4161/cc.7.9.5784
- Todd TW, Shao W, Zhang Y-J, et al. The endolysosomal pathway and ALS/FTD. Trends Neurosci. 2023;46(12):1025–1041. doi: 10.1016/j.tins.2023.09.004
- Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3(1):17071. doi: 10.1038/nrdp.2017.71
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–615. doi: 10.1038/nrneurol.2011.150
- Ferrari R, Kapogiannis DD, Huey E, et al. FTD and ALS: a tale of two diseases. Curr Alzheimer Res. 2011;8(3):273–294. doi: 10.2174/156720511795563700
- Geser F, Martinez-Lage M, Kwong LK, et al. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256(8):1205–1214. doi: 10.1007/s00415-009-5069-7
- Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27(10):1918–1929. doi: 10.1111/ene.14393
- Wheaton MW, Salamone AR, Mosnik DM, et al. Cognitive impairment in familial ALS. Neurology. 2007;69(14):1411–1417. doi: 10.1212/01.wnl.0000277422.11236.2c
- Antonioni A, Raho EM, Lopriore P, et al. Frontotemporal Dementia, where do we stand? A narrative review. IJMS. 2023;24(14):11732. doi: 10.3390/ijms241411732
- Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386(10004):1672–1682. doi: 10.1016/S0140-6736(15)00461-4
- Boeve BF, Boxer AL, Kumfor F, et al. Advances and controversies in frontotemporal dementia: diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022;21(3):258–272. doi: 10.1016/S1474-4422(21)00341-0
- Ringholz GM, Greene SR. The relationship between amyotrophic lateral sclerosis and frontotemporal dementia. Curr Neurol Neurosci Rep. 2006;6(5):387–392. doi: 10.1007/s11910-996-0019-6
- DeJesus-Hernandez M, Mackenzie I, Boeve B, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011
- Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010
- Clayton EL, Mizielinska S, Edgar JR, et al. Frontotemporal dementia caused by CHMP2B mutation is characterised by neuronal lysosomal storage pathology. Acta Neuropathol. 2015;130(4):511–523. doi: 10.1007/s00401-015-1475-3
- Cox LE, Ferraiuolo L, Goodall EF, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLOS One. 2010;5(3):e9872.
- Roos P. et al. CHMP2B frontotemporal dementia, in GeneReviews® M.P. Adam, et al., Editors. 1993, University of Washington, Seattle copyright © 1993-2020 GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved. Seattle: WA.
- Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. doi: 10.1038/ng1609
- van der Zee J, Urwin H, Engelborghs S, et al. CHMP2B C-truncating mutations in frontotemporal lobar degeneration are associated with an aberrant endosomal phenotype in vitro. Hum Mol Genet. 2008;17(2):313–322. doi: 10.1093/hmg/ddm309
- Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018;14(9):544–558. doi: 10.1038/s41582-018-0047-2
- Ghasemi M, Brown RH Jr. Genetics of amyotrophic lateral sclerosis. Cold Spring Harb Perspect Med. 2018;8(5):a024125. doi: 10.1101/cshperspect.a024125
- Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075–2086. doi: 10.1007/s00415-019-09363-4
- Gregory JM, Fagegaltier D, Phatnani H, et al. Genetics of amyotrophic lateral sclerosis. Current Genet Med Rep. 2020;8(4):121–131. doi: 10.1007/s40142-020-00194-8
- Olszewska DA, Lonergan R, Fallon EM, et al. Genetics of frontotemporal dementia. Curr Neurol Neurosci Rep. 2016;16(12):107. doi: 10.1007/s11910-016-0707-9
- Orr HT. FTD and ALS: genetic ties that bind. Neuron. 2011;72(2):189–190. doi: 10.1016/j.neuron.2011.10.001
- Irwin KE, Jasin P, Braunstein KE, et al. Author correction: a fluid biomarker reveals loss of TDP-43 splicing repression in presymptomatic ALS–FTD. Nat Med. 2024. doi: 10.1038/s41591-024-02966-z
- Seddighi S, Qi YA, Brown AL, et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Sci Transl Med. 2024:eadg7162.
- Bodansky A, Kim JMH, Tempest L, et al. TDP-43 and ubiquitinated cytoplasmic aggregates in sporadic ALS are low frequency and widely distributed in the lower motor neuron columns independent of disease spread. Amyotroph Lateral Scler. 2010;11(3): 321–327. DOI:10.3109/17482961003602363
- Brown AL, Wilkins OG, Keuss MJ, et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022;603(7899):131–137. doi: 10.1038/s41586-022-04436-3
- Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17(11):659–667. doi: 10.1016/j.molmed.2011.06.004
- de Boer EMJ, Orie VK, Williams T, et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2020;92(1):86–95. doi: 10.1136/jnnp-2020-322983
- Highley JR, Kirby J, Jansweijer JA, et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol Appl Neurobiol. 2014;40(6):670–685. doi: 10.1111/nan.12148
- Klim JR, Williams LA, Limone F, et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22(2):167–179. doi: 10.1038/s41593-018-0300-4
- Lagier-Tourenne C, Cleveland DW. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136(6):1001–1004. doi: 10.1016/j.cell.2009.03.006
- Ling JP, Pletnikova O, Troncoso JC, et al. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349(6248):650–655. doi: 10.1126/science.aab0983
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79(3):416–438. doi: 10.1016/j.neuron.2013.07.033
- Neumann M. Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci. 2009;10(1):232–246. doi: 10.3390/ijms10010232
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108
- Prudencio M, Humphrey J, Pickles S, et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J Clin Invest. 2020;130(11):6080–6092. doi: 10.1172/JCI139741
- Suk TR, Rousseaux MWC. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15(1):45.
- Barmada SJ. Linking RNA dysfunction and neurodegeneration in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12(2):340–351. doi: 10.1007/s13311-015-0340-3
- Boeynaems S, Bogaert E, Van Damme P, et al. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132(2):159–173. doi: 10.1007/s00401-016-1586-5
- Fiesel FC, Kahle PJ. TDP-43 and FUS/TLS: cellular functions and implications for neurodegeneration. FEBS J. 2011;278(19):3550–3568. doi: 10.1111/j.1742-4658.2011.08258.x
- Hutten S, Dormann D. Nucleocytoplasmic transport defects in neurodegeneration — cause or consequence? Semin Cell Dev Biol. 2019;99:151–162. doi: 10.1016/j.semcdb.2019.05.020
- Janssens J, Van Broeckhoven C. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum Mol Genet. 2013;22(R1):R77–87. doi: 10.1093/hmg/ddt349
- Kapeli K, Martinez FJ, Yeo GW. Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. 2017;136(9):1193–1214. doi: 10.1007/s00439-017-1830-7
- Kim HJ, Taylor JP. Lost in transportation: Nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron. 2017;96(2):285–297. doi: 10.1016/j.neuron.2017.07.029
- Kumar V, Sami N, Kashav T, et al. Protein aggregation and neurodegenerative diseases: from theory to therapy. Eur J Med Chem. 2016;124:1105–1120. doi: 10.1016/j.ejmech.2016.07.054
- Peters OM, Ghasemi M, Brown RH. Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125(5):1767–1779. doi: 10.1172/JCI71601
- Rossi S, Cozzolino M, Carri MT. Old versus new mechanisms in the pathogenesis of ALS. Brain Pathol. 2016;26(2):276–286. doi: 10.1111/bpa.12355
- Ruegsegger C, Saxena S. Proteostasis impairment in ALS. Brain Res. 2016;1648(Pt B):571–579. doi: 10.1016/j.brainres.2016.03.032
- Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi: 10.1038/nature20413
- Baskerville V, Rapuri S, Mehlhop E, et al. SUN1 facilitates CHMP7 nuclear influx and injury cascades in sporadic amyotrophic lateral sclerosis. Brain. 2023;147(1):109–121. doi: 10.1093/brain/awad291
- Chou CC, Zhang Y, Umoh ME, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21(2):228–239. doi: 10.1038/s41593-017-0047-3
- Cornelison GL, Levy SA, Jenson T, et al. Tau-induced nuclear envelope invagination causes a toxic accumulation of mRNA in Drosophila. Aging Cell. 2019;18(1):e12847. doi: 10.1111/acel.12847
- Coyne AN, Baskerville V, Zaepfel BL, et al. Nuclear accumulation of CHMP7 initiates nuclear pore complex injury and subsequent TDP-43 dysfunction in sporadic and familial ALS. Sci Transl Med. 2021;13(604). 10.1126/scitranslmed.abe1923
- Coyne AN, Zaepfel BL, Hayes L, et al. G(4)C(2) repeat RNA initiates a POM121-mediated reduction in specific nucleoporins in C9orf72 ALS/FTD. Neuron. 2020;107(6):1124–1140.e11. doi: 10.1016/j.neuron.2020.06.027
- Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron. 2018;99(5):925–940.e7. doi: 10.1016/j.neuron.2018.07.039
- Frost B, Bardai FH, Feany MB. Lamin dysfunction mediates neurodegeneration in Tauopathies. Curr Biol. 2016;26(1):129–136. doi: 10.1016/j.cub.2015.11.039
- Grima JC, Daigle JG, Arbez N, et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron. 2017;94(1):93–107.e6. doi: 10.1016/j.neuron.2017.03.023
- Schöneberg J, Lee I-H, Iwasa JH, et al. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol. 2017;18(1):5–17. doi: 10.1038/nrm.2016.121
- Babst M, Katzmann DJ, Snyder WB, et al. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3(2): 271–282. doi: 10.1016/S1534-5807(02)00219-8
- Babst M, Katzmann DJ, Snyder WB, et al. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell. 2002;3(2): 283–289. DOI:10.1016/S1534-5807(02)00219-8
- Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106(2):145–155. doi: 10.1016/S0092-8674(01)00434-2
- Ren X, Kloer DP, Kim YC, et al. Hybrid structural model of the complete human ESCRT-0 complex. Structure. 2009;17(3):406–416. doi: 10.1016/j.str.2009.01.012
- Hirano S, Kawasaki M, Ura H, et al. Double-sided ubiquitin binding of hrs-UIM in endosomal protein sorting. Nat Struct Mol Biol. 2006;13(3):272–277. doi: 10.1038/nsmb1051
- Mizuno E, Kawahata K, Kato M, et al. STAM proteins bind ubiquitinated proteins on the early endosome via the VHS domain and ubiquitin-interacting motif. Mol Biol Cell. 2003;14(9): 3675–3689. doi: 10.1091/mbc.e02-12-0823
- Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it’s all in the neck. Nat Rev Mol Cell Biol. 2010;11(8):556–566. doi: 10.1038/nrm2937
- Christ L, Raiborg C, Wenzel EM, et al. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci. 2017;42(1):42–56. doi: 10.1016/j.tibs.2016.08.016
- Bajorek M, Schubert HL, McCullough J, et al. Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol. 2009;16(7):754–762. doi: 10.1038/nsmb.1621
- Lata S, Roessle M, Solomons J, et al. Structural basis for autoinhibition of ESCRT-III CHMP3. J Mol Biol. 2008;378(4):818–827. doi: 10.1016/j.jmb.2008.03.030
- McCullough J, Clippinger AK, Talledge N, et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science. 2015;350(6267):1548–1551. doi: 10.1126/science.aad8305
- McMillan BJ, Tibbe C, Jeon H, et al. Electrostatic interactions between elongated monomers drive filamentation of drosophila shrub, a metazoan ESCRT-III protein. Cell Rep. 2016;16(5):1211–1217. doi: 10.1016/j.celrep.2016.06.093
- Tang S, Henne WM, Borbat PP, et al. Structural basis for activation, assembly and membrane binding of ESCRT-III Snf7 filaments. Elife. 2015;4:4. doi: 10.7554/eLife.12548
- Shim S, Kimpler LA, Hanson PI. Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic. 2007;8(8):1068–1079.
- Tang S, Buchkovich NJ, Henne WM, et al. ESCRT-III activation by parallel action of ESCRT-I/II and ESCRT-0/Bro1 during MVB biogenesis. Elife. 2016;5:5. doi: 10.7554/eLife.15507
- Gu M, LaJoie D, Chen OS, et al., LEM2 recruits CHMP7 for ESCRT-mediated nuclear envelope closure in fission yeast and human cells. Proc natl acad sci USA, 2017. 114(11): p. E2166–e2175.
- Thaller DJ, Allegretti M, Borah S, et al. An ESCRT-LEM protein surveillance system is poised to directly monitor the nuclear envelope and nuclear transport system. Elife. 2019;8:8. doi: 10.7554/eLife.45284
- von Appen A, LaJoie D, Johnson IE, et al. LEM2 phase separation promotes ESCRT-mediated nuclear envelope reformation. Nature. 2020;582(7810):115–118. doi: 10.1038/s41586-020-2232-x
- Webster BM, Thaller DJ, Jäger J, et al. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. Embo J. 2016;35(22):2447–2467. doi: 10.15252/embj.201694574
- Pfitzner AK, Mercier V, Jiang X, et al. An ESCRT-III polymerization sequence drives membrane deformation and fission. Cell. 2020;182(5):1140–1155.e18.
- Ader NR, Chen L, Surovtsev IV, et al. An ESCRT grommet cooperates with a diffusion barrier to maintain nuclear integrity. Nat Cell Biol. 2023;25(10):1465–1477. doi: 10.1038/s41556-023-01235-4
- Adell MA, Teis D. Assembly and disassembly of the ESCRT-III membrane scission complex. FEBS Lett. 2011;585(20):3191–3196. doi: 10.1016/j.febslet.2011.09.001
- Adell MAY, Migliano SM, Upadhyayula S, et al. Recruitment dynamics of ESCRT-III and Vps4 to endosomes and implications for reverse membrane budding. Elife. 2017;6:6. doi: 10.7554/eLife.31652
- Pfitzner AK, Moservon Filseck J, Roux A. Principles of membrane remodeling by dynamic ESCRT-III polymers. Trends Cell Biol. 2021;31(10):856–868.
- Cada AK, Pavlin MR, Castillo JP, et al. Friction-driven membrane scission by the human ESCRT-III proteins CHMP1B and IST1. Proc Natl Acad Sci USA. 2022;119(29):e2204536119. doi: 10.1073/pnas.2204536119
- Henne WM, Stenmark H, Emr SD. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol. 2013;5(9). doi: 10.1101/cshperspect.a016766
- Wollert T, Wunder C, Lippincott-Schwartz J, et al. Membrane scission by the ESCRT-III complex. Nature. 2009;458(7235):172–177. doi: 10.1038/nature07836
- Allison R, Lumb JH, Fassier C, et al. An ESCRT–spastin interaction promotes fission of recycling tubules from the endosome. J Cell Bio. 2013;202(3):527–543. doi: 10.1083/jcb.201211045
- McMahon HT, Boucrot E. Membrane curvature at a glance. J Cell Sci. 2015;128(6):1065–1070. doi: 10.1242/jcs.114454
- Caillat C, Macheboeuf P, Wu Y, et al. Asymmetric ring structure of Vps4 required for ESCRT-III disassembly. Nat Commun. 2015;6(1):8781. doi: 10.1038/ncomms9781
- Davies BA, Azmi IF, Payne J, et al. Coordination of substrate binding and ATP hydrolysis in Vps4-mediated ESCRT-III disassembly. Mol Biol Cell. 2010;21(19):3396–3408.
- Maity S, Caillat C, Miguet N, et al. VPS4 triggers constriction and cleavage of ESCRT-III helical filaments. Sci Adv. 2019;5(4):eaau7198. doi: 10.1126/sciadv.aau7198
- Stuchell-Brereton MD, Skalicky JJ, Kieffer C, et al. ESCRT-III recognition by VPS4 ATPases. Nature. 2007;449(7163):740–744. doi: 10.1038/nature06172
- Yang B, Stjepanovic G, Shen Q, et al. Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat Struct Mol Biol. 2015;22(6):492–498. doi: 10.1038/nsmb.3015
- Lata S, Schoehn G, Jain A, et al. Helical structures of ESCRT-III are disassembled by VPS4. Science. 2008;321(5894): 1354–1357. DOI:10.1126/science.1161070
- Scott A, Gaspar J, Stuchell-Brereton MD, et al. Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc Natl Acad Sci USA. 2005;102(39):13813–13818. doi: 10.1073/pnas.0502165102
- Gatta AT, Olmos Y, Stoten CL, et al. CDK1 controls CHMP7-dependent nuclear envelope reformation. Elife. 2021;10:10. doi: 10.7554/eLife.59999
- Maciejowski J, Hatch EM. Nuclear membrane rupture and its consequences. Annu Rev Cell Dev Biol. 2020;36(1):85–114. doi: 10.1146/annurev-cellbio-020520-120627
- Meinke P, Mattioli E, Haque F, et al. Muscular dystrophy-associated SUN1 and SUN2 variants disrupt nuclear-cytoskeletal connections and myonuclear organization. PLOS Genet. 2014;10(9):e1004605. doi: 10.1371/journal.pgen.1004605
- Wang JY, Yu I-S, Huang C-C, et al. Sun1 deficiency leads to cerebellar ataxia in mice. dis Model mech. Dis Models Mech. 2015;8(8):957–967. doi: 10.1242/dmm.019240
- Dutta S, Bhattacharyya M, Sengupta K. Changes in the nuclear envelope in Laminopathies. Adv Exp Med Biol. 2018;1112:31–38.
- Kang SM, Yoon MH, Park BJ. Laminopathies; mutations on single gene and various human genetic diseases. BMB Rep. 2018;51(7):327–337. doi: 10.5483/BMBRep.2018.51.7.113
- Stiekema M, van Zandvoort MAMJ, Ramaekers FCS, et al. Structural and mechanical aberrations of the nuclear lamina in disease. Cells. 2020;9(8):1884. doi: 10.3390/cells9081884
- Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226(2):316–325. doi: 10.1002/path.2999
- Robijns J, Houthaeve G, Braeckmans K, et al. Loss of nuclear envelope integrity in aging and disease. Int Rev Cell Mol Biol. 2018;336:205–222.
- Andreu I, Granero-Moya I, Chahare NR, et al. Mechanical force application to the nucleus regulates nucleocytoplasmic transport. Nat Cell Biol. 2022;24(6):896–905. doi: 10.1038/s41556-022-00927-7
- Enyedi B, Niethammer P. Nuclear membrane stretch and its role in mechanotransduction. Nucleus. 2017;8(2):156–161. doi: 10.1080/19491034.2016.1263411
- Janota CS, Calero-Cuenca FJ, Gomes ER. The role of the cell nucleus in mechanotransduction. Curr Opin Cell Biol. 2020;63:204–211. doi: 10.1016/j.ceb.2020.03.001
- Lusk CP, King MC. The nucleus: keeping it together by keeping it apart. Curr Opin Cell Biol. 2017;44:44–50. doi: 10.1016/j.ceb.2017.02.001
- King MC, Lusk CP. A model for coordinating nuclear mechanics and membrane remodeling to support nuclear integrity. Curr Opin Cell Biol. 2016;41:9–17. doi: 10.1016/j.ceb.2016.03.009
- Halfmann CT, Sears RM, Katiyar A, et al. Repair of nuclear ruptures requires barrier-to-autointegration factor. J Cell Bio. 2019;218(7):2136–2149. doi: 10.1083/jcb.201901116
- Barger SR, Penfield L, Bahmanyar S. Nuclear envelope assembly relies on CHMP-7 in the absence of BAF-LEM-mediated hole closure. J Cell Sci. 2023;136(21). doi: 10.1242/jcs.261385
- Wallis SS, Ventimiglia LN, Otigbah E, et al. The ESCRT machinery counteracts nesprin-2G-mediated mechanical forces during nuclear envelope repair. Dev Cell. 2021;56(23):3192–3202.e8. DOI:10.1016/j.devcel.2021.10.022
- Penfield L, Shankar R, Szentgyörgyi E, et al. Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J Cell Bio. 2020;219(5): doi: 10.1083/jcb.201908179
- Thaller DJ, Tong D, Marklew CJ, et al. Direct binding of ESCRT protein Chm7 to phosphatidic acid-rich membranes at nuclear envelope herniations. J Cell Biol. 2021;220(3):074880.
- Vietri M, Schultz SW, Bellanger A, et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat Cell Biol. 2020;22(7):856–867. doi: 10.1038/s41556-020-0537-5
- Ventimiglia LN, Cuesta-Geijo MA , Martinelli N, et al. CC2D1B coordinates ESCRT-III activity during the mitotic reformation of the nuclear envelope. Dev Cell. 2018;47(5):547–563.e6.
- Capella M, Martín Caballero L, Pfander B, et al. ESCRT recruitment by the S. cerevisiae inner nuclear membrane protein Heh1 is regulated by Hub1-mediated alternative splicing. J Cell Sci. 2020;133(24):jcs250688.
- Frost B, Hemberg M, Lewis J, et al. Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci. 2014;17(3):357–366. doi: 10.1038/nn.3639
- Paonessa F, Evans LD, Solanki R, et al. Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 2019;26(3):582–593.e5. doi: 10.1016/j.celrep.2018.12.085
- Prissette M, Fury W, Koss M, et al. Disruption of nuclear envelope integrity as a possible initiating event in tauopathies. Cell Rep. 2022;40(8):111249. doi: 10.1016/j.celrep.2022.111249
- Giampetruzzi A, Danielson EW, Gumina V, et al. Modulation of actin polymerization affects nucleocytoplasmic transport in multiple forms of amyotrophic lateral sclerosis. Nat Commun. 2019;10(1):3827. doi: 10.1038/s41467-019-11837-y
- Coyne AN, Rothstein JD. The ESCRT-III protein VPS4, but not CHMP4B or CHMP2B, is pathologically increased in familial and sporadic ALS neuronal nuclei. Acta Neuropathol Commun. 2021;9(1):127. doi: 10.1186/s40478-021-01228-0
- Sontag EM, Morales-Polanco F, Chen J-H, et al. Nuclear and cytoplasmic spatial protein quality control is coordinated by nuclear–vacuolar junctions and perinuclear ESCRT. Nat Cell Biol. 2023;25(5):699–713. doi: 10.1038/s41556-023-01128-6
- Toyama BH, Arrojo e Drigo R, Lev-Ram V, et al. Visualization of long-lived proteins reveals age mosaicism within nuclei of postmitotic cells. J Cell Bio. 2018;218(2):433–444. doi: 10.1083/jcb.201809123
- Beck M, Hurt E. The nuclear pore complex: understanding its function through structural insight. Nat Rev Mol Cell Biol. 2017;18(2):73–89. doi: 10.1038/nrm.2016.147
- Capelson M, Hetzer MW. The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 2009;10(7):697–705. doi: 10.1038/embor.2009.147
- D’Angelo MA. Nuclear pore complexes as hubs for Gene Regulation. Nucleus. 2018 Jan 1;9(1):142–148. doi: 10.1080/19491034.2017.1395542
- Dickmanns A, Kehlenbach RH, Fahrenkrog B. Nuclear pore complexes and nucleocytoplasmic transport: from structure to function to disease. Int Rev Cell Mol Biol. 2015;320:171–233.
- Fahrenkrog B, Koser J, Aebi U. The nuclear pore complex: a jack of all trades? Trends Biochem Sci. 2004;29(4):175–182. doi: 10.1016/j.tibs.2004.02.006
- Hampoelz B, Andres-Pons A, Kastritis P, et al. Structure and assembly of the nuclear pore complex. Annu Rev Biophys. 2019;48(1):515–536. doi: 10.1146/annurev-biophys-052118-115308
- Ibarra A, Hetzer MW. Nuclear pore proteins and the control of genome functions. Genes Dev. 2015;29(4):337–349. doi: 10.1101/gad.256495.114
- Kabachinski G, Schwartz TU. The nuclear pore complex–structure and function at a glance. J Cell Sci. 2015;128(3):423–429. doi: 10.1242/jcs.083246
- Knockenhauer KE, Schwartz TU. The nuclear pore complex as a flexible and dynamic gate. Cell. 2016;164(6):1162–1171. doi: 10.1016/j.cell.2016.01.034
- Lin DH, Hoelz A. The structure of the nuclear pore complex (an update). Annu Rev Biochem. 2019;88(1):725–783. doi: 10.1146/annurev-biochem-062917-011901
- Pascual-Garcia P, Capelson M. Nuclear pores in genome architecture and enhancer function. Curr Opin Cell Biol. 2019;58:126–133. doi: 10.1016/j.ceb.2019.04.001
- Raices M, D’Angelo MA. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol. 2012;13(11):687–699. doi: 10.1038/nrm3461
- Raices M, D’Angelo MA. Nuclear pore complexes and regulation of gene expression. Curr Opin Cell Biol. 2017;46:26–32. doi: 10.1016/j.ceb.2016.12.006
- Strambio-De-Castillia C, Niepel M, Rout MP. The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol. 2010;11(7):490–501. doi: 10.1038/nrm2928
- Antonin W, Ellenberg J, Dultz E. Nuclear pore complex assembly through the cell cycle: regulation and membrane organization. FEBS Lett. 2008;582(14):2004–2016. doi: 10.1016/j.febslet.2008.02.067
- Bodoor K, Shaikh S, Salina D, et al. Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J Cell Sci. 1999;112(Pt13):2253–2264. doi: 10.1242/jcs.112.13.2253
- Dultz E, Ellenberg J. Live imaging of single nuclear pores reveals unique assembly kinetics and mechanism in interphase. J Cell Bio. 2010;191(1):15–22. doi: 10.1083/jcb.201007076
- Dultz E, Wojtynek M, Medalia O, et al. The nuclear pore complex: birth, life, and death of a cellular behemoth. Cells. 2022;11(9):1456. doi: 10.3390/cells11091456
- Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol. 2009;10(3):178–191. doi: 10.1038/nrm2641
- Hampoelz B, Mackmull MT, Machado P, et al. Pre-assembled nuclear pores insert into the nuclear Envelope during early development. Cell. 2016;166(3):664–678.
- Hampoelz B, Schwarz A, Ronchi P, et al. Nuclear pores assemble from nucleoporin condensates during oogenesis. Cell. 2019;179(3):671–686.e17. doi: 10.1016/j.cell.2019.09.022
- Otsuka S, Bui KH, Schorb M, et al. Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. Elife. 2016;5:5. doi: 10.7554/eLife.19071
- Otsuka S, Ellenberg J. Mechanisms of nuclear pore complex assembly – two different ways of building one molecular machine. FEBS Lett. 2018;592(4):475–488. doi: 10.1002/1873-3468.12905
- Otsuka S, Tempkin JOB, Zhang W, et al. A quantitative map of nuclear pore assembly reveals two distinct mechanisms. Nature. 2023;613(7944):575–581. doi: 10.1038/s41586-022-05528-w
- Weberruss M, Antonin W. Perforating the nuclear boundary – how nuclear pore complexes assemble. J Cell Sci. 2016;129(24):4439–4447. doi: 10.1242/jcs.194753
- Jokhi V, Ashley J, Nunnari J, et al. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013;3(4):988–995. doi: 10.1016/j.celrep.2013.03.015
- Mettenleiter TC, Müller F, Granzow H, et al. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol. 2013;15(2): 170–178. DOI:10.1111/cmi.12044
- Speese SD, Ashley J, Jokhi V, et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic wnt signaling. Cell. 2012;149(4): 832–846. DOI:10.1016/j.cell.2012.03.032
- Lee CW, Wilfling F, Ronchi P, et al. Selective autophagy degrades nuclear pore complexes. Nat Cell Biol. 2020;22(2):159–166. doi: 10.1038/s41556-019-0459-2
- Tomioka Y, Kotani T, Kirisako H, et al. TORC1 inactivation stimulates autophagy of nucleoporin and nuclear pore complexes. J Cell Bio. 2020 Jul 6;219(7):e201910063. doi: 10.1083/jcb.201910063
- Toyama BH, Savas J, Park S, et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154(5): 971–982. DOI:10.1016/j.cell.2013.07.037
- Coyne AN, Rothstein JD. Nuclear pore complexes — a doorway to neural injury in neurodegeneration. Nat Rev Neurol. 2022;18(6):348–362. doi: 10.1038/s41582-022-00653-6
- Cristi AC, Rapuri S, Coyne AN. Nuclear pore complex and nucleocytoplasmic transport disruption in neurodegeneration. FEBS Lett. 2023;597(20):2546–2566. doi: 10.1002/1873-3468.14729
- D’Angelo MA, Raices M, Panowski SH, et al. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136(2): 284–295. doi: 10.1016/j.cell.2008.11.037
- Fallini C, Khalil B, Smith CL, et al. Traffic jam at the nuclear pore: all roads lead to nucleocytoplasmic transport defects in ALS/FTD. Neurobiol Dis. 2020;140:104835. doi: 10.1016/j.nbd.2020.104835
- Dubey SK, Maulding K, Sung H, et al. Nucleoporins are degraded via upregulation of ESCRT-III/Vps4 complex in drosophila models of C9-ALS/FTD. Cell Rep. 2022;40(12):111379. doi: 10.1016/j.celrep.2022.111379
- Megat S, Mora N, Sanogo J, et al. Integrative genetic analysis illuminates ALS heritability and identifies risk genes. Nat Commun. 2023;14(1):342. doi: 10.1038/s41467-022-35724-1
- Hurley JH, Emr SD. The ESCRT complexes: structure and mechanism of a membrane-trafficking network. Annu Rev Biophys Biomol Struct. 2006;35(1):277–298. doi: 10.1146/annurev.biophys.35.040405.102126
- Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol. 2014;31:2–10. doi: 10.1016/j.semcdb.2014.03.034
- Zhang J, Jiang Z, Shi A. Rab GTPases: the principal players in crafting the regulatory landscape of endosomal trafficking. Comput Struct Biotechnol J. 2022;20:4464–4472. doi: 10.1016/j.csbj.2022.08.016
- Ahmed I, Akram Z, Iqbal HMN, et al. The regulation of endosomal sorting complex required for transport and accessory proteins in multivesicular body sorting and enveloped viral budding – an overview. Int J Biol Macromol. 2019;127:1–11. doi: 10.1016/j.ijbiomac.2019.01.015
- Katzmann DJ, Stefan CJ, Babst M, et al. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Bio. 2003;162(3):413–423. doi: 10.1083/jcb.200302136
- Metcalf D, Isaacs AM. The role of ESCRT proteins in fusion events involving lysosomes, endosomes and autophagosomes. Biochem Soc Trans. 2010;38(6):1469–1473. doi: 10.1042/BST0381469
- Williams RL, Urbé S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8(5):355–368. doi: 10.1038/nrm2162
- Hanson PI, Roth R, Lin Y, et al. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J Cell Bio. 2008;180(2):389–402. doi: 10.1083/jcb.200707031
- Saksena S, Wahlman J, Teis D, et al. Functional reconstitution of ESCRT-III assembly and disassembly. Cell. 2009;136(1): 97–109. DOI:10.1016/j.cell.2008.11.013
- Teis D, Saksena S, Judson BL, et al. ESCRT-II coordinates the assembly of ESCRT-III filaments for cargo sorting and multivesicular body vesicle formation. Embo J. 2010;29(5):871–883. doi: 10.1038/emboj.2009.408
- Hurley JH. ESCRT complexes and the biogenesis of multivesicular bodies. Curr Opin Cell Biol. 2008;20(1):4–11. doi: 10.1016/j.ceb.2007.12.002
- Liou W, Geuze HJ, Geelen MJH, et al. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Bio. 1997;136(1):61–70. doi: 10.1083/jcb.136.1.61
- Lucocq J, Walker D. Evidence for fusion between multilamellar endosomes and autophagosomes in HeLa cells. Eur J Cell Biol. 1997;72(4):307–313.
- Lee JA, Beigneux A, Ahmad ST, et al. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr Biol. 2007;17(18):1561–1567. doi: 10.1016/j.cub.2007.07.029
- Filimonenko M, Stuffers S, Raiborg C, et al. Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Bio. 2007;179(3):485–500. doi: 10.1083/jcb.200702115
- Urwin H, Authier A, Nielsen JE, et al. Disruption of endocytic trafficking in frontotemporal dementia with CHMP2B mutations. Hum Mol Genet. 2010;19(11): 2228–2238. doiI: 10.1093/hmg/ddq100
- Shim JH, Xiao C, Hayden MS, et al. CHMP5 is essential for late endosome function and down-regulation of receptor signaling during mouse embryogenesis. J Cell Bio. 2006;172(7):1045–1056. doi: 10.1083/jcb.200509041
- Ghazi-Noori S, Froud KE, Mizielinska S, et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. brain. 2012;135(3):819–832. doi: 10.1093/brain/aws006
- Krasniak CS, Ahmad ST. The role of CHMP2B(Intron5) in autophagy and frontotemporal dementia. brain res, 2016. 1649(Pt B):151–157. 10.1016/j.brainres.2016.02.051
- Ugbode C, West RJH. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis. 2021;147:105144. doi: 10.1016/j.nbd.2020.105144
- Niekamp P, Scharte F, Sokoya T, et al. Ca(2+)-activated sphingomyelin scrambling and turnover mediate ESCRT-independent lysosomal repair. Nat Commun. 2022;13(1):1875. doi: 10.1038/s41467-022-29481-4
- Radulovic M, Schink KO, Wenzel EM, et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. Embo J. 2018;37(21): doi: 10.15252/embj.201899753
- Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19(12):918–931. doi: 10.1111/tra.12613
- Alves-Rodrigues A, Gregori L, Figueiredo-Pereira ME. Ubiquitin, cellular inclusions and their role in neurodegeneration. Trends Neurosci. 1998;21(12):516–520. doi: 10.1016/S0166-2236(98)01276-4
- Arai T, Hasegawa M, Nonoka T, et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30(2):170–181. doi: 10.1111/j.1440-1789.2009.01089.x
- Ciechanover A, Kwon YT. Protein quality control by molecular chaperones in neurodegeneration. Front Neurosci. 2017;11:185. doi: 10.3389/fnins.2017.00185
- Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7). doi: 10.1101/cshperspect.a028035
- Gotzl JK, Lang CM, Haass C, et al. Impaired protein degradation in FTLD and related disorders. Ageing Res Rev. 2016;32:122–139. doi: 10.1016/j.arr.2016.04.008
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(S7):S10–S17. doi: 10.1038/nm1066
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9(11):826–838. doi: 10.1038/nrn2499
- Malik BR, Maddison DC, Smith GA, et al. Autophagic and endo-lysosomal dysfunction in neurodegenerative disease. Mol Brain. 2019;12(1):100. doi: 10.1186/s13041-019-0504-x
- Giovedì S, Ravanelli MM, Parisi B, et al. Dysfunctional autophagy and endolysosomal system in neurodegenerative diseases: relevance and therapeutic options. Front Cell Neurosci. 2020;14:602116. doi: 10.3389/fncel.2020.602116
- Wang G, Mao Z. Chaperone-mediated autophagy: roles in neurodegeneration. Transl Neurodegener. 2014;3(1):20. doi: 10.1186/2047-9158-3-20
- Wang YT, Lu JH. Chaperone-mediated autophagy in neurodegenerative diseases: molecular mechanisms and pharmacological opportunities. Cells. 2022;11(14):2250. doi: 10.3390/cells11142250
- Zhang W, Xu C, Sun J, et al. Impairment of the autophagy–lysosomal pathway in Alzheimer’s diseases: pathogenic mechanisms and therapeutic potential. Acta Pharm Sin B. 2022;12(3):1019–1040. doi: 10.1016/j.apsb.2022.01.008
- Benyair R, Panapakkam Giridharan SS, Rivero-Ríos P, et al. Upregulation of the ESCRT pathway and multivesicular bodies accelerates degradation of proteins associated with neurodegeneration. Autophagy Rep. 2023;2(1): doi: 10.1080/27694127.2023.2166722
- Chen JJ, Nathaniel DL, Raghavan P, et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J Biol Chem. 2019;294(50):18952–18966. doi: 10.1074/jbc.RA119.009432
- Feng Q, Luo Y, Zhang X-N, et al. Mapt/Tau accumulation represses autophagy flux by disrupting IST1-regulated ESCRT-III complex formation: a vicious cycle in Alzheimer neurodegeneration. Autophagy. 2020;16(4):641–658. doi: 10.1080/15548627.2019.1633862
- Deng HX, Chen W, Hong S-T, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353
- Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–636. doi: 10.1038/nn.4000
- Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5): 857–864. doi: 10.1016/j.neuron.2010.11.036
- Nishimura AL, Mitne-Neto M, Silva HCA, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75(5):822–831. doi: 10.1086/425287
- Allegretti M, Zimmerli CE, Rantos V, et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature. 2020;586(7831):796–800. doi: 10.1038/s41586-020-2670-5
- Li Z, Nakatogawa H. Degradation of nuclear components via different autophagy pathways. Trends Cell Biol. 2022;32(7):574–584. doi: 10.1016/j.tcb.2021.12.008
- Luo M, Zhao X, Song Y, et al. Nuclear autophagy: an evolutionarily conserved mechanism of nuclear degradation in the cytoplasm. Autophagy. 2016;12(11):1973–1983. doi: 10.1080/15548627.2016.1217381
- Chandra S, Mannino PJ, Thaller DJ, et al. Atg39 selectively captures inner nuclear membrane into lumenal vesicles for delivery to the autophagosome. J Cell Bio. 2021;220(12): doi: 10.1083/jcb.202103030
- Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67(6):1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b
- Holm IE, Englund E, Mackenzie IRA, et al. A reassessment of the neuropathology of frontotemporal dementia linked to chromosome 3. J Neuropathol Exp Neurol. 2007;66(10):884–891. doi: 10.1097/nen.0b013e3181567f02
- Deng X, Sun X, Yue W, et al. CHMP2B regulates TDP-43 phosphorylation and cytotoxicity independent of autophagy via CK1. J Cell Bio. 2022;221(1): doi: 10.1083/jcb.202103033
- Liu G, Coyne AN, Pei F, et al. Endocytosis regulates TDP-43 toxicity and turnover. Nat Commun. 2017;8(1):2092. doi: 10.1038/s41467-017-02017-x
- Lee SM, Asress S, Hales CM, et al. TDP-43 cytoplasmic inclusion formation is disrupted in C9orf72-associated amyotrophic lateral sclerosis/frontotemporal lobar degeneration. Brain Commun. 2019;1(1):fcz014. doi: 10.1093/braincomms/fcz014
- Nana AL, Sidhu M, Gaus SE, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137(1):27–46. doi: 10.1007/s00401-018-1942-8
- Rothstein JD, Warlick C, Coyne AN. Highly variable molecular signatures of TDP-43 loss of function are associated with nuclear pore complex injury in a population study of sporadic ALS patient iPsns. bioRxiv. 2023;571299.
- Baughn MW, Melamed Z, López-Erauskin J, et al. Mechanism of STMN2 cryptic splice-polyadenylation and its correction for TDP-43 proteinopathies. Science. 2023;379(6637):1140–1149. doi: 10.1126/science.abq5622
- Melamed Z, López-Erauskin J, Baughn MW, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22(2):180–190. doi: 10.1038/s41593-018-0293-z
- Ma XR, Prudencio M, Koike Y, et al. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature. 2022;603(7899):124–130. doi: 10.1038/s41586-022-04424-7
- Zhang Y, Schmid B, Nikolaisen NK, et al. Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep. 2017;8(3):648–658. doi: 10.1016/j.stemcr.2017.01.012
- West RJH, Ugbode C, Fort-Aznar L, et al. Neuroprotective activity of ursodeoxycholic acid in CHMP2B(Intron5) models of frontotemporal dementia. Neurobiol Dis. 2020;144:105047. doi: 10.1016/j.nbd.2020.105047
- Chen Y, Krishnan G, Parsi S, et al. The enhanced association between mutant CHMP2B and spastin is a novel pathological link between frontotemporal dementia and hereditary spastic paraplegias. Acta Neuropathol Commun. 2022;10(1):169. doi: 10.1186/s40478-022-01476-8