
ABSTRACT
Carbon monoxide poisoning has been documented in literature to cause severe neurological and tissue toxicity within the body. However, cardiotoxicity is often overlooked, but not uncommon. Previous research studies and case reports have revealed a significant relationship between carbon monoxide intoxication and myocardial ischemic events. We report a case of a 48-year-old male, who was exposed to severe smoke inhalation due to a house fire and subsequently developed a non-ST elevation myocardial infarction. Ischemic changes were evident on electrocardiogram, which demonstrated T-wave inversion in lead III and ST-segment depression in leads V4-V6. Elevated cardiac enzymes were also present. After standard treatment for an acute cardiac event, the patient fully recovered. This case demonstrates that myocardial ischemic changes due to carbon monoxide poisoning may be reversible if recognized in early stages and treated appropriately, thus reminding physicians that a proper cardiovascular examination and diagnostic testing should be performed on all patients with carbon monoxide poisoning.
Abbreviations: NSTEMI: Non-ST elevation myocardial infarction
1. Introduction
Carbon monoxide (CO) intoxication causes impaired oxygen delivery and utilization at the cellular level, causing tissue hypoxia at different sites within the body, while having profound impact on high-oxygen demand organs, and causing changes at a neurological level. As a result of the high affinity of CO to hemoglobin, even small concentrations of CO can result in significant levels of carboxyhemoglobin (HbCO). As CO binds to cardiac myoglobin with an even greater affinity than to hemoglobin, myocardial depression and hypotension can further exacerbate the tissue hypoxia, leading to myocardial ischemia and damage [Citation1,Citation2]. The following case report demonstrates the significant clinical relationship between acute CO poisoning and myocardial ischemic damage presenting as a non-ST elevation myocardial infarction (NSTEMI) in a patient who was exposed to sever smoke inhalation in a house fire. Post-treatment, the cardiac abnormality fully resolved.
2. Case presentation
A 48-year-old male was found outside his apartment building in acute respiratory distress, leaning against a fence, after escaping from a fire that was taking place in a neighboring apartment. The patient suffered severe smoke inhalation and cuts on his legs from shattered glass but denied any chest pain or numbness of extremities. He was immediately taken by ambulance to a local hospital for first aid management and treatment. Patient has a history of hypertension, no coronary artery disease, and no history of smoking. On arrival (Day 1), patient was placed immediately on a non-rebreather mask for high-concentration oxygen delivery at 100%. Vital signs showed a heart rate (HR) of 98 beats/min, blood pressure (BP) of 129/78 mmHg, and a respiratory rate (RR) of 19 breaths/min. The initial venous blood gas (VBG) revealed COHb of 12.5%, pH 7.32, pCO2 44.7 mmHg, pO2 76.3 mmHg, SaO2 100%, and HCO3- 22 mmol/L. Basic biochemistry data showed glucose 110 mg/dL, BUN 14 mg/dL, Cr 1.2 mg/dL, Na 139 mmol/L, K 4.2 mmol/L, Cl 103 mEq/L, CO2 23 mEq/L, Ca 9.8 mmol/L, and Troponin I of 0.039 ng/mL with normal lipid profile. Electrocardiogram (ECG) revealed sinus rhythm with ST-wave depression in leads V4-V6 which was similar to EKG done by EMS and there was no previous EKG to compare. At the time, despite the patient being stable, conscious, and denying any chest pain, he was kept on continuous high-flow oxygen delivery, and monitored on the medical floor. Later in the evening, a second set of cardiac enzymes was drawn, revealing an elevated Troponin I of 3.06 ng/mL. EKG was repeated and showed T-wave inversion in lead III, mild left ventricular hypertrophy (LVH), and no ST-T wave segment changes as compared to earlier EKG. On physical exam patient was stable, alert, awake, oriented, afebrile, but complained of progressive left-sided stabbing type chest pain 9/10 that was non-radiating. He was loaded with oral aspirin 325 mg, Plavix 75 mg, lisinopril, atorvastatin 80 mg and enoxaparin 1 mg/kg. His initially measured serum COHb of 12.5% decreased dramatically within the first 24 hours to 0.8%.
The following day (Day 2), a third set of cardiac enzymes revealed a Troponin I of 1.68 ng/mL. Repeat ECG showed sinus rhythm, with T-wave inversion in lead III, and ST-wave depression in leads V4-V6 (). Echocardiography showed a hypercontractile left ventricle, no structural or valvular abnormalities. Patient was taken to the coronary care unit for further care.
Figure 1. EKG showing T wave inversion in lead III, ST depression in I,II and V4-V6.
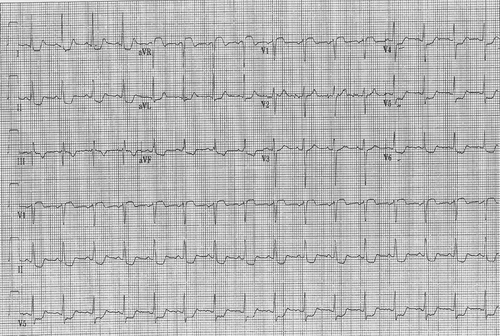
After stabilizing the patient (Day 3), a follow-up ABG showed a pO2 of 93.8 mmHg, and a downtrending of cardiac enzymes with a Troponin I of 0.416 ng/mL. Cardiac catheterization revealed normal coronaries, left ventricular ejection fraction (EF) of 59%, and a mildly elevated left ventricular end diastolic pressure (LVEDP). Because the electrocardiographic changes were attributed to CO intoxication, no thrombolytic or percutaneous coronary intervention was needed.
After continuous oxygen therapy and fluid resuscitation, the patient was transferred back to the medical floor on Day 7. A repeat ECG showed T-wave inversion in lead III, with a resolution of the ST-wave depression in leads V4-V6 that was noted on the previous EKG. The abnormal biochemistry and hemogram data returned to normal values. The patient was discharged on Day 8 with low dose aspirin and statins. On follow-up at the pulmonary clinic one month later, the patient’s general condition was found to be good, without any cognitive dysfunction or neurological deficits. Since cardiac catheterization revealed normal coronaries, the cardiologist felt that no other interventions are needed and to follow up with his primary care physician.
3. Discussion
CO is an odorless, colorless, non-irritating gas, produced by incomplete combustion of carbonaceous material. It is a leading cause of poison-related deaths in the United States (Citation3–Citation5). Its high affinity for hemoglobin is 210 times that of oxygen, thus interfering with oxygen release and delivery to cells due to a shift in the oxygen-hemoglobin dissociation curve and possibly impairing electron transport, as a result of the reversible inhibition of mitochondrial respiration and oxidative stress which results in tissue inflammation and hypoxia [Citation1,Citation3,Citation6]. A wide array of acute symptoms has been documented, ranging from mild viral-like symptoms to more severe respiratory depression, cerebral edema, and fatal arrhythmias [Citation7].
As CO binds to cardiac myoglobin with an even greater affinity than to hemoglobin, myocardial depression and hypotension can further exacerbate the tissue hypoxia, leading to myocardial ischemia and damage [Citation8]. As a result, moderate to severe CO intoxication can result in myocardial injury and present an increased risk of mortality [Citation9]. Therefore, a decrease in oxygen transport capacity of the blood leads to a decreased amount of oxygen delivery to the tissues, resulting in myocardial ischemia and damage, even in patients with normal coronary arteries [Citation10,Citation11], which was the case with our patient who had no cardiac risk factors with a normal coronary angiogram. Even though the main mechanism by which CO causes tissue damage is via production of hypoxia, effects of CO are more profound in the myocardium than in peripheral tissues because of very high oxygen extraction by the myocardium at rest [Citation12]. CO may also have direct myocardial effects. Chen demonstrated that in isolated rat hearts, CO caused a greater decrease in heart rate and pulse pressure compared to the same degree of anoxia produced by the inhalation of nitrogen [Citation13]. The role of CO in platelet aggregation is controversial, but some studies have indicated increased platelet activation and aggregation [Citation14] in animals.
With regards to treatment of CO poisoning with hyperbaric oxygen therapy (HBOT), it has been found to be beneficial in reducing the risk of cognitive deficits [Citation15]. Hyperbaric oxygen (HBO2) hastens carboxyhemoglobin (COHb) elimination and favorably modulates inflammatory processes instigated by CO poisoning, an effect not observed with breathing normobaric oxygen. Hyperbaric oxygen improves mitochondrial function, inhibits lipid peroxidation transiently, impairs leukocyte adhesion to injured microvasculature, and reduces brain inflammation caused by the CO-induced adduct formation of myelin basic protein [Citation16]. Animal studies demonstrate HBOT to reduce myocardial infarct size in ischemic rabbit hearts when hyperbaric oxygen is administered at reperfusion [Citation5]. Other investigators have shown no beneficial effect for HBOT on infarct size in dogs [Citation17]. However, the effects of HBOT in human myocardial ischemia are still poorly understood and needs further investigation. Based on limited data, the indications for HBOT include moderate to severe CO poisoning [Citation18,Citation19]. However, in our case report, the patient’s presenting symptoms of respiratory distress were significantly improved with high-concentration oxygen under normal pressure. His initially measured serum COHb of 12.5% decreased dramatically within the first 24 hours to 0.8%. Therefore, HBOT was not administered to the patient, and his symptoms and cardiac function fully recovered. Though with his clinical presentation and EKG changes as described above with high troponins he was given aspirin, clopidogrel, statins, lisinopril and enoxaparin.
In conclusion, considering the potential mediating role of CO intoxication in tissue hypoxia, it is of extreme importance to examine the influence of CO poisoning in myocardial ischemia and injury. Research studies by Henry & Satran, et al. (2006) have investigated and linked the role and degree of CO intoxication as being both a predictor of myocardial injury and long-term mortality and as an impediment to successful recovery from tissue damage imposed by the body’s hypoxic state. This case further supports and demonstrates the significant relationship between CO poisoning and myocardial ischemic injury mimicking a NSTEMI, which may be fully reversible with a timely diagnosis and proper treatment. It is therefore, fundamental to do a full cardiac work-up on patients exposed to CO poisoning to prevent permanent myocardial injury, and decrease the risk of long-term mortality.
Disclosure statement
The authors alone are responsible for the content and writing of the paper. The authors have no disclosures or conflict of interest to report. This study did not receive any research funding. This study has not been submitted or pending publication in any other journal.
References
- Hardy KR, Thom SR. Pathophysiology and treatment of carbon monoxide poisoning. J Toxicol Clin Toxico. 1994;32(6):613–629.
- Szponar J, Kolodziej MMajewska M, et al. Myocardial injury in the course of carbon monoxide poisoning. Przegl Lek 2012; 69:528–534.
- Ganong WF. Review of medical physiology. Norwalk (CT): Appleton & Lange; 1995.
- Carbon Monoxide Exposures — United States August 5. 2011 / 60(30). p. 1014–1017.
- Sterling DL, Thornton JD, Swafford A, et al. Hyperbaric oxygen limits infarct size in ischemic rabbit myocardium in vivo. Circulation. 1993;12:1931–1936.
- Weaver LK. Clinical practice. Carbon monoxide poisoning. N Engl J Med. 2009;360:1217–1225.
- Tintinalli JE, Kelen GD, Stapczynski JS. Carbon monoxide poisoning. In: Tintinalli JE, Kelen GD, Stapczynski JS, editors. Emergency medicine: a comprehensive study guide. 5th ed. McGraw-Hill; 2000. p. 1302–1306.
- Raub JA, Mathieu-Nolf M, Hampson NB, et al. Carbon monoxide poisoning - a public health perspective. Toxicology. 2000;145:1–14.
- Henry CR, Satran D, Lindgren B, et al. Myocardial injury and long-term mortality following moderate to severe carbon monoxide poisoning. JAMA. 2006;295:398–402.
- Marius-Nunez AL. Myocardial infarction with normal coronary arteries after acute exposure to carbon monoxide. Chest. 1990;97:491–494.
- Lee D, Hsu TL, Chen CH, et al. Myocardial infarction with normal coronary arteries after acute exposure to carbon monoxide exposure: a case report. Chin Med J-Taipei. 1996;57:355–359.
- Ayres SM. Myocardial and systemic responses to carboxyhemoglobin. Ann NY Acad Sci. 1970;174:268–293.
- Chen KC. Response of the isolated heart to carbon monoxide and nitrogen anoxia. Toxicol Appl Pharmacol. 1985;81:363–370.
- Gering SA. Exacerbation of acute platelet thrombus formation in stenosed dog coronary arteries with smoke from a non-tobacco-burning cigarette. J Lab Clin Med. 1990;116:728–736.
- Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347:1057–1067.
- Weaver LK. Hyperbaric oxygen therapy for carbon monoxide poisoning. Undersea Hyperb Med. 2014 Jul-Aug;41(4):339–354.
- Mogelson S, Davidson J, Sobel BE, et al. The effect of hyperbaric oxygen on infarct size in the conscious animal. Eur J Cardiol. 1980;12:135–136.
- Ernst A, Zibrak JD. Carbon monoxide poisoning. N Engl J Med. 1998;339:1603–1608.
- Satran D, Henry CR, Adkinson C, et al. Cardiovascular manifestations of moderate to severe carbon monoxide poisoning. J Am Coll Cardiol. 2005;45:1513–1516.