
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Extracellular vesicles (EVs) are mediators of cell communication during health and disease, and abundantly released by platelets upon activation or during ageing. Platelet EVs exert modulatory effects on immune and vascular cells. Platelet EVs may modulate the function of vascular smooth muscle cells (SMC). Platelet EVs were isolated from platelet-rich plasma and incubated with SMC in order to assess binding, proliferation, migration and pro-inflammatory phenotype of the cells. Platelet EVs firmly bound to resting SMC through the platelet integrin αIIbβ3, while binding also occurred in a CX3CL1–CX3CR1-dependent manner after cytokine stimulation. Platelet EVs increased SMC migration comparable to platelet derived growth factor or platelet factor 4 and induced SMC proliferation, which relied on CD40- and P-selectin interactions. Flow-resistant monocyte adhesion to platelet EV-treated SMC was increased compared with resting SMC. Again, this adhesion depended on integrin αIIbβ3 and P-selectin, and to a lesser extent on CD40 and CX3CR1. Treatment of SMC with platelet EVs induced interleukin 6 secretion. Finally, platelet EVs induced a synthetic SMC morphology and decreased calponin expression. Collectively, these data indicate that platelet EVs exert a strong immunomodulatory activity on SMC. In particular, platelet EVs induce a switch towards a pro-inflammatory phenotype, stimulating vascular remodelling.
Responsible Editor Suresh Mathivanan, La Trobe University, Australia
Introduction
Besides their pivotal role in haemostasis and thrombosis, platelets have been recognised as immune cells, modulating inflammatory reactions and immune responses [Citation1,Citation2]. Platelets firmly adhere to the inflamed vessel wall through interactions of glycoprotein Ib (GPIb) or integrin αIIbβ3 with van Willebrand factor (VWF) or fibrinogen bound to the endothelium [Citation3]. Platelets furthermore interact with leukocytes initiated by P-selectin-PSGL-1 interactions and reinforced by integrins [Citation4]. Platelets also propagate the inflammatory response by engagement of CD40 and its ligand CD154 (CD40L), mediating the secretion of cytokines in leukocytes and endothelial cells [Citation5,Citation6]. Platelets furthermore facilitate the interaction of leukocytes to exposed smooth muscle cells (SMC) of the denuded vessel wall via the CX3CL1-CX3CR1 axis [Citation7]. Through the deposition of chemokines such as CCL5 and CXCL4 onto the vessel wall, platelets promote the recruitment of leukocytes to sites of inflammation [Citation8].
In addition to chemokines, platelets release extracellular vesicles (EVs) upon activation and during storage [Citation9]. In the blood plasma, platelet-derived EVs (platelet EVs) constitute the major fraction (70–80%) of EVs. The platelet EVs are highly accumulated during the prolonged storage of platelet concentrates destined for transfusion. These platelet EVs retain several properties of their parent cells, as they carry a similar but not identical repertoire of surface molecules. Evidence is accumulating that platelet EVs can confer inflammatory signals to a variety of vascular and immune cells [Citation10]. Thus, platelet EV binding to injured endothelium or subendothelial matrix triggers the recruitment of activated platelets and increases their thrombogenicity [Citation11]. Similar to platelets, platelet EVs deposit CCL5 and CXCL4 onto the endothelial surface [Citation12]. Also outside the blood stream, e.g. in synoviocytes of inflamed joints, platelet EVs induce the expression of pro-inflammatory cytokines and chemokines, i.e. IL-6, CXCL1 and CXCL8, thereby contributing to rheumatoid arthritis [Citation13]. Platelet EVs can also transfer the chemokine receptor CXCR4 to angiogenic early outgrowth cells, inducing the release of pro-angiogenic factors and increase their vasoregenerative potential [Citation14]. Work from our group and others has demonstrated that naturally released platelet EVs induce a phenotypic switch in monocytes directing these towards a phagocytic phenotype [Citation15,Citation16]. These interactions of platelet EVs are of possible (patho)physiological relevance, as elevated levels of platelet EVs have been associated with vascular pathologies such as atherosclerosis along with diseases, e.g. diabetes mellitus, hypertension, hyperlipidaemia and hypercholesterolaemia [Citation11,Citation17–Citation21].
In the vessel wall, SMC essentially contribute to the pro-inflammatory environment following vessel injury and remodelling [Citation22]. It is well established that phenotypic switching of SMC drives the progression of vascular diseases such as atherosclerosis and restenosis. In a healthy artery, the medial SMC express high levels of contractile proteins such as α-smooth muscle actin (αSMA), calponin, SM22α and other differentiation markers (reviewed in [Citation23]), which is accompanied by a low rate of proliferation. In contrast, during vascular injury or disease, the SMC modulate their phenotype and support vascular remodelling by downregulating differentiation markers, and by increasing proliferation, migration, and producing matrix proteins and cytokines. The knowledge that platelet EVs are potent modulators of immune cells has prompted us to investigate the interaction of naturally released platelet EVs with SMC, and the consequences thereof. We therefore hypothesised that platelet EV induce a pro-inflammatory phenotype in SMC, by modulating their functions with implications for vascular remodelling.
Materials and methods
Reagents and cells
The antibodies that were used in this study are listed in . PDGF-BB, 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE), heparinase III (hep III) and chondroitinase ABC were obtained from Sigma (St. Louis, MO). CCL5 and the CX3CR1 inhibitor (F1-Fk) were produced recombinantly as described [Citation7,Citation24]. CD40L and tumour necrosis factor α (TNFα) were purchased from Peprotech (Rocky Hill, NJ) and CXCL4 was ordered from ChromaTec GmbH (Greifswald, Germany). P-selectin inhibitor was from Bio-Techne (Minneapolis, MN). Syto 13 was from Thermofisher Scientific (Waltham, MA). Unfractionated heparin, human albumin and eptifibatide were purchased from the MUMC+ hospital pharmacy. Bovine endothelial cell growth factor (ECGF) was ordered from ReliaTech. All other reagents were obtained from Sigma.
Table 1. Antibody specifications.
Human SMC were isolated from coronary arteries [Citation25] and cultured at passages 5–9 at 37°C in smooth muscle cell culture medium, VascuLife (Cell Systems GmbH, Troisdorf, Germany) supplemented with 5% foetal bovine serum, 5 ng/ml fibroblast growth factor, 5 µg/ml insulin, 50 µg/ml ascorbic acid, 10 mM l-glutamine and 5 ng/ml epidermal growth factor and 1% penicillin and streptomycin. Human THP-1 cells (DSMZ, ACC-16) were cultured at 37°C in RPMI medium supplemented with 10% FCS and 1% penicillin and streptomycin. Human umbilical vein endothelial cells (HUVEC, Promocell, C-12203) were cultured at passages 2–8 at 37°C in endothelial cell growth medium constituted with endothelial supplement mix and 1% penicillin and streptomycin. Human glioma cells (ATCC, HTB-14) were cultured in Eagle’s minimum essential medium (EMEM) containing 10% FCS and 1% penicillin and streptomycin. Platelet concentrates were obtained from Sanquin blood services (Amsterdam, the Netherlands). Each concentrate (250–300 ml) consisted of randomly pooled leucocyte-reduced buffy coat-derived platelets (300–370 × 109/concentrate) from five healthy donors, resuspended in plasma from one donor. Platelet concentrates were stored in CompoFlex storage bags from Fresenius Kabi (Bad Homburg, Germany) for 8 days or longer at room temperature (RT).
Platelet EV isolation, quantification and characterisation
Platelet EVs were isolated from expired and anonymised human platelet concentrates by differential centrifugation steps. First, platelets were removed by centrifugation at 4000g for 5 min at RT. The platelet-free plasma preparations were centrifuged at 20,000g for 1 h at 16°C. Platelet EV pellets were resuspended in Hepes buffer pH 7.45 (136 mM NaCl, 10 mM Hepes, 2.7 mM KCl, 2 mM MgCl2, 0.1% bovine serum albumin and 0.1% glucose). Impurities of residual platelets, platelet EV aggregates and other contaminants were removed from the resuspended pellet by filtration (0.8 µm pore size). The filtered platelet EV suspensions were centrifuged again at 20,000g for 40 min at 16°C. Pellets with purified platelet EVs were resuspended in Hepes buffer pH 7.45. This protocol has been optimised and standardised according to the recommendations by Witwer et al. [Citation26]. EVs from glioma cells, which abundantly release EVs, were isolated by the same procedure and used as non-specific control. As a further control, platelet EVs were isolated by size exclusion chromatography (SEC) using Sepharose CL-2B (GE healthcare) according to the protocol of Böing et al. [Citation27] with some adaptations. 15 ml of the platelet concentrate were centrifuged at 4000g for 5 min at RT to remove the platelets. The platelet-free plasma preparations were concentrated with a 100 kDa Amicon® Ultra concentrator (Merck) by centrifugation at 5000g for 30 min at RT. 1.5 ml (3× 500 µl) of the concentrated platelet-free plasma were loaded on the column, followed by elution by PBS containing 0.32% citrate (pH 7.4, 0.22 µm filtered). The eluate was collected in 25 sequential fractions. Both, platelet EVs isolated by centrifugation and SEC were snap frozen in liquid nitrogen and stored at −80°C until use. Prior to use, platelet EVs were thawed at RT.
Flow cytometry
Platelet EVs were characterised and quantified by flow cytometry (BD Accuri) and the FCS Express 4 software (De Novo Software, Glendale, CA). Platelet EVs were stained with 1 µg/ml annexin A5-FITC (Annexin Pharmaceuticals) and 1.25 µg/ml mouse monoclonal anti-human CD41a-Ab-PE (eBioscience) in Hepes buffer containing 0.5% HSA and 5 mM CaCl2 for 30 min protected from light. As a negative control, platelet EVs were stained with 1 µg/ml annexin A5-FITC and 1.25 µg/ml mouse IgG-PE isotype control (eBioscience) in Hepes buffer containing 0.5% HSA and 2 mM EDTA. Single staining controls were prepared accordingly for fluorescence compensation. Platelet EVs were characterised as single events positive for FITC-conjugated annexin A5 and PE-conjugated anti-CD41a Ab. Platelet EVs were quantified by counting beads (AccuCount, Spherotech). All samples were recorded at a threshold of 104 on FSC. First, water and beads (200 beads/µl) were recorded and the background noise and the beads were gated morphologically. Next, platelet EV suspensions in presence of 200 beads/µl were recorded with limit set on bead gate. Platelet EV isotype control and platelet EVs positive for annexin A5 (phosphatidylserine exposure) and CD41a were quantified by dot plots. Doublet correction was performed by analysing width over annexin A5-FITC-area. Platelet EV concentrations were calculated by the following equation:
Bead-coupled flow cytometry was performed as described previously [Citation25], with some adaptations. 3.5 × 108 beads/ml (4 µM aldehyde/sulfate latex beads 5% (w/v); Molecular Probes Life Technologies, Waltham, MA, USA) were coated with 0.125 mg/ml unlabelled anti-CD63, anti-CD81 or anti-CD9 antibody by overnight incubation in MES buffer. These beads were stored in PBS containing 0.1% (m/v) glycine and 0.1% (m/v) sodium azide at 4⁰C until use. All vesicle suspensions were diluted to 1 × 108 EV/ml and incubated with 2 × 105 of the different beads overnight. Beads were washed twice with PBS containing 2% (m/v) BSA and resuspended in 50 µl PBS-2% (m/v) BSA containing 0.01 mg/ml of the respective PE-labeled antibodies and incubated for 1 h. Following incubation, beads were washed twice with PBS – 2% (m/v) BSA, resuspended in PBS and analysed by flow cytometry using a BD FACSCanto (BD Biosciences) with FACS Diva V8.0.1 software (BD Biosciences). All incubations were carried out at RT and with vigorous shaking (1000 min−1). The quantity of EVs in relative fluorescent units (RFU) was calculated by multiplying the percentage of PE-positive beads with the median fluorescent intensity (MFI) of the positive bead population.
Size distribution analysis
Vesicle size distribution was determined by nanoparticle tracking analysis (NTA) using a Malvern NanoSight NS300 (Malvern Technologies, Malvern, UK) with a high-sensitivity sCMOS camera and a 488 nm laser [Citation28]. Platelet EV suspensions were diluted 1:200 in particle-free PBS (Gibco, Waltham, MA, USA) to an acceptable concentration according to manufacturer’s instructions. Samples were analysed under constant flow conditions (flow rate = 50) at 25°C. Fifteen 60 s videos were captured with a camera level of 16. Data were analysed using NTA 3.1.54 software with a detection threshold of 5. Alternatively, tunable resistive pulse sensing (TRPS) was performed using a qNano Gold with Izon Control Suite 3.2 Software and SKP400 calibration beads and an NP300 nanopore (Izon, Christchurch, New Zealand). Platelet EV suspensions were analysed after a single freeze–thaw cycle at −80°C. The NP300 nanopore was used at a stretch of approximately 45 mm and a pressure of 12 mbar, keeping the current between 120 and 130 nA for all measurements. Recordings were stopped after detection of 500 blockades. A concentration fraction from 140 to 600 nm was applied prior to determining the particle concentration.
Cryo-transmission electron microscopy (cryo-TEM)
The preparations of platelet EVs were visualised by the cryo-TEM method. A thin aqueous film was formed by applying a 5 µl droplet of the suspension to a bare specimen grid. Glow-discharged holey carbon grids were used. After the application of the suspension the grid was blotted against filter paper, leaving thin sample film spanning the grid holes. These films were vitrified by plunging the grid into ethane, which was kept at its melting point by liquid nitrogen, using a Vitrobot (FEI Company, Eindhoven, Netherlands) and keeping the sample before freezing at 95% humidity. The vitreous sample films were transferred to a microscope Tecnai T12 Spirit (FEI Company, Eindhoven, Netherlands) using a Gatan cryotransfer. The images were taken at 120 kV with a 4096×4096 pixel CCD Eagle camera (FEI Company, Eindhoven, Netherlands) at a temperature between −170ºC and −175ºC and using low-dose imaging conditions.
Proteomic analysis
Platelet EV suspensions (100 µl) were incubated with 300 µl of 10% trichloroacetic acid (TCA)/20 mM DTT in ice-cold acetone overnight at −20ºC, centrifuged, washed with ice cold aceton, recentrifuged, air-dried and resuspended in 50 µl 50 mM ammonium bicarbonate (ABC) with 5 M urea. To a total of 125 µg protein in 50 µl ABC/urea, DTT (20 mM) was added and incubated at RT for 45 min; iodoacetamide (40 mM) was subsequently added to alkylate for a further 45 min at RT until quenching by a second addition of DTT. The digestion was initiated by the addition of trypsin/lysC (2 µg) mix and incubation at 37ºC for 2 h. The mixture was diluted with ABC without urea and incubation was continued at 37ºC for 18 h. The digestion mix was briefly centrifuged and the digest/peptide mixture was diluted fourfold for LCMS analysis. A nanoflow HPLC instrument (Dionex ultimate 300) was coupled in-line with a Q Exactive (Thermo Scientific) with a nano-electrospray Flex ion source (Proxeon). Of the digest/peptide mixture, 5 μl was loaded onto a C18-reversed phase column (Thermo Scientific Acclaim PepMap C18 column, 75 μm inner diameter × 15 cm, 5 μm particle size). The peptides were separated with a 90 min linear gradient of 4–45% buffer B (80% acetonitrile and 0.08% formic acid) at a flow rate of 300 nl/min.
Mass spectrometry data were acquired using a data-dependent top10 method, dynamically choosing the most abundant precursor ions from the survey scan (250–1250 m/z) in positive mode. Survey scans were acquired at a resolution of 70,000. Dynamic exclusion duration was 30 s. Isolation of precursors was performed with a 4.0 m/z window. Resolution for HCD spectra was set to 17,500 and the Normalised collision energy was 30 eV. The under fill ratio was defined as 1.0%. The instrument was run with peptide recognition mode enabled, but exclusion of singly charged and charge states of more than five.
Database search
The data were analysed using Sequest HT Proteome Discoverer 1.4 search engine (Thermo Scientific), against the UniProt database. The false discovery rate (FDR) was set to 0.01 for proteins and peptides, which had to have a minimum length of 6 amino acids. The precursor mass tolerance was set at 10 ppm and the fragment tolerance at 0.2 Da. One miss-cleavage was tolerated, oxidation of methionine was set as a dynamic modification and carbamidomethylation of cysteines was a fixed modification. Biological pathway analysis was performed by Functional Enrichment analysis tool (FunRich) using the UniProt database [Citation29].
Analysis of platelet EV interactions with SMC
Platelet EVs (2 × 108/ml) were labelled with 10 µM CFSE and pre-incubated with F1-Fk (10 µg/ml), eptifibatide (50 µg/ml), P-selectin inhibitor, anti-CD40- and anti-CD40L blocking antibodies (all at 20 µg/ml) for 30 min at RT. Pre-incubated labelled platelet EVs were co-incubated with SMC for 40 min at RT. Control incubations were performed by co-incubation of CFSE-labelled platelet EVs and SMC in the presence of EDTA. Non-bound platelet EVs were removed by centrifugation and washing. The binding and the effects of blocking agents of platelet EV to SMC were presented as median SMC-fluorescein intensity compared to CFSE-platelet EVs in presence of EDTA and CFSE-platelet EVs, respectively (FCS Express 4).
Cell migration assay
A 12-well chemotaxis chamber with a 8 µm polycarbonate membrane (Neuroprobe, Gaithersburg, MD) was used to assess the migration of SMC or HUVEC (as additional primary cell type control) towards a chemoattractant. The factors PDGF, CCL5, CD40L (all at 50 ng/ml), CXCL4 (1 µg/ml) and ECGF (200 µg/ml) in combination with 2 × 108/ml platelet EVs or SEC-platelet EVs were added into the lower wells, and SMC (2.5 × 104 cells) were seeded in the upper wells. Chemotaxis induced by PDGF was inhibited by 2.5 µg/ml PDGF-BB neutralising antibody. CXCL4- and platelet EV-triggered chemotaxis was inhibited by heparin (10 µg/ml) or by preincubation of SMC with heparinase III (1 U/ml) or chondroitinase ABC (0.5 U/ml) for 1 h at 37°C. After incubation of 4 h at 37°C, the non-migrated cells were carefully removed and the membrane was stained with Diff-Quick stain (Eberhard Lehmann GmbH, Berlin, Germany). Migrated cells were visualised by light microscopy, counted manually in three fields and expressed as cells/mm2.
Cell proliferation assay
SMC proliferation was measured by the BrdU assay kit (Merck, Darmstadt, Germany). 96-well plates were pre-coated with collagen (40 µg/ml) for 30 min at 37°C and 2.5 × 104 cells/ml were seeded. SMC were left for attachment (3 h at 37°C). Growth medium was then replaced by RPMI-1640 medium supplemented with 5% foetal calf serum. Cells were stained with BrdU label, and incubated with control or platelet EV (7.5 × 106/ml) in the presence or absence of P-selectin inhibitor and anti-CD40 blocking antibody (both at 10 µg/ml). After 48 h incubation at 37°C, BrdU staining was analysed following the manufacturer’s protocol.
Flow chamber adhesion assay
For flow chamber adhesion, SMC or HUVEC (as additional primary cell type control) were seeded in 35-mm, pre-coated collagen (30 µg/ml) dishes and activated with TNFα and CD40L (both 10 ng/ml) for 4 h or platelet EV (7.5 × 106/ml) for 72 h, respectively. THP-1 cells were labelled with 1 µM green fluorescent nucleic acid stain (Syto 13) for 30 min at 37°C, washed and perfused in Hank’s buffer pH 7.45, containing 10 mM Hepes, 3 mM CaCl2, 2 mM MgCl2 and 0.2% human albumin for 2–6 min at 3 dynes/cm2. Adherent fluorescent cells were manually counted in >6 fields and expressed as cells/mm2.
Immunocytochemistry
Expression of contractile proteins of SMC was assessed by immunofluorescence microscopy. SMC were seeded on 8-well culture slides pre-coated with collagen (30 µg/ml). SMC were incubated with control, 7.5 × 106/ml platelet EVs, PDGF or CD40L at 20 ng/ml or CXCL4 (1 µg/ml) for 96 h. Following incubation, SMC were fixed in 4% paraformaldehyde for 10 min at 37°C and washed with PBS. Coverslips were blocked in PBS with 2% bovine serum albumin and 0.1% Triton X-100 for 1 h at RT. Cells were washed with PBS and incubated with primary antibodies (anti-α smooth muscle actin or anti-calponin mAb) overnight at 4°C. Culture slides were washed with PBS and incubated with secondary FITC-conjugated antibodies for 1 h at RT. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Coverslips incubated with the secondary antibody only served as negative controls. Cells from at least six micrographs were manually outlined and surface area was determined using ImageJ v1.8 (NIH). Corrected total cell fluorescence (CTCF) was calculated as described elsewhere [Citation30].
Enzyme linked-immunosorbent-assay (ELISA)
The interleukin 6 (IL-6) ELISA kit was purchased from eBioscience (San Diego, CA). The CCL5 and CXCL4 ELISA kits were from Bio-Techne. Cell culture supernatants and platelet EV suspensions in Hepes buffer were measured by ELISA according to the manufacturer’s instructions. Chemokine amounts were expressed as mean±SD.
Statistical analyses
Statistical analysis was performed using Prism 6.0. Data are presented as means ± SEM and were compared by one-way analysis of variance (ANOVA) with Tukey or Sidak’s post-test. Differences with p < 0.05 were considered as statistically significant. Each experiment was independently repeated at least three or more times, as indicated for each experiment in the figure legends.
Results
Platelet EVs interact with resting and activated SMC
Platelet EV suspensions were obtained from platelet concentrates and quantified as separate, double positive events for annexin A5 (phosphatidylserine-exposing) and CD41a (74.2%) (). NTA revealed an average diameter of 153.1 ± 0.5 nm ()). Platelet EV suspensions contained heterogeneously-sized populations of particles. Cryo-TEM revealed different sizes and structural types of platelet EVs. Both the smaller and larger platelet EVs are membrane enclosed and have electron-dense structures ()). Platelet EV suspensions did not contain exosome markers CD63 and CD81, but they did express CD9 ()).
Figure 1. Characterisation and quantification of platelet EVs. SSC-H over FSC-H colour dot plots: water and 200 beads/µl were recorded. Background noise (light blue) and bead gate (red) was set (a). Platelet EV suspensions without or with CaCl2 were recorded with a limit set on bead gate (b). Colour dot plots of platelet EV isotype control (blue) (c) and annexin A5/CD41a positive platelet EVs (green) (d). Size distribution of platelet EV suspensions measured by advanced nanoparticle tracking analysis (n = 10) (e).
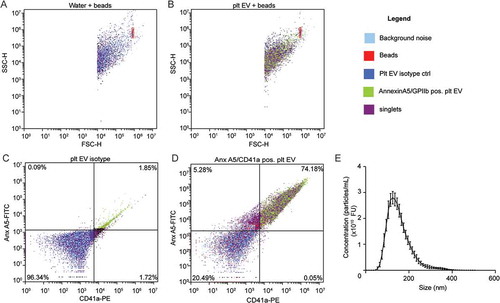
Figure 2. Cryo-transmission electron microscopy and bead coupled flow cytometry of platelet EVs. Representative micrographs from cryo-TEM of platelet EV suspensions. Scale bar 200 nm (a). Representative scatterplots and quantification of bead coupled flow cytometry for CD9 (b), CD63 (c) and CD81 (d) (n = 3).
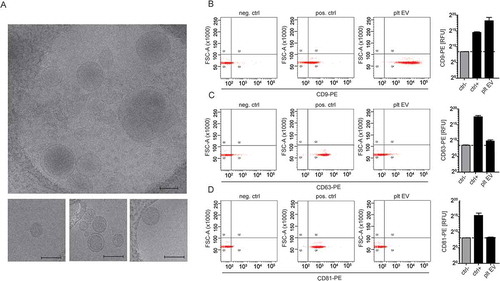
The ability of these platelet EVs to bind to resting SMC was assessed by flow cytometry. Therefore, the platelet EVs were labelled with CFSE, and the calcium- and magnesium-dependent binding to SMC was examined by flow cytometry and expressed as median fluorescence intensity. SMC co-incubated with platelet EVs in presence of EDTA was used as a negative control ()). With unstimulated SMC, platelet EVs firmly bind to the cells as depicted by an increase in median fluorescence intensity. This interaction was dependent on integrin αIIbβ3, as the fluorescence intensity was attenuated in the presence of the αIIbβ3 inhibitor eptifibatide ()). P-selectin appeared not to be involved in the binding of platelet EVs to SMC, neither was CD40L ()) nor CD40 (data not shown). After stimulation of SMC with TNFα for 4 h, platelet EVs similarly interacted with the cells through integrin αIIbβ3. Interestingly, blocking the CX3CL1 receptor CX3CR1 with F1-Fk attenuated the SMC–platelet EV interaction ()). Thus, the CX3CR1–CX3CL1 axis can additionally support this interaction under inflammatory conditions.
Figure 3. Platelet EVs interact with resting or TNFα stimulated SMC via platelet integrin αIIbβ3 or CX3CR1. CFSE-platelet EVs bound to SMC were measured by flow cytometry. Representative histograms of CFSE- platelet EVs bound to resting SMC in the presence of EDTA (filled histogram) or CaCl2 (line histogram) (a). CFSE-labelled platelet EVs (2 × 108/ml) were co-incubated with resting or TNFα-stimulated SMC in presence or absence of indicated inhibitors (eptifibatide, F1-Fk, P-selectin inhibitor) or blocking antibodies (CD40L) for 30 min at room temperature. Median fluorescence intensity of CFSE-platelet EV bound to resting (b) or TNFα-stimulated SMC (c) and platelet receptors implicated in platelet EV–SMC interaction. p-values were calculated by ANOVA with Sidak’s post-test (n = 3–7).
The EVs from platelets were found to contain CXCL4 (640 ± 31 pg/107 platelet EV, n = 3). Of relevance, CXCL4 has been recently described to stimulate pro-inflammatory responses by triggering SMC migration, proliferation, cytokine production and decline of differentiation markers [Citation31]. The platelet EVs also contained relevant amounts of CCL5 (450 ± 4.6 pg/108 platelet EV, n = 3).
Platelet EV influence the phenotype of SMC
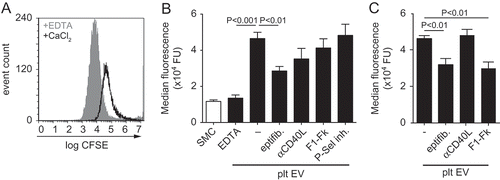
Normal healthy SMC predominantly show a contractile phenotype that is characterised by the expression of contractile differentiation markers and a spindle-shaped morphology [Citation23]. In contrast, SMC within atherosclerotic lesions lose their contractile markers, gain synthetic markers and change their morphology into a rhomboid SMC phenotype [Citation32]. The effects of platelet EV on SMC phenotype change were assessed by immunocytochemistry. SMC-specific morphology was assessed by filamentous (F)-actin-, α-smooth muscle actin- (αSMA) and calponin staining. Immunofluorescent F-actin- ()) and αSMA-staining ()) revealed a rhomboid SMC morphology and an increase in cell surface area after incubation with platelet EVs or CXCL4 compared to untreated spindle shaped SMC (). Immunofluorescent staining for calponin revealed a decreased expression after treatment with platelet EVs and PDGF, whereas CXCL4 did not induce a reduction in calponin expression ().
Figure 4. Platelet EVs change SMC morphology. Micrographs of SMC stained with antibodies against F-actin (a), αSMA (b) or calponin (c) after control, platelet EV-, PDGF- or CXCL4-stimulation for 96 h. Nuclei were stained with DAPI. Scale bars: 50 µm. Cell surface area (µm2) (d,e) and corrected total cell fluorescence (CTCF) (f) were quantified and displayed as bar graphs or box-and-whiskers plots, respectively. p-values were calculated by ANOVA with Tukey’s (d,e) or Kruskal–Wallis (f) post-tests (n = 3).
Platelet EVs induce migration and proliferation of SMC
In a proliferation assay based on BrdU incorporation, co-incubation of platelet EVs with SMC for 48 h resulted in an increase in SMC proliferation. This proliferative effect by platelet EVs was reduced by blocking P-selectin as well as CD40 ()). The potential influence of platelet EV on the SMC function was assessed in a cell migration assay. For this purpose, platelet EVs were compared to various bioactive compounds, with buffer medium or EVs isolated from glioma cells (Glioma-EV), that abundantly shed EV as negative or non-specific controls, respectively. In the presence of platelet EVs or alternatively-isolated platelet EVs by size exclusion (SEC-platelet EV), SMC showed an increased migration comparable to that induced by soluble CXCL4 or the positive control PDGF ()). While CCL5 is known to trigger the recruitment of leukocytes, recent studies have shown that SMC possess functional receptors (CCR5) for this chemokine [Citation33]. This prompted us to determine the chemotactic activity of CCL5. However, the presence of CCL5 did not affect the migration of SMC, nor did soluble CD40L ()). To investigate whether PDGF or CXCL4 derived from platelet EV were able to trigger the migration of SMC, a PDGF neutralising antibody or heparin was used, respectively. Whereas the neutralising antibody inhibited PDGF-triggered SMC migration, no effects were observed on platelet EV-triggered migration. Markedly, heparin did suppress CXCL4- and platelet EV-mediated, but not PDGF-induced SMC migration, thus indicating that CXCL4 presented on the platelet EV surface can functionally influence SMC ()). Unlike other chemokines that bind specific receptors, it is known that CXCL4 binds with high affinity to glycosaminoglycans (GAGs), particularly heparan sulfate and chondroitin sulfate. Removal of GAGs by heparinase III treatment strongly reduced the CXCL4- or platelet EV-triggered SMC migration ()), while migration towards PDGF was less affected (data not shown). Chondroitinase ABC treatment only showed minor effects on platelet EV-induced SMC migration (data not shown). To further investigate a possible role of CXCL4, platelet EV-membrane bound CXCL4 was depleted by washing in the presence of heparin. Subsequently, CXCL4-depleted platelet EV were reconstituted with CXCL4 in increasing concentrations. Reconstitution of platelet EV with CXCL4 did recover SMC migration, albeit to a maximum of about 60% of the original platelet EV activity ()). We further assessed the influence of platelet EV on the migration of endothelial cells. HUVEC showed an increased migration in presence of ECGF, platelet EV and SEC-platelet EV, compared to buffer medium or glioma-EV as negative or non-specific controls ().
Figure 5. Platelet EVs induce migration and proliferation of SMC. Proliferation of SMC was measured following platelet EV–SMC co-incubation in absence and presence of indicated blocking antibodies; n = 3 (a). SMC migration was assessed in a Boyden chamber in the presence of medium, Glioma-EV, platelet EVs, SEC-platelet EVs, PDGF, CXCL4, CCL5 or CD40L; n = 4–12 (b) or blocking antibodies (PDGF), heparin or heparinase III; n = 3–8 (c). Dose-dependent SMC migration of CXCL4 reconstituted platelet EVs (d). P-values were calculated by ANOVA with Tukey’s post-test.
Platelet EVs increase adhesiveness of monocytic cells to SMC and stimulate cytokine release
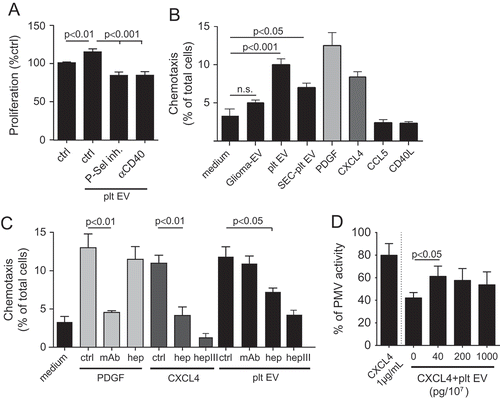
The effects of platelet EV on the adhesion of monocytic cells onto a confluent monolayer of human SMC was studied under flow conditions. Firm adhesion of monocytic cells to SMC was significantly increased by platelet EV treatment, when compared to the resting SMC (). Remarkably, the adhesion of monocytic cells to platelet EV-treated SMC was comparable to TNFα- (data not shown) or CD40L-stimulated SMC. The number of stably adherent monocytic cells to SMC was significantly reduced by blocking P-selectin, integrin αIIbβ3, CD40L and CX3CR1, whereas an IgG1 isotype control did not have a significant effect ()). Thus, platelet EV stimulation can provide an additional manner for monocytes to interact with SMC. Moreover, we investigated the effect of platelet EV on the adhesion of monocytic cells to HUVEC under flow conditions. TNFα stimulation increased monocytic cell adhesion, whereas there was no effect of platelet EVs, platelet SEC-EV or the respective controls ().
Figure 6. Platelet EVs increase monocytic cell adhesion and cytokine release. SMC were incubated without or with platelet EV or CD40L in the presence of indicated inhibitors (P-selectin inhibitor, eptifibatide, F1-Fk) or blocking antibody (α-CD40L). Syto13 fluorescently labelled THP-1 were perfused at 0.15 ml/min (3 dynes/cm2) and adherent cells were quantified in six different fields; n = 5–9 (a). Representative micrographs of THP-1 cells adherent to non- or platelet EV-stimulated SMC (b). SMC were incubated with indicated agonists for 24 h and IL-6 release was measured by ELISA; n = 6 (c). P-values were calculated by ANOVA with Tukey’s post-test. Scale bar: 100 µm.
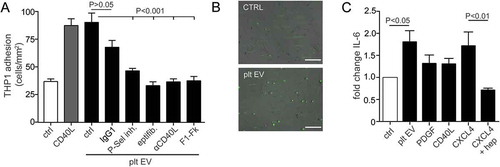
Unlike PDGF and CD40L, the presence of platelet EV triggered the production and release of the cytokine IL-6 by SMC to a similar extent as CXCL4 ()). The effects of platelet EV were mimicked by the addition of CXCL4, however not in the presence of heparin.
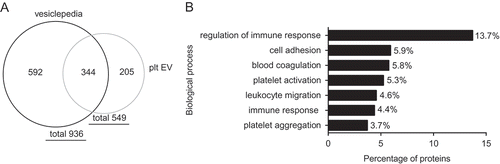
Moreover, proteomic analysis of platelet EV identified in total 549 proteins. From these, 344 proteins overlap with the in total 936 identified proteins of vesiclepedia [Citation34] ()). Pathway analysis by a Functional Enrichment analysis tool (FunRich) using the UniProt database identified these proteins involved in the regulation of immune response, cell adhesion, leucocyte migration, platelet activation and aggregation and coagulation ()). In concert, our data indicate that platelet EV can induce an inflammatory phenotypic change in SMC.
Figure 7. Proteomic analysis of platelet EV preparations. Venn diagram shows 549 identified proteins from which 344 proteins overlap with the in total 936 identified proteins of vesiclepedia (a). Pathway analysis identified proteins involved in biological processes (b).
Discussion
Increasing evidence points towards an immunomodulating role of cell- and platelet-derived EVs with vascular and immune cells [Citation13,Citation15,Citation35]. In this study we have explored a novel and likely physiologically important role of platelet EVs that are spontaneously released from platelets, e.g. to modulate the functional properties of vascular SMC. The spontaneous release of platelet EVs from platelets has been demonstrated previously [Citation9] and accumulates over time; reaching the maximum of released platelet EV/µl after 3 days in stored platelet concentrates destined for transfusion [Citation15]. The isolation and characterisation of EV remains among the most critical steps in the EV research field, particularly for downstream analyses and comparability between studies. In the present study, differential centrifugation and filtration has been used to isolate platelet EVs. Thorough characterisation by flow cytometry, NTA, cryo-TEM and proteomic analysis was performed to identify EVs in our preparations. Platelet EVs were found to express CD41a, PS and CD9, but not CD63 or CD81. Although CD9 has been identified as a marker for exosomes [Citation36], it is a membrane protein abundantly expressed on the surface of platelets and is also localised intracellularly in their α-granules [Citation37]. Moreover, CD9 is involved in the release of platelet EVs from thrombin- and convulxin- stimulated platelets [Citation38]. Other cells, e.g. prostate cancer cells also release CD9-positive EVs [Citation39]. The average size of platelet EVs in suspensions determined by NTA measurement of 153 nm could be confirmed by cryo TEM. The present study is the first to demonstrate the interaction of such platelet EVs to primary vascular cells evoking a switch towards a pro-inflammatory phenotype. Firm adhesion of platelet EV to resting SMC is divalent metal ion-dependent and facilitated by the integrin αIIbβ3. High expression levels of activated integrin αIIbβ3 on spontaneously released platelet EV have been observed earlier [Citation15]. Moreover, platelet EV can adhere to the subendothelial matrix thereby promoting platelet interaction via an integrin αIIbβ3 dependent mechanism [Citation40]. Likewise, from the present data, platelet EV interact with SMC through the interaction of integrin αIIbβ3 with VWF, fibrinogen or fibronectin presented on SMC. Another binding partner of integrin αIIbβ3 might be CD40L, as it is expressed on SMC and on activated platelets to stabilise arterial thrombi [Citation41]. However, blockade of CD40L did not lead to a strong reduction of platelet EV binding to SMC, suggesting that this is not the main αIIbβ3 ligand on SMC. Markedly, platelet EV also interact with TNFα-stimulated SMC via integrin αIIbβ3. However in this case, the CX3CR1–CX3CL1 axis is additionally involved in the platelet EV–SMC interaction. Previous studies have demonstrated high expression levels of CX3CL1 present on SMC upon TNFα or INFγ stimulation [Citation42,Citation43]. Thus, CX3CR1 present on platelet EVs might interact with CX3CL1 expressed by SMC under inflammatory conditions, thereby strengthening the SMC–platelet EV binding.
Experimental and clinical studies suggest an important role of CD40 ligand and its receptor CD40 in the pathophysiology of neointima formation and arterial remodelling [Citation44–Citation46] involving SMC proliferation, extracellular matrix remodelling and inflammation. In the present study, we show an increased platelet EV-induced SMC proliferation, which could be inhibited by blocking CD40 ligation on SMC. The relevance of the CD40-CD40L axis for the vasculature has been confirmed by platelet EV-induced proliferation of endothelial cells [Citation47]. Moreover, it has been demonstrated that platelet EV induce SMC proliferation, where the effects of platelet EV vastly exceeded the effects of PDGF-BB [Citation48]. Interestingly, a later study found that sCD40L activates mitogenic signalling and DNA synthesis of SMC, but not proliferation [Citation49]. Our results indicate that platelet EV-induced SMC proliferation might be triggered by ligation of CD40. In addition, P-selectin appeared to contribute to the proliferative effect of SMC, and such an effect has been described before for activated P-selectin-expressing platelets [Citation50]. Since CD40L in its soluble form did not induce proliferation in previous studies, it can be envisioned that receptor clustering by platelet EV-membrane-bound CD40L or co-stimulation by additional molecules, e.g. P-selectin is necessary for effective induction of proliferation.
In atherogenesis, SMC within the vessel migrate from the media into the intima. Our present study shows that platelet EV can stimulate the migration of SMC to a similar extent as PDGF. Interestingly, unlike CCL5 and CD40L, the non-classical chemokine CXCL4 induced SMC migration to a similar extent as platelet EV or PDGF. platelet EV-induced chemotaxis was attenuated by the CXCL4 neutraliser heparin, but not by a neutralising anti-PDGF antibody. Additionally, the presence of CXCL4 on platelet EVs was confirmed by ELISA, indicating that platelet EV-derived CXCL4 can be responsible for inducing SMC migration. Moreover, CXCL4-depleted platelet EVs that were reconstituted with concentrations of CXCL4 in the same order of original platelet EVs induced SMC migration, although not to a full extent. This indicates that CXCL4 is not the only mediator of the observed effects of platelet EV and that other yet to be identified mechanisms are relevant. Moreover, platelet EVs induced HUVEC migration, which is in line with a previous study [Citation51] that showed increased HUVEC migration towards platelet EVs released from thrombin-stimulated platelets. Taken together, this points towards the presence of other relevant mediators, since CXCL4 inhibits endothelial cell proliferation and migration [Citation52]. Nevertheless, CXCL4 is relevant since cleavage of CXCL4-binding GAGs by heparinase III on SMC greatly reduced CXCL4 or platelet EV-triggered SMC migration. PDGF-induced SMC migration was slightly reduced by heparinase III treatment, which is in line with a previous study that identified inhibition of PDGF-BB- and bFGF-induced proliferation of rat SMC by heparinase III [Citation53]. The above findings emphasise the contribution of membrane-bound CXCL4 and appear in line with the data from the group of Morrell that identified CXCL4 as an important effector of the platelet-mediated injury response of SMC [Citation31].
Remarkably, the presence of platelet EVs caused a change of morphology from contractile, spindle-shaped into synthetic and rhomboid SMC. In addition, co-culture of platelet EVs with SMC led to the loss of the contractile SMC marker calponin. Previous work has shown a heterogeneity of SMC populations with an enrichment of rhomboid SMC isolated from pig and rat neointimal explants [Citation54,Citation55]. Besides platelet EVs, treatment of the SMC with PDGF or CXCL4 also led to a change in phenotype, while the presence of PDGF also reduced the expression of calponin. Since CXCL4 did not induce a reduction of calponin expression, this might indicate that the presence of CXCL4 leads to alternative routes of differentiation, in a way as previously shown for monocytes [Citation56,Citation57].
As a consequence of phenotypic changes, we show that platelet EV binding increases the adhesiveness of monocytic THP-1 cells to SMC under flow, thereby increasing the amount of possibilities to interact with the SMC. The adhesion strongly depended on integrin αIIbβ3 and P-selectin, which are both exposed on platelet EVs. The interaction of platelet EV with monocytic cells via P-selectin has been demonstrated [Citation58,Citation59]. CX3CL1 is an atypical chemokine that functions either as chemoattractant or as adhesion molecule and CX3CL1 expressed by endothelial cells and SMC can directly mediate leucocyte adhesion [Citation60]. Blocking CX3CR1 using a modified ligand F1-Fk [Citation61], resulted in a decrease of monocytic cell adhesion. In line with these observations, we have previously shown that CX3CR1 expressed on activated platelets supports the complex formation of platelets and monocytes during hyperlipidaemia in mice and facilitated platelet leucocyte interaction with SMC [Citation7]. Also the CD40-CD40L axis was involved in the adhesion of monocytic cells to SMC. Blocking the CD40-CD40L axis resulted in less adhesion, which might be due to a reduction in the interaction of platelet EV with either leukocytes or SMC [Citation5]. When cultured with SMC, platelet EV induced cytokine IL-6 production, which might further contribute to platelet EV-mediated increased proliferation, migration and monocytic cell recruitment. Taken together, our present data indicate that platelet EVs spontaneously released from platelets induce a pro-inflammatory SMC phenotype. These findings correspond well with the proteomic analysis of the platelet EV preparations that identified proteins involved in pro-inflammatory (patho)physiological processes such as cell adhesion, leucocyte migration, platelet activation and aggregation and coagulation.
We conclude that platelet EV confer inflammatory signals between platelets and vascular cells by presenting a source of chemokine CXCL4 and membrane-bound effectors e.g. P-selectin, CD40L and CD40 thereby changing their properties and behaviour. These findings are relevant for transfusion medicine, since platelet concentrates destined for patients are stored for up to 7 days and accumulate EV from platelets. Moreover, spontaneous release of EV by platelets might occur as a physiologic process during the ageing of circulating platelets. Our findings collectively demonstrate that these platelet EV can induce phenotypic changes in different cell types, and thus harbour the potential to contribute to the progression of vascular diseases in vulnerable patients, e.g. with unstable angina pectoris, diabetes or auto-immune disease [Citation15,Citation62]. Notably, our observations might also be relevant for intra-plaque haemorrhage, a risk factor for adverse cardiovascular events, where newly formed vessels, particularly those close to atherosclerotic plaques are susceptible to leakage and might constitute a source of platelets and platelet EVs in the inflamed vessel wall. The present data demonstrate the importance and relevance of platelet EVs capable to reprogram the recipient cell and improves the understanding of communication between platelets and vascular cells.
ZJEV_A_Supplementary.pdf
Download PDF (155.3 KB)Acknowledgements
The authors thank Ms. Sandra Rouwette for assistance with the platelet products and Ms. Annemiek Dickhout, Mr. Brecht Willems, Mr. Rick van Gorp and Mr. Armand Jaminon for kind assistance. The authors would also like to thank Dr. Freek Bouwman and Prof. Edwin C.M. Mariman of the Maastricht Proteomics Centre for their kind assistance with the acquisition of the LCMS data and Dr. Carmen Lopez-Iglesias and Hans Duimel from the Maastricht Multimodal Molecular Imaging institute (M4I) for assistance with the electron microscopy experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed here.
Additional information
Funding
Notes on contributors
Tanja Vajen
T.V., B.J.B, A.C.A.H., E.M.V. performed and analysed experiments and T.V. wrote the manuscript. M.P. performed nanoparticle tracking analysis, Y.H., P.B.M., F.R.S, L.J.S. and J.W.M.H. provided intellectual and material input, R.R.K. supervised study, analysed data, obtained funding and wrote the manuscript.
References
- Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res. 2013;112(11):1–14.
- Koenen RR. The prowess of platelets in immunity and inflammation. Thromb Haemost. 2016;116(4):605–612.
- Gawaz M, Neumann FJ, Dickfeld T, et al. Vitronectin receptor (alpha(v)beta3) mediates platelet adhesion to the luminal aspect of endothelial cells: implications for reperfusion in acute myocardial infarction. Circulation. 1997;96(6):1809–1818.
- da Costa Martins PA, van Gils JM, Mol A, et al. Platelet binding to monocytes increases the adhesive properties of monocytes by up-regulating the expression and functionality of beta1 and beta2 integrins. J Leukoc Biol. 2006;79(3):499–507.
- Gerdes N, Seijkens T, Lievens D, et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler Thromb Vasc Biol. 2016;36(3):482–490.
- Willecke F, Tiwari S, Rupprecht B, et al. Interruption of classic CD40L-CD40 signalling but not of the novel CD40L-Mac-1 interaction limits arterial neointima formation in mice. Thromb Haemost. 2014;112(2):379–389.
- Postea O, Vasina EM, Cauwenberghs S, et al. Contribution of platelet CX(3)CR1 to platelet-monocyte complex formation and vascular recruitment during hyperlipidemia. Arterioscler Thromb Vasc Biol. 2012;32(5):1186–1193.
- von Hundelshausen P, Schmitt MM. Platelets and their chemokines in atherosclerosis-clinical applications. Front Physiol. 2014;5:294.
- Cauwenberghs S, Feijge MA, Harper AG, et al. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006;580(22):5313–5320.
- Vajen T, Mause SF, Koenen RR. Microvesicles from platelets: novel drivers of vascular inflammation. Thromb Haemost. 2015;114(2):228–236.
- Suades R, Padró T, Vilahur G, et al. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb Haemost. 2012;108(6):1208–1219.
- Mause SF, von Hundelshausen P, Zernecke A, et al. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol. 2005;25(7):1512–1518.
- Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327(5965):580–583.
- Mause SF, Ritzel E, Liehn EA, et al. Platelet microparticles enhance the vasoregenerative potential of angiogenic early outgrowth cells after vascular injury. Circulation. 2010;122(5):495–506.
- Vasina EM, Cauwenberghs S, Feijge MA, et al. Microparticles from apoptotic platelets promote resident macrophage differentiation. Cell Death Dis. 2011;2:e211.
- Sadallah S, Eken C, Martin PJ, et al. Microparticles (ectosomes) shed by stored human platelets downregulate macrophages and modify the development of dendritic cells. J Immunol. 2011;186(11):6543–6552.
- Cohen Z, Gonzales RF, Davis-Gorman GF, et al. Thrombin activity and platelet microparticle formation are increased in type 2 diabetic platelets: a potential correlation with caspase activation. Thromb Res. 2002;107(5):217–221.
- Trzepizur W, Martinez MC, Priou P, et al. Microparticles and vascular dysfunction in obstructive sleep apnoea. Eur Respir J. 2014;44(1):207–216.
- Sabatier F, Darmon P, Hugel B, et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes. 2002;51(9):2840–2845.
- Preston RA, Jy W, Jimenez JJ, et al. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension. 2003;41(2):211–217.
- Suades R, Padró T, Alonso R, et al. Lipid-lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb Haemost. 2013;110(2):366–377.
- Hao H, Gabbiani G, Bochaton-Piallat M-L. Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler Thromb Vasc Biol. 2003;23(9):1510–1520.
- Chaabane C, Coen M, Bochaton-Piallat M-L. Smooth muscle cell phenotypic switch: implications for foam cell formation. Curr Opin Lipidol. 2014;25(5):374–379.
- Projahn D, Simsekyilmaz S, Singh S, et al. Controlled intramyocardial release of engineered chemokines by biodegradable hydrogels as a treatment approach of myocardial infarction. J Cell Mol Med. 2014;18(5):790–800.
- Kapustin AN, Chatrou ML, Drozdov I, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116(8):1312–1323.
- Witwer KW, Buzas EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2:doi: 10.3402/jev.v2i0.20360.
- Boing AN, van der Pol E, Grootemaat AE, et al. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J Extracell Vesicles. 2014;3:doi: 10.3402/jev.v3.23430.
- Ostenfeld MS, Jensen SG, Jeppesen DK, et al. miRNA profiling of circulating EpCAM+ extracellular vesicles: promising biomarkers of colorectal cancer. J Extracell Vesicles. 2016;5:31488.
- Pathan M, Keerthikumar S, Ang CS, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–2601.
- McCloy RA, Rogers S, Caldon CE, et al. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13(9):1400–1412.
- Shi G, Field DJ, Long X, et al. Platelet factor 4 mediates vascular smooth muscle cell injury responses. Blood. 2013;121(21):4417–4427.
- Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007;15(3):100–108.
- Schecter AD, Calderon TM, Berman AB, et al. Human vascular smooth muscle cells possess functional CCR5. J Biol Chem. 2000;275(8):5466–5471.
- Kalra H, Simpson RJ, Ji H, et al. Vesiclepedia: a compendium for extracellular vesicles with continuous community annotation. Plos Biol. 2012;10(12):e1001450.
- Laffont B, Corduan A, Ple H, et al. Activated platelets can deliver mRNA regulatory Ago2*microRNA complexes to endothelial cells via microparticles. Blood. 2013;122(2):253–261.
- Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113(8):E968–E977.
- Brisson C, Azorsa DO, Jennings LK, et al. Co-localization of CD9 and GPIIb-IIIa (alpha IIb beta 3 integrin) on activated platelet pseudopods and alpha-granule membranes. Histochem J. 1997;29(2):153–165.
- Dale GL, Remenyi G, Friese P. Tetraspanin CD9 is required for microparticle release from coated-platelets. Platelets. 2009;20(6):361–366.
- Soekmadji C, Riches JD, Russell PJ, et al. Modulation of paracrine signaling by CD9 positive small extracellular vesicles mediates cellular growth of androgen deprived prostate cancer. Oncotarget. 2016: doi: 10.18632/oncotarget.11111.
- Merten M, Pakala R, Thiagarajan P, et al. Platelet microparticles promote platelet interaction with subendothelial matrix in a glycoprotein IIb/IIIa-dependent mechanism. Circulation. 1999;99(19):2577–2582.
- André P, Prasad KS, Denis CV, et al. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8(3):247–252.
- Ludwig A, Berkhout T, Moores K, et al. Fractalkine is expressed by smooth muscle cells in response to IFN-gamma and TNF-alpha and is modulated by metalloproteinase activity. J Immunol. 2002;168(2):604–612.
- Dragomir E, Manduteanu I, Calin M, et al. High glucose conditions induce upregulation of fractalkine and monocyte chemotactic protein-1 in human smooth muscle cells. Thromb Haemost. 2008;100(6):1155–1165.
- Li G, Sanders JM, Bevard MH, et al. CD40 ligand promotes Mac-1 expression, leukocyte recruitment, and neointima formation after vascular injury. Am J Pathol. 2008;172(4):1141–1152.
- Hristov M, Gümbel D, Lutgens E, et al. Soluble CD40 ligand impairs the function of peripheral blood angiogenic outgrowth cells and increases neointimal formation after arterial injury. Circulation. 2010;121(2):315–324.
- Donners MM, Beckers L, Lievens D, et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008;111(9):4596–4604.
- Leroyer AS, Rautou PE, Silvestre JS, et al. CD40 ligand+ microparticles from human atherosclerotic plaques stimulate endothelial proliferation and angiogenesis a potential mechanism for intraplaque neovascularization. J Am Coll Cardiol. 2008;52(16):1302–1311.
- Weber A, Köppen HO, Schrör K. Platelet-derived microparticles stimulate coronary artery smooth muscle cell mitogenesis by a PDGF-independent mechanism. Thromb Res. 2000;98(5):461–466.
- Hermann A, Schrör K, Weber A-A. CD40 ligand (CD40L) does not stimulate proliferation of vascular smooth muscle cells. Eur J Cell Biol. 2002;81(4):213–221.
- Cirillo P, Golino P, Ragni M, et al. Activated platelets and leucocytes cooperatively stimulate smooth muscle cell proliferation and proto-oncogene expression via release of soluble growth factors. Cardiovasc Res. 1999;43(1):210–218.
- Kim HK, Song KS, Chung J-H, et al. Platelet microparticles induce angiogenesis in vitro. Br J Haematol. 2004;124(3):376–384.
- Bikfalvi A. Platelet factor 4: an inhibitor of angiogenesis. Semin Thromb Hemost. 2004;30(3):379–385.
- Silver PJ, Moreau JP, Denholm E, et al. Heparinase III limits rat arterial smooth muscle cell proliferation in vitro and in vivo. Eur J Pharmacol. 1998;351(1):79–83.
- Walker LN, Bowen-Pope DF, Ross R, et al. Production of platelet-derived growth factor-like molecules by cultured arterial smooth muscle cells accompanies proliferation after arterial injury. Proc Natl Acad Sci U S A. 1986;83(19):7311–7315.
- Hao H, Ropraz P, Verin V, et al. Heterogeneity of smooth muscle cell populations cultured from pig coronary artery. Arterioscler Thromb Vasc Biol. 2002;22(7):1093–1099.
- Scheuerer B, Ernst M, Durrbaum-Landmann I, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. 2000;95(4):1158–1166.
- Gleissner CA, Shaked I, Little KM, et al. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184(9):4810–4818.
- Vasina EM, Cauwenberghs S, Staudt M, et al. Aging- and activation-induced platelet microparticles suppress apoptosis in monocytic cells and differentially signal to proinflammatory mediator release. Am J Blood Res. 2013;3(2):107–123.
- Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000;95(4):1317–1323.
- Barlic J, Zhang Y, Murphy PM. Atherogenic lipids induce adhesion of human coronary artery smooth muscle cells to macrophages by up-regulating chemokine CX3CL1 on smooth muscle cells in a TNFalpha-NFkappaB-dependent manner. J Biol Chem. 2007;282(26):19167–19176.
- Dorgham K, Ghadiri A, Hermand P, et al. An engineered CX3CR1 antagonist endowed with anti-inflammatory activity. J Leukoc Biol. 2009;86(4):903–911.
- Laffont B, Corduan A, Rousseau M, et al. Platelet microparticles reprogram macrophage gene expression and function. Thromb Haemost. 2016;115(2):311–323.