
ABSTRACT
Background
Endometrial cancer is a malignant tumour with a high incidence and mortality rate, and obesity is one of the most significant risk factors for the disease. However, it remains unclear whether leptin affects cell activity, proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT).
Materials and methods
Samples of endometrial cancer tissue were obtained from clinical patients and nude mice Enzyme-linked immunosorbent assays (ELISAs) were performed to assess leptin levels. Western blotting, immunohistochemical (IHC) and immunofluorescence (IF) analyses were conducted to detect EMT, JAK2/STAT3 signalling pathway proteins, and cell proliferation biomarkers. Cell Counting Kit-8 (CCK-8) assays, 5-ethynyl-2’-deoxyuridine (EdU) staining, and Transwell assays were used to evaluate cell activity, proliferation, migration, and invasion, respectively.
Results
ELISA, western blot and immunohistochemistry (IHC) analyses showed that leptin was highly expressed, and the JAK2/STAT3 signalling pathway was activated in endometrial cancer patients. Cell-based experiments showed that adipocytes secreted leptin, which increased the levels of leptin, and also promoted cell migration and invasion, EMT transition, and cell activity and proliferation. Leptin accelerated cell progression and promoted EMT via the JAK2/STAT3 signalling pathway in a dose-dependent manner. The tumour-promoting effect of leptin on endometrial cancer cells was further verified by in vivo experiments, in which leptin promoted tumour growth and activated the JAK2/STAT3 signalling pathway.
Conclusion
Leptin secreted by adipocytes promotes EMT transition and endometrial cancer progression via the JAK2/STAT3 signalling pathway in a dose-dependent manner.
Highlights
Endometrial cancer patients have high levels of leptin
Leptin promotes EMT transition via the JAK2/STAT3 signalling pathway
Leptin promotes endometrial cancer progression via the JAK2/STAT3 signalling pathway
Leptin promotes endometrial cancer in a dose-dependent manner
Introduction
Endometrial cancer is a malignant tumour that originates in the endometrium gland, and is also known as uterine carcinoma. Most cases of endometrial cancer are adenocarcinoma. Endometrial cancer has a high incidence and mortality rate, and approximately 76,000 females die from the disease each year [Citation1,Citation2]. In recent years, the incidence of endometrial cancer has risen, making it the second most prevalent malignancy in women, after breast cancer [Citation3]. Studies have shown that long-term continuous oestrogen stimulation, obesity, hypertension, diabetes, infertility, menopause, and genetic factors are risk all factors for endometrial cancer [Citation4].
As one of the risk factors, obesity, especially after menopause, considerably increases the risk for developing endometrial cancer. A higher body mass index (BMI) disrupts the balance between adipocytokines and hormones, and is strongly correlated with endometrial cancer [Citation5,Citation6]. The underlying process is thought to involve insulin resistance and adipokine release. Insulin resistance increases insulin levels, which stimulates endometrial cell mitogenesis; furthermore, adipokines such as leptin and adiponectin also contribute to the development of endometrial cancer. While additional studies are needed to fully understand the complicated relationships between obesity, insulin resistance, and endometrial cancer, it is evident that maintaining a healthy body weight and treating insulin resistance can help minimize the risk for endometrial cancer [Citation7].
Leptin is an adipokine polymer with a MW of 16 kDa that is secreted by white adipose tissue and transcribed by the leptin gene. Several studies have shown that a high leptin level is an independent risk factor for endometrial cancer [Citation8]. Therefore, the mechanism by which leptin affects endometrial cancer has attracted increased attention. Leptin stimulates signal transducer and activator of transcription 3 (STAT3) proteins in endometrial cancer that enhance the efficiency of tumour formation by modulating cell proliferation, neoangiogenesis, and the ability for immune evasion [Citation9]. It was reported that activation of STAT3 proteins increases the expression of anti-apoptotic genes, including recombinant human B-cell leukaemia/lymphoma xL (Bcl-xL) and myeloid cell leukaemia sequence 1 (Mcl-1) in endometrial cancer cell lines, making the epithelial cells immortal and chemo-resistant [Citation10]. Ahn et al. [Citation11] demonstrated that leptin could stimulate human endometrial cancer cell progression via the Janus kinase 2 (JAK2)/STAT3 signalling pathway. Gorrab et al. [Citation12] showed that a high dose of leptin promoted prostate cancer cell migration and epithelial-mesenchymal transition (EMT) by stimulating the STAT3 pathway. In order to further understand the expression and function of leptin, we referred to the leptin gene record in the NCBI database (https://www.ncbi.nlm.nih.gov/gene/3952). However, it remains unknown whether leptin affects cell activity, proliferation, migration, invasion, and EMT.
In our present study, we investigated the role played by leptin in endometrial cancer by assessing its effects on cell activity, proliferation, migration, invasion, and EMT. In addition, we also evaluated whether leptin regulates endometrial cancer cell progression via the JAK2/STAT3 signalling pathway.
Materials and methods
Clinical samples
Samples of blood and tissue were collected from endometrial cancer patients (T, n = 20) and patients with normal endometrial tissue (N, n = 20) who were hospitalized at Hainan General Hospital. All patients provided their written informed consent for study participation, and the study protocol was approved by the Ethical Committee of Hainan General Hospital (No. 2022–597).
Cell culture and co-culture experiments
A mouse preadipocyte cell line (3T3-L1) and two human endometrial cancer cell lines (Ishikawa and MFE-280) were obtained from Sigma-Aldrich (Sigma, Burlington, MA, USA). To induce the differentiation of 3T3-L1 preadipocytes, 3T3-L1 cells were incubated in DMEM containing 10% FBS, 0.5 mM 3-isobutyl-1-methylxanthine, 1 μM dexamethasone, and 10 μg/mL insulin for 3 days. The cells were then cultured in DMEM containing 10% FBS and insulin for another 2 days; after which, they were grown in DMEM containing 10% FBS. Ishikawa and MFE-280 cells were cultured with leptin at concentrations of 25 ng/mL (Leptin-L), 50 ng/mL (Leptin-M), and 100 ng/mL (Leptin-H), a JAK2/STAT3 signalling pathway inhibitor (AG490) (Leptin+AG490), and JSI-124 (Leptin+JSI-124). Ishikawa cells were cultured in RPMI 1640 medium supplemented with 1% penicillin-streptomycin and 10% foetal bovine serum (FBS). MFE-280 cells were cultured in MEM supplemented with 1% insulin-transferrin-selenium, 10% FBS, and 1% penicillin-streptomycin. Adipocytes were co-cultured with Ishikawa cells (Ishikawa+adipocytes) or MFE-280 (MFE-280+adipocytes) in a Transwell plate, with adipocytes in the lower layer and cancer cells in the upper layer.
Vector construction and transfection
Truncated leptin promoter and inhibitor fragments were amplified and inserted into a pGL3-Basic vector (Promega, Madison, WI, USA) for vector construction. T4 DNA ligase (TaKaRa, Japan) was used to link the amplified leptin promoter and suppressor fragments to pGL3-Basic vectors. In order to achieve specificity of the connection, the following specific primer sequence was designed for amplification: F: 5’-GGGTACCAAGCTTATGCATTGGGGAACC-3“, R: 5”-CCGCAGCTGTCAGCACCCAGGGCTGAGG −3“. The pGL3-Basic vector, amplified leptin promoter sequence, and suppressor fragments were digested using HindIII and PvuII restriction endonuclease. In addition, a lentiviral small hairpin RNA (shRNA) targeting leptin (GenePharma, Shanghai, China) was used to inhibit leptin gene expression. The shRNA sequence was as follows: F:5”-CCGGGCAGTCAGTCTCCTCCAAACACTCGAGTGTTTGGAGGAGACTGACTGCTTTTTG-3’, R: 5’-AATTCAAAAAGCAGTCAGTCTCCTCCAAACACTCGAGTGTTTGGAGGAGACTGACTGC-3’. When the cultured adipocytes reached 80%-90% confluence, leptin expression was modulated using shRNA for knockdown and pcDNA 3.0 vector (NM_000230.3) for overexpression. Transfection was performed using the Lipofectamine 2000 Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Finally, the transfected adipocytes were co-cultured with Ishikawa and MFE-280 cells (+shRNA-adipocytes, +Leptin-adipocytes).
Enzyme-linked immunosorbent assay (ELISA)
The levels of leptin in tissue and plasma samples were measured using a commercial ELISA kit according to the manufacturer’s instructions. First, an HRP-labelled antibody was added to the sample and incubated for 60 min at 37°C. Next, 100 µL of TMB substrate was added and incubated for 30 min at 25°C in the dark. Finally, Stop Solution was added, and the absorbance at 405 nm was measured with an ELISA instrument (Thermo Fisher Scientific Inc.).
Western blotting
The total proteins in tissue samples and cells were extracted using RIPA lysis buffer (#R0278, Sigma) and the protein concentration in each extract was quantified using a BCA Protein Assay Kit (Beyotime, Jiangsu, China). Next, a sample of protein from each extract was separated by standard sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), and the separated protein bands were transferred onto polyvinylidene fluoride (PVDF) membranes, which were subsequently washed with TBST (Tween-20: TBS = 1:1000), and then blocked with 5% powdered skimmed milk at 4°C overnight. Next, the membranes were incubated with primary antibodies against JAK2, p-JAK2, STAT3, p-STAT3, E-cadherin (E-cad), N-cadherin (N-cad), Vimentin, Ki67, PCNA, and β-actin for 1 h, and washed three times with TBST. The membranes were then incubated with a secondary antibody (HRP-labelled goat anti-rabbit IgG; #cat: ab672, Abcam) for 40 min at room temperature, washed with 25 mL of TBST (5 min per wash), and placed into an electrogenerated chemiluminescence (ECL) solution (ECL808–25, Biomiga, San Diego, CA) for 1 min. Finally, the membranes were photographed and observed using an X-ray machine (36209ES01, Shanghai Qcbio Science and Technologies Inc., Shanghai, China). The net density value of each protein band was calculated using Image-Pro Plus 6.0 software, with β-actin serving as an internal reference protein. All experiments were conducted in triplicate. The antibodies used were purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunohistochemical (IHC) staining
Samples of tumour tissue from patients and nude mice were immediately fixed with 4% paraformaldehyde for 24 h, embedded in paraffin, and cut into 5 μm-thick sections. The sections were decarbonized with xylene, rehydrated with ethanol, and repaired with citric acid. After blocking endogenous peroxidase activity, the tissue sections were incubated with primary antibodies against JAK2 (#cat: ab32101, Abcam), STAT3 (#cat: ab68153, Abcam), Ki-67 (#cat: Ab16667, Abcam), and PCNA (the primary antibody against ab29, Abcam), at 4°C overnight, and then washed with PBS. Next, the sections were incubated with a goat anti-rabbit secondary antibody at 37°C for 40 minutes, stained with haematoxylin, and examined under an optical microscope (Olympus, Tokyo, Japan).
Transwell assays
Transwell assays were conducted to evaluate cell migration and invasion abilities. Cell migration and invasion experiments were conducted in a similar manner, except for the use of DMEM and cell culture in the cell invasion studies. The studies were performed using 24-well Transwell plates and 8.0-μm pore membranes (Corning, Corning, NY, USA); the plates were coated with Matrigel (BD Biosciences, San Jose, CA, USA). Approximately 1.0 × 105 cells in 100 mL of serum-free DMEM were seeded into the top chamber of each plate and 500 mL of culture medium containing 10% FBS was added to each lower chamber. After incubation for 6 h at 37°C, the chambers were washed, the cells were fixed, and then stained with 0.1% crystal violet. Finally, the cells were observed under a ×400microscope.
Cell counting kit-8 (CCK-8) assay
The CCK-8 assay (Solarbio, Beijing, China) was used to evaluate cell viability. Approximately 1 × 104 Ishikawa or MFE-280 cells were seeded into a 96-well plate and cultured with adipocytes, shRNA-adipocyte, leptin-adipocyte, 25 ng/mL of leptin, 50 ng/mL of leptin, 100 ng/mL of leptin, leptin+AG490, or leptin+JSI-124 at 37°C. Next, CCK-8 solution was added to each well and the plates were incubated for 0, 24, 48, or 72 hrs; after which, they were incubated for an additional 2 h. Finally, the number of viable cells in each well was determined by measuring absorbance at 450 nm.
5-ethynyl-2’-deoxyuridine (EdU) staining assay
Ishikawa and MFE-280 cells treated with adipocytes, shRNA-adipocyte, leptin-adipocyte, 25 ng/mL of leptin, 50 ng/mL of leptin, 100 ng/mL of leptin, leptin+AG490, or leptin+JSI-124 were incubated in starvation medium containing 50 nM EdU (Life, China) for 2 h. Next, the co-culture systems were fixed in 4% polyformaldehyde for 1 h and incubated with 0.5% Triton X-100 for 15 min. After treatment, the co-culture systems were stained with reagents in a one Cell-Light™ EdU Cell Proliferation Detection Assay Kit (Life, USA). A fluorescent microscope (Nikon, Germany) was used to detect cells with positive staining.
Immunofluorescence (IF) analysis
Cells that had been cultured to 60%–70% confluence were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked with 1% BSA, incubated with primary antibodies against p-JAK2 and p-STAT3, and then incubated with a goat anti-rabbit secondary antibody (Sigma, Darmstadt, DE). For nuclear/cytosolic localization, the cells were stained with 4’, 6-diamidino-2-phenylindole (DAPI). Finally, p-JAK2 and p-STAT3 expression were detected under a confocal laser microscope (Olympus Optical, Tokyo, Japan).
Animal experiments
A total of 20 male BALB/c nude mice (age, 10 weeks) were purchased from The First Affiliated Hospital of Sun Yat-sen University (Guangzhou, China) and subsequently housed in polypropylene cages in a room with a 12 h dark/light cycle. Food and water were available ad libitum. Next, the mice were assigned to four separate groups, and the nude mice in the treatment groups were injected with Ishikawa or MFE-280 cells that had been pre-treated with leptin (1 mg/kg) every three days. After 35 days, the animals were anesthetized and sacrificed. Tumour volume was calculated as length × (width)2/2. The weight of each tumour was also measured.
Statistical analysis
GraphPad Prism 9 software (San Diego, CA, USA) was used for all statistical analyses and the preparation of images. The unpaired student’s t-test or one-way ANOVA was used to compare differences between groups, depending on the number of groups. All experiments were performed in triplicate, and results are presented as a mean value ± standard deviation (SD). A p-value <0.05 was considered to be statistically significant.
Results
Leptin was highly expressed and the JAK2/STAT3 pathway was activated in endometrial cancer patients
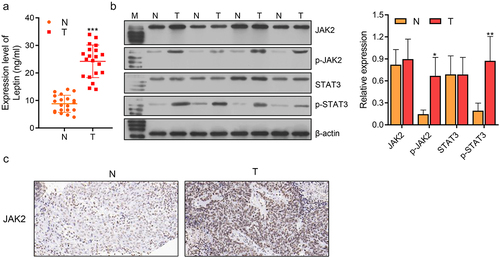
To investigate the role of leptin in endometrial cancer patients, its levels in normal endometrial tissue (N) and endometrial cancer tissue (T) were investigated. ELISA results showed that the levels of leptin were significantly higher in the endometrial cancer patients when compared to the control patients (***p < 0.001, ). As for the JAK2/STAT3 pathway, the levels of JAK2, p-JAK2, STAT3, and p-STAT3 proteins were assessed by western blotting, which showed that p-JAK2 and p-STAT3 were expressed higher levels in endometrial cancer tissue than in normal endometrial tissue (*p < 0.05, **p < 0.01, ). IHC staining showed that JAK2 was more highly expressed in endometrial cancer tissue when compared to normal tissue (). Therefore, we concluded that leptin and JAK2/STAT3 were highly expressed in endometrial cancer patients. However, it remained unknown whether leptin expression was associated with the JAK2/STAT3 signalling pathway.
Figure 1. Leptin was highly expressed and the JAK2/STAT3 pathway was activated in endometrial cancer patients. (a) ELISA was used to assess the expression levels of leptin in normal and tumour tissues. (b, c) Western blotting and IHC staining showed the expression levels of JAK2/STAT3 in normal and tumour tissues. N: normal endometrial tissue; T: endometrial cancer tissue. *p < 0.05, ** p < 0.01, *** p < 0.001.
Adipocytes increased the levels of leptin, cell migration and invasion, cell activity and proliferation, and promoted EMT
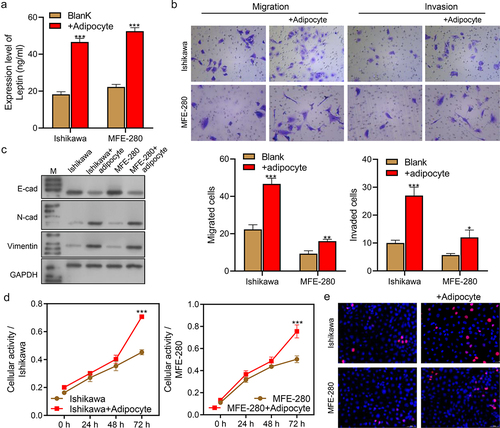
We next evaluated the effect of adipocytes on endometrial cancer cells. ELISA assays showed that leptin levels were increased in the co-culture systems (Ishikawa+Adipocytes and MFE-280+Adipocytes) when compared to their levels in single culture Ishikawa and MFE-280 cells (Blank system, ***p < 0.001, ). Transwell assays showed that cell migration and invasion abilities were enhanced in the Ishikawa+Adipocyte co-culture system and MFE-280+Adipocyte co-culture system when compared with Blank systems (***p < 0.001, ). We also evaluated the effect of adipocytes on the EMT of endometrial cancer cells. Western blot results showed that E-cad expression was significantly decreased, while N-cad and Vimentin expression were dramatically increased in the co-culture systems (), indicating that the EMT of cancer cells was promoted by co-culture with adipocytes. CCK-8 assays showed that cellular activity was significantly increased in the co-culture systems when compared with the Blank systems after 72 h of culture (***p < 0.001, ). Furthermore, EdU assays showed that the percentages of EdU-positive proliferating cells were significantly higher in the co-culture systems than in the blank systems (). Taken together, the above findings indicated that adipocytes increased the levels of leptin, promoted cell migration, invasion, and EMT, and increased cellular activity and proliferation.
Figure 2. Adipocytes increased the levels of leptin, cell migration and invasion, cell activity and proliferation, and promoted the EMT process. (a) ELISA assays were performed to examine the levels of leptin in the Blank and co-cultured systems. (b) Transwell assays were performed to examine cell migration and invasion in the Blank and co-culture systems. (c) Western blotting was used to assess the expression levels of EMT biomarkers (E-cad, N-cad, and Vimentin) in the Blank and co-culture systems. (d) CCK-8 assays were performed to evaluate the activity levels of Ishikawa and MFE-280 cells in the Blank and co-culture systems groups at time points of 0 h, 24 h, 48 h, and 72 h. (e) EdU staining was used to evaluate cell proliferation in the Blank and co-culture systems. *p < 0.05, **p < 0.01, ***p < 0.001.
Leptin increased cell migration and invasion, cell activity and proliferation, and promoted the EMT process
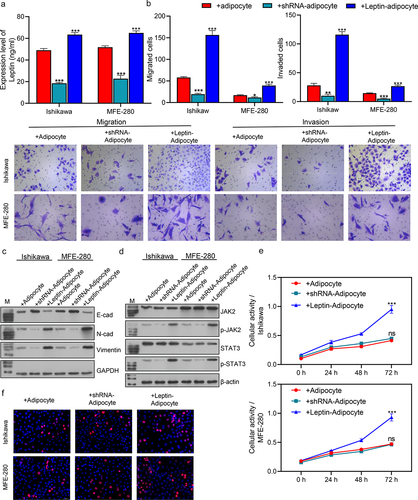
When compared with the Blank groups, the levels of leptin were significantly increased in the +Leptin-adipocyte (***p < 0.001) groups and decreased in the +shRNA-adipocyte groups (***p < 0.001; ). ELISA results showed that the levels of leptin in the co-culture systems were also reduced or increased after a reduced or excessive expression of leptin. We next evaluated the migration and invasion abilities of the transfected co-cultured cells and found that the migration and invasion abilities of the cells were significantly increased in the +Leptin-adipocyte groups (***p < 0.001) and decreased in +shRNA-adipocyte groups (***p < 0.001) when compared with the +Adipocyte groups (). As for the effect of leptin on EMT, western blot results showed that E-cad expression was increased in the co-culture systems while N-cad and Vimentin expression were significantly decreased by inhibition of leptin. Furthermore, E-cad expression in the co-culture systems was decreased, while N-cad and Vimentin expression were significantly increased by overexpression of leptin (). These results suggested that leptin promotes the EMT of cancer cells. We also evaluated the effect of leptin on the JAK2/STAT3 signalling pathway. Western blot studies showed that the levels of p-JAK2 and p-STAT3 expression were significantly increased in the +Leptin-adipocyte groups (***p < 0.001) and decreased in the +shRNA-adipocyte groups (***p < 0.001) when compared with the +Adipocyte groups (), suggesting that leptin is involved in regulating JAK2/STAT3 in endometrial cancer. As shown in the levels of cell activity and proliferation were elevated by overexpression of leptin and decreased by inhibition of leptin in the co-culture systems. In summary, leptin was found to promote cell migration, invasion, and EMT, and increase cell activity and proliferation.
Figure 3. Leptin increased cell migration and invasion, cell activity and proliferation, and promoted the EMT process. (a) ELISA assays were performed to evaluate the levels of leptin in transfected cells. (b) Transwell assays were performed to assess the migration and invasion abilities of transfected cells. (c, d) Western blotting was used to assess the expression levels of EMT biomarkers (E-cad, N-cad, and Vimentin) and JAK2/STAT3 signaling pathway proteins in transfected cells. (e) CCK-8 assays were performed to evaluate the activity of transfected Ishikawa and MFE-280 cells at time points of 0 h, 24 h, 48 h, and 72 h. (f) EdU staining was used to assess the proliferation of transfected cells. *p < 0.05, **p < 0.01, ***p < 0.001 vs. +Adipocytes.
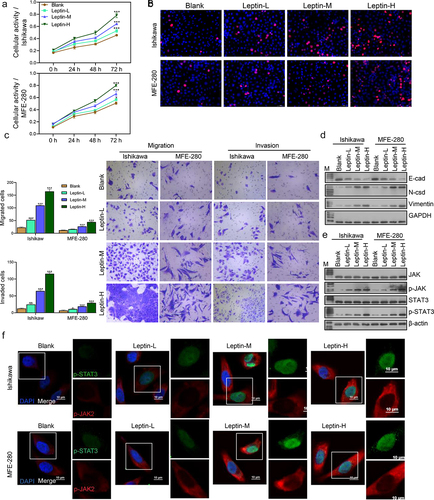
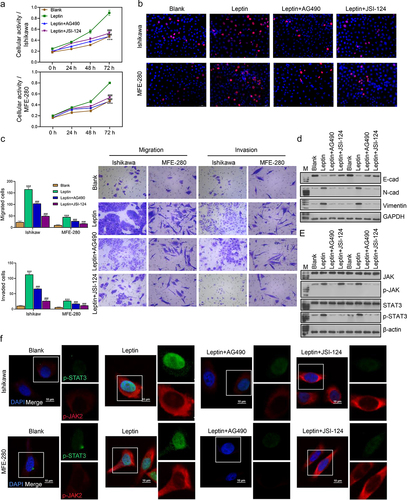
Next, we further investigated how different concentrations of leptin (25 ng/mL, 50 ng/mL, and 100 ng/mL) affected endometrial cancer cells. We found that the activity and proliferation of both Ishikawa and MFE-280 cells gradually increased with an increase in leptin concentration. Cells treated with a higher concentration of leptin showed higher cell activity (). When compared with cells in the Blank groups, the migration and invasion abilities of cells were gradually enhanced in the Leptin-L, Leptin-M, and Leptin-H groups (). A western blot analysis of EMT and JAK2/STAT3 signalling pathway proteins revealed that higher leptin concentrations significantly promoted EMT and the JAK2/STAT3 signalling pathway (). We then further assessed the expression levels of p-JAK2 and p-STAT3 by IF (), and the results were consistent with those of western blotting. Taken together, our data showed that leptin increased endometrial cancer cell migration and invasion, promoted EMT, and increased cellular activity and proliferation.
Figure 4. Leptin increased cell migration and invasion, cell activity and proliferation, and promoted the EMT process. (a) CCK-8 assays were performed to evaluate the activity of leptin-treated Ishikawa and MFE-280 cells at time points of 0 h, 24 h, 48 h, and 72 h. (b) EdU staining was used to assess the proliferation of Ishikawa and MFE-280 cells treated with graded concentrations of leptin. (c) Transwell assays were performed to evaluate the migration and invasion abilities of Ishikawa and MFE-280 cell treated with graded concentrations of leptin. (d, e) Western blotting was used to assess the expression levels of EMT biomarkers (E-cad, N-cad, and Vimentin) and JAK2/STAT3 signaling pathway-related proteins in Ishikawa and MFE-280 cells treated with graded concentrations of leptin. (f) If analyses were performed to evaluate the expression levels of p-JAK2 and p-STAT3 in Ishikawa and MFE-280 cells treated with graded concentrations of leptin. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Blank.
Leptin increased cell progression and promoted EMT via the JAK2/STAT3 signaling pathway
Next we further investigated whether JAK2/STAT3 regulated the effect of leptin on endometrial cancer cells. The levels of cell activity, cell proliferation, migration, invasion, EMT, and JAK2/STAT3 signalling pathway proteins were assessed in the Blank, leptin, leptin+AG490, and leptin+JSI-124 groups. As shown in the cell activity and proliferation rates were higher in the Leptin than in the leptin+AG490 and leptin+JSI-124 groups, demonstrating that leptin can improve cell activity and cell proliferation. At the same time, addition of the JAK2/STAT3 signalling pathway inhibitors AG490 and JSI-124 restored the increased cellular activity and proliferation induced by leptin. In addition, the migration and invasion abilities of cells were enhanced by leptin addition, and those increases were reversed by a JAK2/STAT3 signalling pathway inhibitor (). As for EMT transition and JAK2/STAT3, the leptin-induced enhancements of EMT and JAK2/STAT3 were reversed by the JAK2/STAT3 signalling pathway inhibitor (). Taken together, these findings showed that leptin increased cell migration and invasion, promoted EMT, and elevated the levels of cell activity and proliferation via the JAK2/STAT3 signalling pathway.
Figure 5. Leptin increased cell progression and promoted EMT via the JAK2/STAT3 signalling pathway. (a) CCK-8 assays were performed to evaluate the activity of Ishikawa and MFE-280 cells treated with Blank, leptin, leptin+AG490, and leptin+JSI-124 at time points of 0 h, 24 h, 48 h, and 72 h. (b) EdU staining was used evaluate the proliferation of Ishikawa and MFE-280 treated with Blank, leptin, leptin+AG490, and leptin+JSI-124. (c) Transwell assays were performed to examine the migration and invasion abilities of Ishikawa and MFE-280 cells treated with Blank, leptin, leptin+AG490, and leptin+JSI-124. (d, e) Western blotting was used to assess the expression levels of EMT biomarkers (E-cad, N-cad, and Vimentin) and JAK2/STAT3 signaling pathway proteins in Ishikawa and MFE-280 cells treated with Blank, leptin, leptin+AG490, and leptin+JSI-124. (f) If analyses were performed to evaluate the expression levels of p-JAK2 and p-STAT3 in Ishikawa and MFE-280 treated with Blank, leptin, leptin+AG490, and leptin+JSI-124. ***p < 0.001 vs. Blank, ###p < 0.001 vs. leptin.
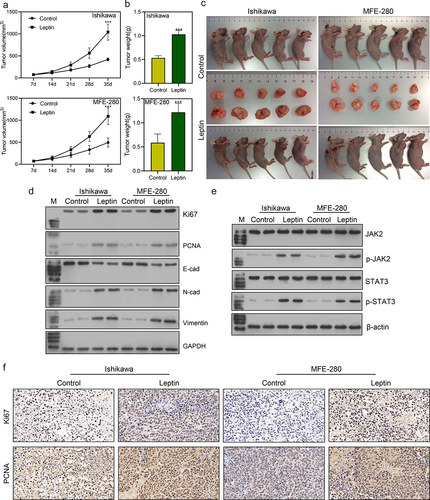
Leptin promoted endometrial cancer tumor growth in vivo
Finally, we examined the effect of leptin on endometrial cancer progression in vivo. Our data showed that when compared with tumours in control mice, tumour size, volume, and weight were all significantly increased by injection with leptin (***p < 0.001; ). The cell proliferation biomarkers Ki67 and PCNA were more highly expressed in the Leptin group than in the Control group (). Moreover, western blot analyses showed that the expression of proteins associated with EMT transition was enhanced, and the JAK2/STAT3 signalling pathway was activated by leptin addition (). IHC analyses showed that the levels of Ki67 and PCNA were higher in the Leptin group than in the Control group (), which was consistent with the results of western blotting.
Figure 6. Leptin promoted endometrial cancer tumour growth in vivo. (a-c) The volumes and weights of tumours were measured in the control and leptin injection groups. (d, e) Western blot studies were performed to assess the expression levels of Ki67, PCNA, EMT biomarkers, and JAK2/STAT3. (f) IHC staining was used to evaluate the expression levels of Ki67 and PCNA. ***p < 0.001 vs. control.
Discussion
Obesity is one of the most significant risk factors for endometrial cancer. Leptin secreted by adipocytes has been demonstrated to regulate cancer cell progression. However, the molecular mechanism of leptin in endometrial cancer remains unclear. In our present study, we assessed the effects of leptin on endometrial cancer cell activity, proliferation, migration, invasion, and EMT. In addition, we investigated whether leptin regulates endometrial cancer cell progression via the JAK2/STAT3 signalling pathway. Our data showed that leptin was expressed at high levels in endometrial cancer patients. Moreover, leptin increased cell migration and invasion, promoted EMT, and elevated the levels of cell activity and proliferation via the AK2/STAT3 signalling pathway in a dose-dependent manner.
Leptin is associated with cancer through various molecular mechanisms. Leptin regulates ovarian cancer cell proliferation by increasing the expression levels of Mcl-1 and cyclin D1 after activating the MAP kinase-ERK kinase 1/2 (MEK/ERK1/2) and phosphatidylinositol 3 kinase (PI3K)/protein kinase B (AKT) signalling axes [Citation13]. Leptin can induce the malignant transformation of breast cancer cells, and that induction effect is attenuated by the JAK2/STAT3 signalling inhibitor AG490, suggesting critical interplay between leptin and JAK2/STAT3 during cancer progression [Citation14]. Adiponectin attenuates leptin-elicited SPEC-2 endometrial cancer metastasis by suppressing the JAK/STAT3 pathway [Citation15]. However, the current studies on the molecular mechanism of leptin in endometrial cancer are insufficient.
A high leptin level is an independent risk factor for endometrial cancer [Citation8]. Several studies have shown a positive correlation between leptin and cancer risk, and especially for prostate cancer [Citation16,Citation17]. Even more interesting, they showed that higher leptin levels were associated with more advanced tumours. Leptin increases the risk for prostate cancer by interacting with humoral factors such as sex insulin, hormones, and insulin-like growth factor 1 (IGF-1), all of which are related to abdominal obesity [Citation18]. However, some studies have indicated that circulating leptin is not associated with prostate cancer [Citation18–20]. The association between leptin and cancer risk has not been explored in some other cancers. Our present study found that a higher concentration (100 ng/mL) of leptin had a stronger promoting effect on endometrial cancer progression than a lower concentration (50 ng/mL and 25 ng/mL), and this is the first time that a dose-dependent effect of leptin on tumour promotion has been reported.
The function of the JAK2/STAT3 signalling pathway has been widely studied in several cancers. Yuan et al. [Citation21] revealed that the JAK2/STAT3 was activated by complement C3 and associated with gastric cancer progression. Similarly, Shen et al. [Citation22] demonstrated that MUC16 facilitates cervical cancer progression via the JAK2/STAT3 signalling pathway. However, the effect of JAK2/STAT3 on endometrial cancer progression has not been adequately studied. The molecular mechanism by which JAK2/STAT3 affects endometrial cancer progression is mainly leptin-related. Gao et al. [Citation10] concluded that leptin activates cyclooxygenase‐2 function via the JAK2/STAT3 pathway in human endometrial cancer. Oh et al. [Citation23] showed that leptin stimulates the development of endometriotic epithelial cells via the JAK2/STAT3 and ERK pathways. Ahn et al. [Citation11] demonstrated that leptin promotes human endometriotic cell migration and invasion by up-regulating matrix metallopeptidase 2 (MMP-2) expression via the JAK2/STAT3 signalling pathway. Leptin also influences ovarian cancer cell phenotypes via the JAK2/STAT3 signalling pathway [Citation24]. Another study showed that leptin induces leiomyoma cell proliferation and extracellular matrix deposition via the JAK2/STAT3 signalling pathway [Citation25]. Our present study has helped to clarify the role of leptin in endometrial cancer.
Olea-Flores et al. [Citation26] demonstrated that leptin is novel actor which drives the EMT process in several cancers, including breast, gastric, prostate, and lung cancer. In breast cancer, leptin can activate EMT and promote cell migration via the JAK/STAT3–Akt signalling pathway [Citation27]. In prostate cancer cells, leptin promotes cell migration and EMT by activating the STAT3 pathway [Citation28]. Similarly, leptin promotes the invasion and metastasis of lung cancer cells by inducing EMT [Citation29]. Our present study, for the first time, revealed the role played by leptin in the EMT of endometrial cancer cells; however, additional studies are required. This study revealed one of the mechanisms by which an adipocyte secretion participates in the pathogenesis of endometrial cancer, and provides a new perspective for our understanding of the disease. In addition, by revealing a new mechanism (the JAK2/STAT3 pathway) by which leptin affects the EMT process, we gain a more thorough understanding of the important role played by leptin in the development of endometrial cancer, which is great significance for the development of relevant treatment strategies.
However, our study does have some limitations. First, due to the small number of patients in the study, our conclusions need to be validated in a larger patient population. Second, we have not fully clarified the specific downstream pathway by which leptin exerts its effect, and identification of that pathway will be one of the focus of our future research.
Conclusion
Our present study assessed the molecular mechanism of leptin in endometrial cancer. Our data showed that leptin secreted by adipocytes promotes the EMT process and endometrial cancer progression via the JAK2/STAT3 signalling pathway in a dose-dependent manner.
Contributions
(I) Study design/planning: Lifan Shen; (II) Data collection/entry: Lifan Shen, Chen Zhang, Xin Liang, Kaiying Cui; (III) Data analysis/statistics: Lifan Shen, Chen Zhang; (IV) Data interpretation: Lifan Shen, Chen Zhang, Genhai Zhu, Lan Hong; (V) Preparation of manuscript: Lifan Shen; (VI) Literature analysis/search: All authors; (VII) Funding acquisition: National Natural Science Foundation Cultivation Project-530 Youth Science Fund Project(2021QNXM20); (VIII) Final approval of manuscript: All authors.
Ethics statement
All the study participants signed an informed consent for the inclusion in the study. The study was approved by Ethical Committee of Hainan General Hospital (No. 2022–597)
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data are available from the figshare with request. (doi: 10.6084/m9.figshare.23742015)
Additional information
Funding
References
- Morice P, Leary A, Creutzberg C, et al. Endometrial cancer. Lancet. 2016;387(10023):1094–13. doi: 10.1016/S0140-6736(15)00130-0
- Urick ME, Bell DW. Clinical actionability of molecular targets in endometrial cancer. Nat Rev Cancer. 2019;19(9):510–521. doi: 10.1038/s41568-019-0177-x
- Crosbie EJ, Kitson SJ, McAlpine JN, et al. Endometrial cancer. Lancet. 2022;399(10333):1412–28. doi: 10.1016/S0140-6736(22)00323-3
- Raglan O, Kalliala I, Markozannes G, et al. Risk factors for endometrial cancer: an umbrella review of the literature. Intl J Cancer. 2019;145(7):1719–30. doi: 10.1002/ijc.31961
- Nagle C, Crosbie E, Brand A, et al. The association between diabetes, comorbidities, body mass index and all-cause and cause-specific mortality among women with endometrial cancer. Gynecol Oncol. 2018;150(1):99–105. doi: 10.1016/j.ygyno.2018.04.006
- Hazelwood E, Sanderson E, Tan VY, et al. Identifying molecular mediators of the relationship between body mass index and endometrial cancer risk: a Mendelian randomization analysis. BMC Med. 2022;20(1):125. doi: 10.1186/s12916-022-02322-3
- Tzenios N, Chahine M, Tazanios M. Obesity and endometrial cancer: the role insulin resistance and adipokines. SJMAS. 2023;1(2). doi: 10.58676/sjmas.v1i2.12
- Wang P-P, He X-Y, Wang R, et al. High leptin level is an independent risk factor of endometrial cancer: a meta-analysis. Cell Physiol Biochem. 2014;34(5):1477–84. doi: 10.1159/000366352
- Deng J, Grande F, Neamati N. Small molecule inhibitors of Stat3 signaling pathway. Curr Cancer Drug Targets. 2007;7(1):91–107. doi: 10.2174/156800907780006922
- Gao J, Tian J, Lv Y, et al. Leptin induces functional activation of cyclooxygenase‐2 through JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways in human endometrial cancer cells. Cancer Sci. 2009;100(3):389–395. doi: 10.1111/j.1349-7006.2008.01053.x
- Ahn J-H, Choi YS, Choi J-H. Leptin promotes human endometriotic cell migration and invasion by up-regulating MMP-2 through the JAK2/STAT3 signaling pathway. Mol Hum Reprod. 2015;21(10):792–802. doi: 10.1093/molehr/gav039
- Gorrab A, Pagano A, Ayed K, et al. Leptin promotes prostate cancer proliferation and migration by stimulating STAT3 pathway. Nutr Cancer. 2021;73(7):1217–1227. doi: 10.1080/01635581.2020.1792946
- Chen C, Chang Y-C, Lan MS, et al. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways corrigendum in/10.3892/ijo.2013.1789. Int J Oncol. 2013;42(3):1113–9. doi: 10.3892/ijo.2013.1789
- Boothby-Shoemaker W, Benham V, Paithankar S, et al. The relationship between leptin, the leptin receptor and FGFR1 in primary human breast tumors. Cells. 2020;9(10):2224. doi: 10.3390/cells9102224
- Wu X, Yan Q, Zhang Z, et al. Acrp30 inhibits leptin-induced metastasis by downregulating the JAK/STAT3 pathway via AMPK activation in aggressive SPEC-2 endometrial cancer cells. Oncol Rep. 2012;27(5):1488–96. doi: 10.3892/or.2012.1670
- Saglam K, Aydur E, Yilmaz MI, et al. Leptin influences cellular differentiation and progression in prostate cancer. J Urol. 2003;169(4):1308–11. doi: 10.1097/01.ju.0000055903.18400.25
- Chang S, Hursting SD, Contois JH, et al. Leptin and prostate cancer. Prostate. 2001;46(1):62–67. doi: 10.1002/1097-0045(200101)46:1<62:AID-PROS1009>3.0.CO;2-V
- Hsing AW, Chua JS, Gao Y-T, et al. Prostate cancer risk and serum levels of insulin and leptin: a population-based study. JNCI. 2001;93(10):783–789. doi: 10.1093/jnci/93.10.783
- Lagiou P, Signorello LB, Trichopoulos D, et al. Leptin in relation to prostate cancer and benign prostatic hyperplasia. Int J Cancer. 1998;76(1):25–8.
- Stattin P, Kaaks R, Johansson R, et al. Plasma leptin is not associated with prostate cancer risk. Cancer Epidemiol Biomark Prev. 2003;12(5):474–5.
- Yuan K, Ye J, Liu Z, et al. Complement C3 overexpression activates JAK2/STAT3 pathway and correlates with gastric cancer progression. J Exp Clin Cancer Res. 2020;39(1):9. PMID:PMC6956509. doi: 10.1186/s13046-019-1514-3
- Shen H, Guo M, Wang L, et al. MUC16 facilitates cervical cancer progression via JAK2/STAT3 phosphorylation-mediated cyclooxygenase-2 expression. Genes Genomics. 2020;42(2):127–133. doi: 10.1007/s13258-019-00885-9
- Oh HK, Choi YS, Yang Y-I, et al. Leptin receptor is induced in endometriosis and leptin stimulates the growth of endometriotic epithelial cells through the JAK2/STAT3 and ERK pathways. Mol Hum Reprod. 2012;19(3):160–8. doi: 10.1093/molehr/gas055
- Kumar J, Fang H, McCulloch DR, et al. Leptin receptor signaling via Janus kinase 2/Signal transducer and activator of transcription 3 impacts on ovarian cancer cell phenotypes. Oncotarget. 2017;8(55):93530. doi: 10.18632/oncotarget.19873
- Reschke L, Afrin S, El Sabah M, et al. Leptin induces leiomyoma cell proliferation and extracellular matrix deposition via JAK2/STAT3 and MAPK/ERK pathways. F S Science. 2022;3(4):383–91. doi: 10.1016/j.xfss.2022.05.001
- Olea-Flores M, Juárez-Cruz JC, Zuñiga-Eulogio MD, et al. New actors driving the epithelial–mesenchymal transition in cancer: the role of leptin. Biomolecules. 2020;10(12):1676. doi: 10.3390/biom10121676
- Wang L, Tang C, Cao H, et al. Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol Ther. 2015;16(8):1220–1230. doi: 10.1080/15384047.2015.1056409
- Xu C, Dong L, Kang X, et al. Leptin promotes proliferation and inhibits apoptosis of prostate cancer cells by regulating ERK1/2 signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24(16):8341–8348. doi: 10.26355/eurrev_202008_22630
- Xu M, Cao F-L, Li N, et al. Leptin induces epithelial-to-mesenchymal transition via activation of the ERK signaling pathway in lung cancer cells. Oncol Lett. 2018;16(4):4782–4788. doi: 10.3892/ol.2018.9230