
ABSTRACT
Converting an immunosuppressive melanoma microenvironment into one that favors the induction of antitumor immunity is indispensable for effective cancer immunotherapy. In the current study we demonstrate that oat-derived β-(1-3)-(1-4)-glucan of 200 kDa molecular size (BG34-200) previously shown to mediate direct interaction with macrophages could alter the immune signature within melanoma microenvironment. Systemic administration of BG34-200 resulted in reversion of tolerant melanoma microenvironment to an immunogenic one that allows M1-type activation of macrophages, the induction of pro-inflammatory cytokines/chemokines including IFN-γ, TNF-α, CXCL9, and CXCL10, and enhanced IRF1 and PD-L1 expression. In turn, BG34-200 induced a superior antitumor response against primary and lung metastatic B16F10 melanoma compared to untreated controls. The enhanced tumor destruction was accompanied with significantly increased tumor infiltration of CD4+ and CD8+ T cells as well as elevated IFN-γ in the tumor sites. Systemic administration of BG34-200 also provoked systemic activation of tumor draining lymph node T cells that recognize antigens naturally expressing in melanoma (gp100/PMEL). Mechanistic studies using CD11b-knockout (KO), CD11 c-DTR transgenic mice and nude mice revealed that macrophages, DCs, T cells and NK cells were all required for the BG34-200-induced therapeutic benefit. Studies using IFN-γ-KO transgenic mice showed that IFN-γ was essential for the BG34-200-elicited antitumor response. Beyond melanoma, the therapeutic efficacy of BG34-200 and its immune stimulating activity were demonstrated in a mouse model of osteosarcoma. Together, BG34-200 is highly effective in modulating antitumor immunity. Our data support the potential therapeutic use of this novel immune modulator in the treatment of metastatic melanoma.
Introduction
Cancer immunotherapy achieves immune-mediated control of tumor growth and metastasis by mounting tumor-reactive T cell responses. Although immunotherapy holds great promise for cancer treatment, its clinical success has so far been limited.Citation1 Increasing studies have demonstrated that cancer cells exploit multiple mechanisms to create an immunosuppressive environment that enable them to escape immune destruction.Citation2,3 Therefore, overcoming immunosuppressive mechanisms and induction of durable antitumor immunity using novel immune modulators are essential goals of cancer immunotherapy.
We and others have previously demonstrated that β-glucan molecules can be exploited as immune modulators for generating antitumor immune responses, which is based on their ability to integrate innate and adaptive immune componentsCitation4 β-glucans (or polysaccharides) are major cell wall components of microbes. Their carbohydrate structures can be recognized as pathogen associated molecular pattern (PMAP) by pattern recognition receptor (PRR) such as dectin-1 and CR3, which are C-type lectin and carbohydrate PRR. Similar to other PRR such as toll-like receptors (TLRs), carbohydrate PRRs are also involved in host defense mechanisms against infection. However, different from TLRs that recognize various PAMPs such as lipopolysaccharide, proteoglycans, DNAs and RNAs, C-type lectins appear to be more specific and mostly recognize carbohydrate structures. Because of the specific recognition, some β-glucans display a capability of stimulating host immune responses via priming macrophage, neutrophil and granulocytes through dectin-1 and/or complement receptor 3 (CR3).Citation5 β-glucan-mediated action on these receptors can further trigger natural killer (NK) cells, dendritic cells (DCs) and T cells to respond to tumor targets.Citation5–9 The glucan molecule-mediated immunomodulation has been attributed to its efficient modulatory function during pathogen recognition and antigen presentation. More importantly, studies on soluble β-glucan have demonstrated that the β-glucan-bound monocytes and neutrophils can mediate direct binding of these cells to complement 3 opsonized targets such as iC3b-bound tumor cells, which provides a cellular mechanism of β-glucan to target tumor environment.Citation10 Therefore, modification of tumor environment using novel glucan-type immune modulator can potentially enhance immunogenicity of tumor.
Mechanism of β-(1-3)-(1-6)-glucan in inducing pro-inflammatory cytokine secretion and stimulating innate and adaptive immune responses has been investigated,Citation4,6,11 and multiple clinical trials of cancer immunotherapy have incorporated β-(1-3)-(1-6)-glucan in therapeutic antibody treatment against cancer (Clinical trial IDs such as NCT01829373, NCT00682032, NCT01727895, NCT00289003 on clinicaltrials.org). Glucan-induced direct manipulation of antibody sensitized tumor microenvironment has also been reported to elicit a potent antitumor response.Citation12–15 However, emerging data suggest that β-glucans from different resources with different impurities, glycosidic linkage, molecular weight, solubility, and route of administration all exhibit different mechanism of actions and potency of antitumor effect.Citation16–19 A current limitation in studies involving β-glucan includes a lack of β-glucan control standard with specific molecular weight and branches available. Most of the β-glucans studies to-date used zymosan, which is a mixture of chitosan, β-glucans and cell wall particles; and the exact immunological actions and signaling pathway induced by β-glucan are still unclear and have to be further defined.Citation4
We have previously engineered oat-derived β-(1-3)-(1-4)-glucans (BG34) with well-characterized molecular weight and chemical structure.Citation20 These glucan samples adopt linear chain structure with no branches. They are highly purified glucan with carbohydrate content over 98% and have been documented to be free of endotoxin. Since studies have demonstrated that the peptide/protein impurities, endotoxin contaminants and broad molecular weight distribution could significantly affect β-glucan-mediated activity, these unique features of BG34 make them excellent candidates for pre-clinical/clinical studies and necessary approval by regulatory agencies.
Previous studies on BG34 have proposed two mechanisms for their anti-cancer action. In vitro studies on low molecular weight oat β-(1-3)-(1-4)-glucans showed that they could mediate tumor cell apoptosis via direct cytotoxic effec.Citation21 A study on barley β-(1-3)-(1-4)-glucans suggested that barley BG34 alone did not mediate any in vivo anticancer action in a murine neuroblastoma model; however, barley BG34 in combination with an anti-ganglioside monoclonal antibody mediated enhanced inhibitory effect on cancer growth.Citation22 In this current study, we show that BG34 with a molecular weight of 200 kDa (BG34-200) does not mediate direct cytotoxicity to murine and human melanoma cells. Instead, systemic administration of BG34-200 results in profound inhibition of malignant primary melanoma B16F10 as well as lung metastases. The superior antitumor efficacy of BG34-200 was supported by enhanced T cell activation both locally at tumor site and regionally within the tumor-draining lymph node. Our results indicate that BG34-200 alters immune signature and promotes the development of highly potent antitumor immune responses by modulating the tumor microenvironment.
Materials and methods
BG34 sample preparation and characterization
Isolation, purification and fractionation of β-(1-3)-(1-4)-glucans (BG34 s) from oat brans were described as our previous work [20]. Chemical structure, molecular weight and polydispersity of BG34 samples were characterized by Infrared Spectroscopy (IR), 13C and 1H Nuclear Magnetic Resonance Spectroscopy (NMR) and Gel Permeation Chromatography (GPC), as previously described [20]. BG34 of 10, 200 and 500 kDa (BG34-10, BG34-200 and BG34-500) were used in this study. Endotoxin of the glucan samples were tested by Endosafe PTS™ system (Charles River, Skokie, Illinois).
Mice and cell lines
Male, age-matched wild-type (WT) C57 BL/6 J, CD11b-knockout (CD11b-KO) (B6.129S4-Itgamtm1Myd/J), IFN-γ-KO (B6.129S7-IFNgtm1Ts/J), CD11 c-DTR (B6.FVB-Tg(Itgax-DTR/EGFP)57Lan/J) transgenic mice on a CD45.2 background (8–10 weeks), C57 BL/6 J (B6.SJL-PtprcaPepcb/BoyJ) mice on a CD45.1 background (8–10 weeks), and PMEL (B6.Cg-Thy1a/CyTg(TcraTcrb)8Rest/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Nude mice were from the Animal Research Center of Case Western Reserve University. CD11 c-venus mice were from Dr. Alex YC Huang's lab at Case Western Reserve University. To generate CD11 c-depleted mouse model, bone marrow (BM) chimeras were generated by reconstitution with BM cells from CD11 c-DTR mice as described.Citation23 All experimental procedures were conducted according to the protocols approved by Case Western Reserve University Animal Research Center. B16F10 and A375 cells were purchased from ATCC. Osteosarcoma K7 M2-Luc2 cells were from Dr. Alex YC Huang's lab. Cells were periodically authenticated by morphologic inspection and animal grafting to assess tumor histology and ability to grow and metastasize. All cell lines were maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and routinely examined for mycoplasma contamination using a PCR-based mycoplasma detection kit.
Reagents and antibodies (Abs)
For fluorescence-activated cell sorting (FACS) analysis, anti-mouse CD45-V450 (30-F11), CD11b-PE (M1/70), CD19-PE-Cy7 (1D3), F4/80-AF647 (T45-2342), CD4-AF488 (RM4-5), CD8-PE (53-6.7), CD86-V450 (GL1), CD11b-PE, CD45-PerCp (30-F11), CD8-V450 (53-6.7), CD62 L-AF700 (MEL-14), CD44-PE (1M7), CD27-APC (3A10), CD19-PE-Cy7 (1D3), CD5-PE (53.7.3), B220-PE (RA3-6 B2), NK1.1-AF700 (PK136), CD11 c-FITC (HL3), CD49b-APC (DX5), CD45.1-PE (A20), CD45.2-APC (104), IFN-γ-PE (XMG1.2), IFN-γ-AF647 (XMG1.2) and IL-2-PE (JES6-5H4) were purchased from BD Biosciences (San Jose, CA), CD3-Percp (17A2) and B220-AF488 (RA3-6 B2) were purchased from R&D Systems (Minneapolis, MN), CD3-AF405 (C415.9 A) was purchased from Santa Cruz Biotechnology (Dallas, TX), GR-1-APC (RB5-8C5) was purchased from Miltenyi Biotec (San Diego, CA), Ly6G-FITC (1A8) and granzyme B-PE (MGZB) were purchased from ThermoFisher Scientific (Waltham, MA). Mouse IFN-γ and IL-2 ELISA kits were purchased from R&D Systems (Minneapolis, MN). FITC-conjugated latex beads were purchased from Sigma Aldrich (St Louis, MO). Thymidine incorporation assay kit was purchased from ThermoFisher Scientific (Waltham, MA). Mouse recombinant GM-CSF and M-CSF proteins were purchased from R&D Systems. GP10025-33 (KVPRNQDWL) peptide was gifted from Prof. Shawn Wang (Virginia Commonwealth University). Cytofix/Cytoperm and fixation buffers were purchased from BD Biosciences (San Jose, CA). Primer sets used for real time PCR are listed in Supplemental Table 1. Cytometric bead array mouse inflammation kit (TNF-α, IL-6, IL-10, MCP-1, IFN-γ and IL-12p70) was purchased from BD Biosciences (San Jose, CA).
Table 1. XXXXX.
Tumor study
B16F10 tumors were established by injecting 5 × 105 B16F10 tumor cells to the flank of mice. Four days after tumor cell inoculation, mice were randomized and received intraperitoneally (i.p.) injected BG34 solutions at ∼ 25mg/kg. Mice were treated by BG34 of three different molecular weight (10, 200 and 500 kDa), respectively, to determine effect of molecular weight on antitumor activity. Mice treated with PBS or dextran served as controls. Treatments were administered daily for five days, or twice or three times a week. Tumor growth was monitored by measuring the length of short (l) and long (L) diameters (volume = l2 x L/2) as described.Citation24 Animals were euthanized when tumors reached 10% of the total body weight. To generate experimental lung metastases, mice were injected intravenously (i.v.) with 1 × 105 B16F10 tumor cells. For analysis of leukocyte infiltration, tumor tissues were digested with tumor dissociation kit (Miltenyi Biotec, San Diego, CA), and cell suspensions were filtered through a 40-μm cell strainer (ThermoFisher Scientific, Waltham, MA) as described.Citation25
For osteosarcoma model, tumors were established by injecting 5 × 105 K7 M2-luc2 cells i.v. to Balb/c mice. Mice received intranasal administered BG34-200 weekly for 7 weeks at 25 mg/kg. Mice treated with PBS served as control. Tumor growth and metastasis were monitored by non-invasive bioluminescent imaging. Images collected via an IVIS Spectrum instrument (Perkin Elmer, Waltham, MA) using their proprietary acquisition and data analysis software (Living Image v4.5).
Real time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen Corp., Carlsbad, CA). Reverse transcriptional and real-time reverse transcriptional PCR were performed using primers and FAM-labeled probe sets from Applied Biosystems (Carlsbad, CA). Gene expression was quantified relative to the expression of β-actin, and normalized to that measured in PBS-treated group by standard 2 (-ΔΔCT) calculation.
Expansion and function assay of bone marrow derived macrophages (BM-Ms) and dendritic cells (BM-DCs)
BM-Ms and BM-DCs were developed by culturing mouse bone marrow cells using M-CSF (20 ng/mL) and GM-CSF (20 ng/mL), respectively.
For BM-M cultures, BG34-200 was added to BM-M cultures at 0, 1, 5 and 20 μM. Proportions of CD11b+F4/80+ cells on day 0, 4, 7 and 14 were quantified by FACS. The day 14 BM-Ms were co-cultured with FITC-conjugated beads for 6 hours. After co-culture, BM-Ms were washed to remove beads and analyzed by FACS to quantify macrophage phagocytosis of fluorescent beads. The day 14 BM-Ms were also co-cultured with B16F10 cells at 5: 1 ratio. After co-culture with tumor cells, cell conditioned media were harvested to determine the concentration of inflammatory cytokines using cytometric bead array mouse inflammation kit (TNF-α, IL-6, IL-10, MCP-1, IFN-γ and IL-12p70).
For BM-DC cultures, BG34-200 was added to BM-DC cultures at 0, 1, 5 and 20 μM. BG34-200-treated macrophage conditioned medium at different dilution were also added to BM-DC cultures. For these cultures, proportions of CD11 c+ cells on day 0, 4, 7 and 14 were quantified by FACS. Function of these BM-DCs were determined by T cell activation assay. BMDCs were pulsed with 25 μg/mL of gp10025-33 peptides. Serially diluted cells were incubated with 1 × 105 purified PMEL CD8+ T cells for 3 days. 3H-thymidine (3H-TdR, 0.5 μCi/well) was added to the wells for the final 16 h of culture. T cell proliferation was measured using 3H-TdR incorporation assays.
T cell activation assays
For intracellular cytokine staining of T cells, gp10025-33 peptide (1 μg/mL)-stimulated splenocytes or tumor draining lymph node cells (TDLNs) from treated or untreated group were treated with PMA (10 nm) plus ionomycin (1μM) in the presence of brefeldin A (5 μg/mL) for 5 h. Cells were stained with anti-CD8 Abs and permeabilized using a Cytofix/Cytoperm kit (BD Biosciences). The cells were then stained with Abs for IFN-γ or granzyme B.
FACS analysis
Multi-parameter FACS analysis of various immune cell phenotype was performed by staining with monoclonal Abs listed in supplemental Table 2 and 3. Flow cytometric analysis was performed on BD LSRII flow cytometer (BD Biosciences, San Jose, CA); compensation and analysis were performed using Winlist 7.0 (Verity Software House, Inc. Topsham, ME).
Table 2. XXXXX.
Statistical analysis
Data are expressed as mean ± S.D. values. Statistical significance between groups within experiments was determined by the Student's t-test or ANOVA test. Values of p <0.05 were considered to be statistically significant.
Results
Systemic treatment using BG34-200 generates potent systemic anti-melanoma response
We have previously engineered BG34 of molecular weight 10 kDa (BG34-10), 200 kDa (BG34-200) and 500 kDa (BG34-500) and observed that BG34-200 could mediate the most effective uptake by murine bone marrow macrophages (BMM) [20]. Compared to BG34-10, BG34-500, and PBS and dextran controls, intraperitoneal (i.p.) treatment of wild type (WT) C57 BL/6 mice bearing established subcutaneous B16F10 tumor with BG34-200 resulted in a potent inhibition of tumor growth (, and ), leading to significantly prolonged survival of the mice (). We also examined the dosing effects by treating tumor-bearing mice with BG34-200 twice, three times or five times a week. Administration of BG34-200 three times or five times a week i.p. resulted in the most effective therapeutic benefits (Supplementary Fig. S1). Mice that responded to BG34-200 treatment did not show any noticeable autoimmune symptoms (e.g., vitiligo) or apparent toxicities in major organs (data not shown).
Figure 1. Systemic administration of BG34-200 induces a potent anti-melanoma response. (A). Schedule for subcutaneous inoculation of B16F10. WT C57BL/6 mice inoculated with subcutaneously injected B16F10 and treated by PBS, BG34-10, BG34-200, BG34-500 and dextran. (B) Tumor volumes in WT C57BL/6 mice bearing established B16F10 and different treatments. Tumor volumes at preclinical endpoint (day 18) for individual animals in each group were graphed. n = 10 per group. ##p < 0.01. Significance was determined by two-way ANOVA with Student's t test. (C) Photographs of tumors of representative mouse of untreated (PBS) and BG34-200 treatment. (D) Survival of mice treated by PBS or BG34-200. (n = 10 per group). (E) Schedule for i.v. inoculation of B16F10. WT C57BL/6 mice inoculated with i.v. injected B16F10 and treated by PBS and BG34-200. (F) Photographs of lungs of representative mouse of untreated (PBS) and BG34-200-treated. (G) The number of metastatic nodules in lungs of untreated (PBS) and BG34-200-treated mouse. n = 8 per group. ##p < 0.01. Significance was determined by two-way ANOVA with Student's t test.
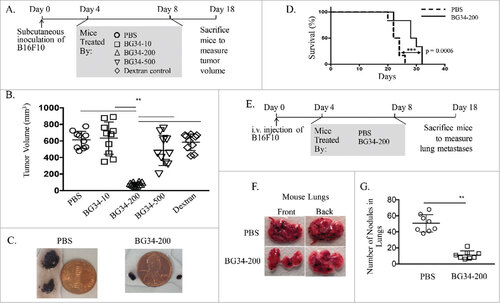
In order to evaluate whether BG34-200 treatment generated systemic protective immunity, we examined the efficacy of BG34-200 in an experimental lung metastasis model by injecting mice with B16F10 melanoma i.v. (). Indeed, mice treated with BG34-200 harbored the fewest lung metastases as compared to PBS treatment group ( and ).
BG34-200 treatment alters immune signature of melanoma microenvironment and induces T-cell activation in the tumor sites
Next, we determined BG34-200 induced changes in immune cellular composition within melanoma tumor microenvironment by FACS (Supplementary Fig. S2 A, S2B). Frequencies of intratumoral CD45highF4/80+CD11b−SSChigh macrophages and CD11 c+B220+ DCs were significantly increased, while frequencies of intratumoral granulocytes were significantly decreased in BG34-200-treated mice as compared to those receiving PBS ( and ). We observed similar BG34-200-induced increases in CD11 c+ DCs when CD11 c-venus mice were used to examine DC infiltration in primary B16F10 tumors (). BG34-200 treatment resulted in a significant increase in the frequency of tumor-infiltrating CD3+ T cells (), which were consisted of ∼30% CD4+ cells and ∼70% CD8+ cells. BG34-200 treatment also resulted in tumor-infiltrating effector T cells (both CD4+CD62 L−CD44+ and CD8+ CD62 L−CD44+) and memory T cells (CD4+CD62 L+CD44+) (), as determined by fluorescence staining and FACS analysis (Supplementary Fig. S2 C). Importantly, tumor-infiltrating CD8+ cells from BG34-200 treated mice expressed high levels of IFN-γ and granzyme B ().
Figure 2. BG34-200 treatment promotes immune activation in the tumor site. The WT C57BJ/6 mice were inoculated with subcutaneously injected B16F10 and treated by PBS or BG34-200. Three days after treatment, tumors from PBS- and BG34-200 treated mice were harvested for FACS analysis and qPCR. (A) The frequency of tumor-infiltrating macrophage and monocytes by FACS. (B) The frequency of tumor-infiltrating granulocytes and DCs by FACS. (C) Fluorescence microscopic imaging of B16F10 tumor tissues from CD11c-venus C57BL/6 mice treated by PBS or BG34-200 (Left). Fluorescence intensity of tumors treated without BG34-200 (-BG34) and with BG34-200 (+BG34-200) (Right). Data were graphed as means ± SD. Each data point represents fluorescence intensity of one region of interest. (D) The frequency of tumor infiltrating T (CD3+) and B (CD19+) cells by FACS. (E) The frequency of tumor-infiltrating CD62L+CD44+ and CD62L-CD44+ cells in CD4+ and CD8+ populations by FACS. (F) The intracellular frequency of IFN-g and Granzyme B by CD8+ populations within tumors. (G) RNA expression in B16F10 tumor tissues from WT C57BL/6 mice treated by PBS or BG34-200. For (A), (B), (D), (E), (F) and (G), n = 9 per group. (A), (B), (D), (E) and (G) data were graphed as means ± SD. For (A), (B), (D) and (E), each data point represents pooled samples from three mice. For (G), each data point represents one of three replicates of RNA samples from individual tumor. #p < 0.05, ##p < 0.01. Significance was determined by two-way ANOVA with Student's t test.
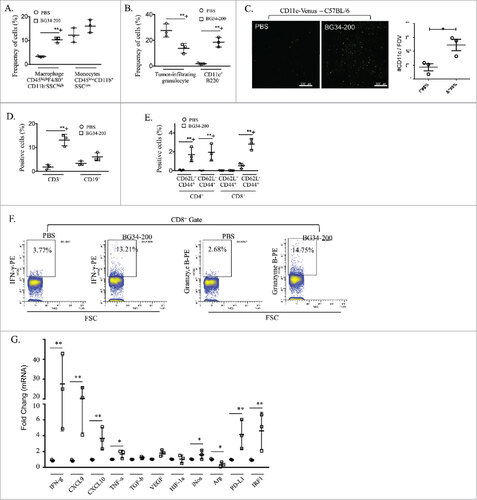
Next, we examined multiple gene expression profiles of tumor tissues by qRT-PCR. We observed that BG34-200 treatment significantly increased the mRNA expression of IFN-γ, TNF-α, CXCL9, CXCL10, PDL-1 and IRF-1 (), suggesting that BG34-200 treatment resulted in a “pro-inflammatory” tumor microenvironment. In addition, mRNA expression of iNOS was increased while that of arginase 1 was decreased in BG34-200 treated mice as compared to PBS control (), suggesting a strong M1-like activation among tumor-associated macrophages as a result of exposure to BG34-200.
BG34-200 treatment augments systemic activation of melanoma-specific T cells
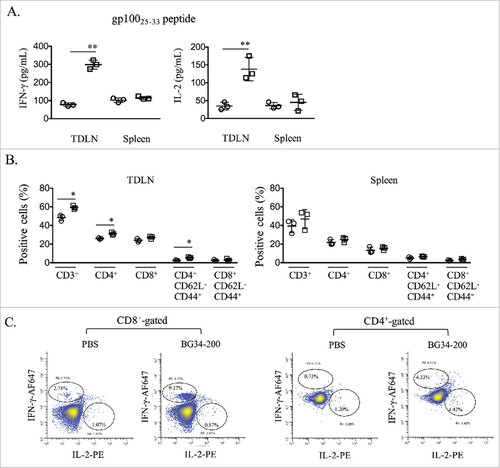
In addition to immune stimulation in the tumor site, BG34-200 treatment led to a strong systemic activation of tumor-reactive T cells, as indicated by enhanced IFN-γ and IL-2 production by T cells from the tumor draining lymph nodes (TDLNs) upon re-stimulation with H-2Db-restricted gp10025-33 in vitro, as determined by ELISA (). We also observed increased frequencies of effector memory and central memory T cells in TDLNs (, Supplementary Fig. S3). Intracellular cytokine staining also showed a higher frequency of IFN-γ and IL-2 production among gp10025-33-specific CD8+ T cells ().
Figure 3. BG34-200 treatment augments systemic activation of T cells recognizing melanoma antigen. (A) Splenocytes or TDLN cells were stimulated with gp10025-33 peptide. IFN-g and IL-2 levels in the culture media were assessed using ELISA. B. Frequencies of CD3, CD4, CD8 T cells and their expression of CD62L and CD44 in spleen and TDLN were determined by FACS analysis. C. TDLN cells were subjected to intracellular cytokine staining for determining the frequency of gp10025-33 -specific T cells. (A) and (B) were graphed as means ± SD. Each data point represents one of three replicates of samples from individual TDLN or spleen. #p < 0.05, ##p < 0.01. Significance was determined using Student's t test.
CD11b+, CD11c+, T cells and IFN-γ are required for BG34-200 induced anti-tumor responses
Given the robust BG34-200-induced anti-melanoma response, we sought to determine the effects of BG34-200 on tumor cells and various immune subsets in vitro. First, direct co-culture of BG34-200 with murine melanoma B16F10 cells or human melanoma A375 cells did not directly affect tumor cell proliferation, as determined by MTT assay (Supplementary Fig. S4). Next, direct co-culture of BG34-200 with murine TDLNs or human melanoma draining lymph nodes in the presence of IL-2 and CD3/CD28 activating beads in vitro demonstrated that BG34-200 did not directly affect T cell expansion, CD4/CD8 ratio or IFN-γ/IL-2 production (Supplementary Fig. S5).
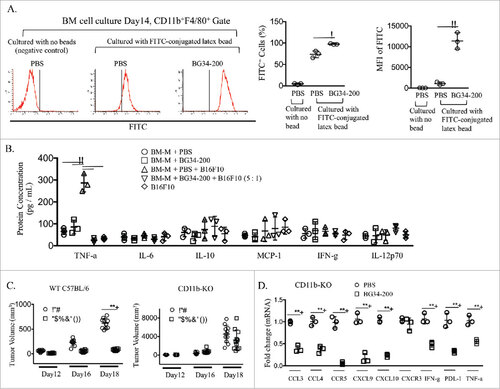
Since our previous study has demonstrated that BG34 could directly engage macrophages via binding to CD11b receptor,Citation4 we co-cultured BG34-200 with bone marrow-derived primary macrophages in vitro. Although direct culture with BG34-200 did not affect macrophage expansion (Supplementary Fig. S6), exposure to BG34-200 significantly enhanced the phagocytic activity of macrophages () and their secretion of TNF-α (, ). To confirm the important functional roles of macrophages and CD11b in BG34-200 mediated effect, we utilized mice that lack CD11b+ myeloid cells (CD11b-KO) to define the potential involvement of CD11b in BG34-200-induced anti-melanoma immune responses. Our results showed that the lack of CD11b+ cells abrogated the in vivo therapeutic efficacy of BG34-200 (). Also, the BG34-200-mediated increase of gene expressions (IFN-γ, TNF-α, CXCL9, CXCL10, PDL-1 and IRF-1) in tumors of WT mice () was abrogated in CD11b-KO mice receiving the same BG34-200 treatment (). These results suggest that CD11b+ cells play an important contributory role in the observed BG34-200-mediated antitumor effect. Furthermore, mRNA levels of CCL3, CCL4, CCR5, CXCL9, CXCL10, IFN-γ, PDL-1 and TNF-α in the primary tumor sites were significantly reduced in CD11b-KO mice compared to WT mice even in the absence of BG34-200 administration (), further supporting the view that CD11b+ cells contribute critically to the production of these factors. Consistent with these views, deletion of CD11b+ cells results in the lack of myeloid-derived monocytes, macrophages, dendritic cells, neutrophils or granulocytes in tumor microenvironment, cells which are principle mediators of inflammatory cytokines.
Figure 4. CD11b+ cells are required for BG34-200-induced anti-melanoma responses. (A). Bone marrow derived macrophages (BM-Ms) were cultured with BG34-200 in the presence of MCSF for 14 days. PBS-treated BM-Ms served as treatment control. The PBS- or BG34-200-treated BM-Ms were then co-cultured with FITC-conjugated latex beads to determine phagocytic activity. PBS-treated BM-Ms with no beads serve as control. Left: FACS histograms showing the frequencies of FITC+ cells in the CD11b+F4/80+-gated BM-M cell populations. Middle: Percentage of FITC+ cells in CD11b+F4/80+ BM-Ms. Right: Mean fluorescence intensity (MFI) of FITC+ cells in CD11b+F4/80+-BM-Ms. (B) Inflammatory cytokine concentration in cell conditioned media. BM-Ms were cultured with BG34-200. PBS-treated BM-Ms served as control. The PBS- and BG34-200-treated BM-Ms were then co-cultured with B16F10 tumor cells at BMM to B16F10 ratio 5:1. BM-M or B16F10 cell conditioned media served as controls. Inflammatory cytokines (TNF-a, IL-6, IL-10, MCP-1, IFN-g and IL-12p70) in cell conditioned media were determined by cytometric bead array. For (A) and (B), data were graphed as means ± SD. Each data point represents one of three replicates of samples from individual BM-M cultures. (C) Tumor volumes in WT C57BJ/6 or CD11b-KO mice bearing established B16F10 with or without BG34-200 treatment. Tumor volumes at day 12, 16 and 18 for individual animals in each group were graphed. n = 9 per group. (D) RNA expression in B16F10 tumor tissues from CD11b-KO mice treated by PBS or BG34-200. Each data point represents one of three replicates of RNA samples from individual tumor. For (A) – (D), #p < 0.05, ##p < 0.01. Significance was determined by two-way ANOVA with Student's t test.
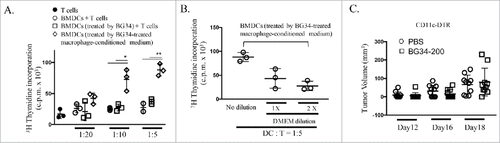
Considering the important role of DCs in coordinating innate and adaptive immune responses, we performed direct co-culture of BG34-200 with BMDCs in vitro. Neither the expansion of BMDCs nor their antigen presenting capability was affected ( and Supplementary Fig. S7). However, when co-cultured with BG34-200-treated macrophage-conditioned medium, BMDCs appeared to exhibit enhanced antigen presenting capability in a dose-dependent manner, as determined by 3 H-thymidine incorporation assay of PMEL CD8+ T cell activation driven by gp10025-33-pulsed BMDCs ( and ). Finally, we utilized CD11 c-DTR transgenic mice to define the potential involvement of CD11 c+ DCs in BG34-200-induced anti-melanoma immune responses. We developed bone marrow chimera in which WT mice were reconstituted with the BM from CD11 c-DTRtg mice. Our results showed that depletion of CD11 c+ cells in diphtheria toxin (DTx)-treated chimera mice abrogated the therapeutic efficacy of BG34-200 () as compared to WT mice receiving PBS and BG34-200 (, left).
Figure 5. CD11c+ cells are required for BG34-200-induced anti-melanoma responses. (A) 3H thymidine assay of PMEL CD8+ T cells co-cultured with gp10025-33 peptide-pulsed BMDCs. T cells alone served as negative control. BMDCs were treated by PBS or BG34-200, or BG34-200-treated macrophage-conditioned medium. BMDCs pulsed with gp10025-33 peptide and co-cultured with PMEL CD8+ T cells at DC : T ratios of 1: 20, 1:10 and 1:5. (B) BG34-200-treated macrophage-conditioned medium at three different dilutions with DMEM were used to co-culture with BMDCs. Then BMDCs were pulsed with gp10025-33 peptide and co-cultured with PMEL CD8+ T cells at DC : T ratios of 1:5. (C) Tumor volumes in CD11c-KO mice bearing established B16F10 with or without BG34-200 treatment. Tumor volumes at day 12, 16 and 18 for individual animals in each group were graphed. n = 9 per group. # p < 0.05, ## p < 0.01. Significance was determined by two-way ANOVA with Student's t test.
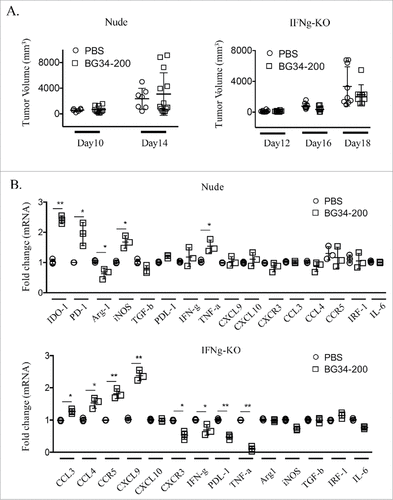
In addition to testing BG34-200 effects in CD11b-KO and CD11 c-DTR mouse models, we also treated tumor-bearing nude mice and IFN-γ-KO transgenic mice with BG34-200. Our results showed that lymphocytes and IFN-γ were required for the observed BG34-200-induced melanoma regression () as compared to WT mice receiving PBS and BG34-200 treatment (, left). Tumors harvested from nude mice treated with BG34-200 showed a significant increase in mRNA levels of IDO-1, PD-1, iNOS, PDL-1 and TNF-α, while the levels of Arg-1 and TGF-β were significantly decreased (). In contrast, tumors from BG34-200 treated IFN-γ-KO mice exhibited a significantly decreased mRNA level of CXCR3, IFN-γ, PDL-1, TNF-α and IL-6 while those of CCL3, CCL4, CCR5 and CXCL9 were significantly increased ().
Figure 6. Lymphocytes and IFN-g are essential for BG34-200-induced antitumor responses. (A) Tumor volumes in nude (left) and IFN-g-KO mice (right) bearing established B16F10 with or without BG34-200 treatment. Tumor volumes at different time points for individual animals in each group were graphed. n = 9 per group. (B) RNA expression in B16F10 tumor tissues from nude mice (up) and IFN-g-KO mice (down) with or without BG34-200 treatment. Each data point represents one of three replicates of RNA samples from individual tumor. # p < 0.05, ## p < 0.01. Significance was determined by two-way ANOVA with Student's t test.
BG34-200 treatment potently inhibits osteosarcoma tumor growth in vivo
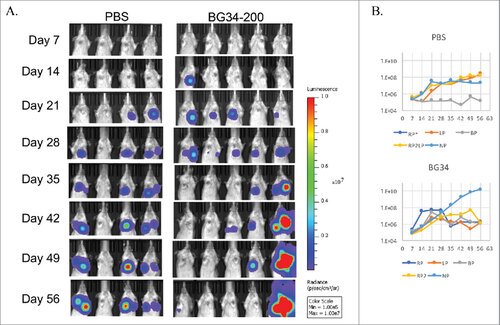
To test anti-tumor enhancing capacity of BG34-200 beyond melanoma, we administered BG34-200 into Balb/c mice inoculated intratibially with metastatic osteosarcoma cell line, K7 M2 (genetic engineered to express luciferase gene Luc2) i.v. to create sarcoma with both primary and lung metastastic lesions. In PBS-treated mice, 75% of the mice developed both primary and metastatic K7 M2-Luc2 osteosarcoma tumors. By contrast, while all of the BG34-200-treated mice developed primary tumors on day 28, 80% of these mice showed diminished tumor burden on day 56, at a time when all of the PBS-control mice reached pre-clinical endpoints ().
Figure 7. Systemic administration of BG34-200 induces antitumor responses in K7M2-Luc2 osteosarcoma model of Balb/c mice. Balb/c mice bearing i.v. injected K7M2-Luc2 osteosarcoma cells were administered with BG34-200 intranasal once a week for 7 weeks. PBS-treated mice bearing i.v. injected K7M2-Luc2 osteosarcoma cells served as control. Tumor volume were quantified by non-invasive imaging of luciferase signal. (A) Weekly whole body imaging of mice treated by PBS or BG34-200. (B) Mean fluorescence intensity of PBS-treated (up) and BG34-200-treated (down) mice at various time points. For PBS group, n = 4; for BG34-200 group, n = 5.
Discussion
Tumor escape can result from the loss of immunogenicity in cancer cells and/or the establishment of an immunosuppressive state within the tumor microenvironment.Citation26 Therefore, it is critical to devise new therapeutic strategies that are capable of effectively and safely restoring immune-mediated tumor recognition and delivery of effector function. In the present study, we demonstrate that an oat-derived β-glucan (BG34) with a specific molecular weight 200kDa (BG34-200) displays a superior immune stimulating activity at the melanoma tumor sites. Furthermore, the antitumor potency of systemic BG34-200 therapy was demonstrated using both murine model of metastatic melanoma () and osteosarcoma in two different genetic strains (). In addition, BG34-200 therapy also augments systemic protective immunity, dramatically reducing distant pulmonary metastases. Therefore, BG34-200 represents a highly potent, non-toxic immune modulator that may be used to overcome cancer-induced immune tolerance.
The systemic administration of BG34-200 has a strong immune modulating impact on the tumor environment, as evidenced by dramatic increases in the levels of inflammatory factors including IFN-γ, TNF-α, CXCL9, CXCL10, IRF1 and PDL-1 in BG34-200-treated tumors ( and ). The ratios of iNos/Arg1 in the BG34-200-treated tumors within WT mice are also dramatically increased (), suggesting M1-type activation of macrophages. These changes were associated with significantly increased tumor infiltrations of IFN-γ and Granzyme B producing T cells (, and ). In addition to immune activation in the tumor site, BG34-200 therapy results in systemic mobilization of tumor-reactive T cells, as indicated by lymphocyte activation in TDLN (). These activated T cells contained those that recognize naturally expressed endogenous melanoma antigens, gp100 ().
Knock-out mice studies clearly demonstrate that CD11b+ cells, CD11 c+ DCs and T (and possibly NK) cells are involved in the BG34-200-induced immune control of B16F10 (, and ). IFN-γ was also critical in the antitumor efficacy of BG34-200 (). Knock-out mice studies demonstrated that CD11b+ myeloid cells, T cells and possibly NK cells are required for IFN-γ production in tumor sites ( and ). In addition, lack of IFN-γ diminished the BG34-200-induced upregulation of CCL3, CCL4, CCR5, CXCL9, CXCL10, PDL-1, TNF-α, and iNos/Arg1 ratio in tumor site (). These results emphasize the requirement of CD11b+ myeloid cells for BG34-200-induced activation of tumor-specific effector cells. Although these findings provide mechanistic insights into the therapeutic activity of BG34-200, future studies are necessary to further examine how DC, T or NK cells regulate the antitumor effect of BG34-200 in vivo.
A recent study reported an FDA-approved ferumoxytol nanoparticles that, when co-injected with MMTV-PyMT cancer cells into mice, could suppress tumor growth by introducing M1-type macrophage responses in tumor site.Citation27 By contrast, our study demonstrates that the systemic administration of BG34-200 to mice bearing established metastatic tumor could result in a robust tumor regression by mediating M1-type macrophage activation and cytotoxic T cell responses in tumor site. This suggests that BG34-200 is much more efficacious than the FDA-approved iron oxide nanoparticles in generating antitumor immunity via modulating macrophages in tumor environment.
On the other hand, glucan-type molecules such as particulate yeast β-glucan have been reported to convert polarized alternatively activated macrophages or immunosuppressive TAM into a classically activated phenotype with potent immunostimulating activity. Nevertheless, particulate yeast glucan exhibits poor water solubility and is administered orally, which limited the bioavailability and clinical efficacy.Citation4 Recent studies on small molecular oat-derived beta glucan demonstrate that they have a direct cytotoxic effect on tumor cells by inducing strong expression of caspase-3, -7 and/or -12 in cancer cell lines, resulting in tumor cell apoptosis.Citation21,28 However, prior to our current study, the in vivo antitumor activity and immune-stimulating effect of oat bran-derived β-glucan containing a well-characterized chemical structure, linear chain (no branches), high purity, free of endotoxin, excellent solubility and specific molecular weight has not been reported. Our study is the first to directly demonstrate that BG34-200 is much more efficacious than particulate yeast glucan in generating antitumor immunity in different cancer models tested.
We hypothesize that the efficient uptake and high capacity of BG34-200 to modulate tumor microenvironment contributes to potent macrophage and DC activation after BG34-200 therapy. In vitro BG34-200-treated BM macrophages exhibit significantly enhanced phagocytosis and secretion of TNF-α (), suggesting a direct effect of BG34-200 on stimulating inflammatory responses of macrophages. Intriguingly, BMDCs exposed to BG34-200-treated macrophage conditioned medium were more potent than BMDCs treated directly with BG34-200 in stimulating gp100 specific T cells in vitro ( and ). This suggests that the BG34-200-treated macrophage conditioned medium contain substances that could potentially enhance DCs antigen processing/presenting function. However, BMDCs cultures with BG34-200 and TNF-α of various concentrations appear not to activate DCs the same way as those with BG34-200 treated BM macrophage condition medium. Indeed, early studies have shown that immune cells including neutrophils, granulocytes and NK cells can interact with macrophage to process and secret bio-active glucan moiety (small fragment of glucan) to participate in immune activation and tumor killing.Citation4 Our future study will focus on determining whether BG34-200-treated macrophages contain altered cytokine profile and/or BG34 fragments that facilitate DC activation and enhanced antigen processing and presentation.
In addition, results of this study inspire us to further examine the working mechanism of BG34-200 for DC trafficking/activation and Th1-type cell infiltration. Indeed, Th-1 type chemokines such as CXCL9 and CXCL10 are known to mediate the trafficking of CXCR3+ cytotoxic T cells and NK cells across tumor vasculature, thus may play key roles in cancer immunotherapy. Recent studies have shown that a melanoma-intrinsic oncogenic signaling pathway (WNT/β-catenin) contribute to lack of T cells in melanoma, and data revealed that deactivation of the WNT/β-catenin of melanoma resulted in the production of CCL4 which is a key chemokine for the infiltration of CCR5+ DCs producing high level of CXCL9 and CXCL10.Citation29,30 Based upon that, our observations that the BG34-200-mediated increase of CXCL9 and CXCL10 in tumor microenvironment and that the DCs, macrophages and T cells involved in the antitumor role of BG34-200 provide strong rational for us to further examine whether BG34-200 can enhance infiltration of CTLs (and NK cells) via the modulation of WNT/β-catenin signaling and CCR5+ DCs infiltration. Observations from this current study also now inspire us to further examine whether BG34-200 can enhance CCR7+ DCs in tumor and draining lymph nodes, as well as antigen presenting capability of these DCs. Results of these studies will determine whether BG34 can enhance DC trafficking to melanoma and draining lymph nodes to enhance Th-1 cell activation and expansion for systemic antitumor effect.
In summary, our studies demonstrate a profound antitumor effect of systemic administration of BG34-200 in vivo. It is capable of “conditioning” an immune-suppressive/tolergenic tumor environment to result in therapeutic antitumor immunity. However, questions remain with regard to the precise molecular and cellular events triggered by BG34-200. Despite that fact that BG34 functions as a ligand for CR3,Citation20 our studies suggest that direct macrophage activation by BG34-200 may further trigger DCs activation through an as yet undefined signaling pathway. BG34-200-mediated antitumor immunity requires CD11b+ myeloid cells, DCs, and T cells, and driven by IFN-γ. More studies are warranted to further investigate the molecular actions of BG34-200 in vivo and the feasibility of exploiting this novel immune modulator to break tumor-induced immune tolerance in clinical application. Since BG34-200 displays excellent capability of inducing melanoma-specific T cells, our future study will also focus on combining BG34 with anti-PD-1, anti-PD-L1 or anti-CTLA-4 checkpoint blockade.
Supp_data.zip
Download Zip (60.2 MB)Acknowledgment
This work is supported by funding from the VeloSano Cancer Research Award (M.Z. and J.A.K.). This work is supported by funding from the National Cancer Institute (R21CA181875 and R21CA218790 to AYH), the St. Baldrick's Foundation, the Alex's Lemonade Stand Foundation, Hyundai Hope-on-Wheels Program, Pediatric Cancer Research Foundation, the Steven G. AYA Cancer Research Fund, Samuel Szabo Foundation, Keira Kilbane Cancer Discovery Fund, Erroll's Cancer Discovery Fund, and the Theresia G. & Stuart F. Kline Family Foundation.
Conflict of interest statement
The authors do not have any conflict of interest to declare.
References
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. doi:10.1038/nm1100. PMID:15340416
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J, Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev. 2006;213:131–45. doi:10.1111/j.1600-065X.2006.00442.x. PMID:16972901
- Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi:10.1126/science.1203486. PMID:21436444
- Chan GC, Chan WK, Sze DM. The effects of beta-glucan on human immune and cancer cells. J Hematol Oncol. 2009;2:25. doi:10.1186/1756-8722-2-25. PMID:19515245
- Yan J, Allendorf DJ, Brandley B. Yeast whole glucan particle (WGP) beta-glucan in conjunction with antitumour monoclonal antibodies to treat cancer. Expert Opin Biol Ther. 2005;5(5):691–702. doi:10.1517/14712598.5.5.691. PMID:15934844
- Li B, Allendorf DJ, Hansen R, Marroquin J, Ding C, Cramer DE, Yan J. Yeast beta-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells ia complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol. 2006;177(3):1661–9. doi:10.4049/jimmunol.177.3.1661. PMID:16849475
- Liu J, Gunn L, Hansen R, Yan J. Combined yeast-derived beta-glucan with anti-tumor monoclonal antibody for cancer immunotherapy. Exp Mol Pathol. 2009;86(3):208–14. doi:10.1016/j.yexmp.2009.01.006. PMID:19454271
- Vetvicka V, Thornton BP, Ross GD. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest. 1996;98(1):50–61. doi:10.1172/JCI118777. PMID:8690804
- Xia, Y, Ross GD. Generation of recombinant fragments of CD11b expressing the functional beta-glucan-binding lectin site of CR3 (CD11b/CD18). J Immunol. 1999;162(12):7285–93. PMID:10358177
- Bose N, Chan AS, Guerrero F, Maristany CM, Qiu X, Walsh RM, Ertelt KE, Jonas AB, Gorden KB, Dudney CM, et al. Binding of Soluble Yeast beta-Glucan to human neutrophils and monocytes is complement-dependent. Front Immunol. 2013;4:230. doi:10.3389/fimmu.2013.00230. PMID:23964276
- Liu M, Luo F, Ding C, Albeituni S, Hu X, Ma Y, Cai Y, McNally L, Sanders MA, Jain D, et al. Dectin-1 activation by a natural product beta-glucan converts Immunosuppressive Macrophages into an M1-like Phenotype. J Immunol. 2015;195(10):5055–65. doi:10.4049/jimmunol.1501158. PMID:26453753
- Hong F, Yan J, Baran JT, Allendorf DJ, Hansen RD, Ostroff GR, Xing PX, Cheung NK, Ross GD. Mechanism by which orally administered beta-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol. 2004;173(2):797–806. doi:10.4049/jimmunol.173.2.797. PMID:15240666
- Vannucci L, Krizan J, Sima P, Stakheev D, Caja F, Rajsiglova L, Horak V, Saieh M. Immunostimulatory properties and antitumor activities of glucans (Review). Int J Oncol. 2013;43(2):357–64. doi:10.3892/ijo.2013.1974. PMID:23739801
- Samuelsen AB, Schrezenmeir J, Knutsen SH. Effects of orally administered yeast-derv ed beta-glucans: a review. Mol Nutr Food Res. 2014;58(1):183–93. doi:10.1002/mnfr.201300338. PMID:24019098
- Muta T. Molecular basis for in ertebrate innate immune recognition of (1->3)-beta-D-glucan as a pathogen-associated molecular pattern. Curr Pharm Des. 2006;12(32):4155–61. doi:10.2174/138161206778743529. PMID:17100618
- Ezekowitz RA, Sim RB, Hill M, Gordon S. Local opsonization by secreted macrophage complement components. Role of receptors for complement in uptake of zymosan. J Exp Med. 1984;159(1):244–60. doi:10.1084/jem.159.1.244. PMID:6319531
- Tani M, Tanimura H, Yamaue H, Tsunoda T, Iwahashi M, Noguchi K, Tamai M, Hotta T, Mizobata S. Augmentation of lymphokine-acti ated killer cell acti ity by lentinan. Anticancer Res. 1993;13(5C):1773–6. PMID:8267381
- Qi C, Cai Y, Gunn L, Ding C, Li B, Kloecker G, Qian K, Vasilakos J, Saijo S, Iwakura Y, et al. Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived beta-glucans. Blood. 2011;117(25):6825–36. doi:10.1182/blood-2011-02-339812. PMID:21531981
- Li B, Cramer D, Wagner S, Hansen R, King C, Kakar S, Ding C, Yan J. Yeast glucan particles activate murine resident macrophages to secrete proinflammatory cytokines ia MyD88- and Syk kinase-dependent pathways. Clin Immunol. 2007;124(2):170–81. doi:10.1016/j.clim.2007.05.002. PMID:17572156
- Zhang M, Kim JA. Effect of molecular size and modification pattern on the internalization of water soluble beta-(1 ->3)-(1 ->4)-glucan by primary murine macrophages. Int J Biochem Cell Biol. 2012;44(6):914–27. doi:10.1016/j.biocel.2012.02.018. PMID:22679629
- Choromanska A, Kulbacka J, Rembialkowska N, Pilat J, Oledzki R, Harasym J, Saczko J. Anticancer properties of low molecular weight oat beta-glucan - An in vitro study. Int J Biol Macromol. 2015;80:23–8. doi:10.1016/j.ijbiomac.2015.05.035. PMID:26092171
- Cheung NK, Modak S. Oral (1->3),(1->4)-beta-D-glucan synergizes with antiganglioside GD2 monoclonal antibody 3F8 in the therapy of neuroblastoma. Clin Cancer Res. 2002;8(5):1217–23. PMID:12006541
- Yi H, Yu X, Gao P, Wang Y, Baek SH, Chen X, Kim HL, Subjeck JR, Wang XY. Pattern recognition sca enger receptor SRA/CD204 down-regulates Toll-like receptor 4 signaling-dependent CD8 T-cell activation. Blood. 2009;113(23):5819–28. doi:10.1182/blood-2008-11-190033. PMID:19349620
- Fisher DT, Chen Q, Skitzki JJ, Muhitch JB, Zhou L, Appenheimer MM, Vardam TD, Weis EL, Passanese J, Wang WC, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. 2011;121(10):3846–59. doi:10.1172/JCI44952. PMID:21926464
- Yi H, Guo C, Yu X, Gao P, Qian J, Zuo D, Manjili MH, Fisher PB, Subjeck JR, Wang X. Targeting the immunoregulator SRA/CD204 potentiates specific dendritic cell accine-induced T-cell response and antitumor immunity. Cancer Res. 2011;71(21):6611–20. doi:10.1158/0008-5472.CAN-11-1801. PMID:21914786
- Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241(1):104–18. doi:10.1111/j.1600-065X.2011.01007.x. PMID:21488893
- Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, Coussens LM, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–994. doi:10.1038/nnano.2016.168. PMID:27668795
- Parzonko A, Makarewicz-Wujec M, Jaszewska E, Harasym J, Kozłowska-Wojciechowska M. Pro-apoptotic properties of (1,3)(1,4)-beta-D-glucan from Avena sativa on human melanoma HTB-140 cells in vitro. Int J Biol Macromol. 2015;72:757–63. doi:10.1016/j.ijbiomac.2014.09.033. PMID:25285849
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. doi:10.1038/nature14404. PMID:25970248
- Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour ascular checkpoints. Nat Commun. 2015;6:7458. doi:10.1038/ncomms8458. PMID:26109379