
ABSTRACT
Purpose of our study was to explore a new immunotherapy for high grade soft tissue sarcomas (STS) based on cytokine-induced killer cells (CIK) redirected with a chimeric antigen receptor (CAR) against the tumor-promoting antigen CD44v6. We aimed at generating bipotential killers, combining the CAR specificity with the intrinsic tumor-killing ability of CIK cells (CAR+.CIK). We set a patient-derived experimental platform. CAR+.CIK were generated by transduction of CIK precursors with a lentiviral vector encoding for anti-CD44v6-CAR. CAR+.CIK were characterized and assessed in vitro against multiple histotypes of patient-derived STS. The anti-sarcoma activity of CAR+.CIK was confirmed in a STS xenograft model. CD44v6 was expressed by 40% (11/27) of patient-derived STS. CAR+.CIK were efficiently expanded from patients (n = 12) and killed multiple histotypes of STS (including autologous targets, n = 4). The killing activity was significantly higher compared with unmodified CIK, especially at low effector/target (E/T) ratios: 98% vs 82% (E/T = 10:1) and 68% vs 26% (1:4), (p<0.0001). Specificity of tumor killing was confirmed by blocking with anti-CD44v6 antibody. CAR+.CIK produced higher amounts of IL6 and IFN-γ compared to control CIK. CAR+.CIK were highly active in mice bearing subcutaneous STS xenografts, with significant delay of tumor growth (p<0.0001) without toxicities.
We report first evidence of CAR+.CIK's activity against high grade STS and propose CD44v6 as an innovative target in this setting. CIK are a valuable platform for the translation of CAR-based strategies to challenging field of solid tumors. Our findings support the exploration of CAR+.CIK in clinical trials against high grade STS.
Introduction
In this study, we explored the anti-sarcoma activity of a new adoptive immunotherapy strategy, based on Cytokine-Induced Killer cells (CIK) redirected with a Chimeric Antigen Receptor (CAR) against the isoform variant 6 of adhesive receptor CD44 (CD44v6). Citation1-3Soft tissue sarcomas (STS) are a heterogeneous group of mesenchymal tumors and, in advanced stages, represent a highly unmet medical need. Conventional treatments usually fail to cure advanced STS patients, in most cases young adults, and there is an urgent need for new therapeutic approaches.Citation4 Adoptive immunotherapy is among the most promising strategies for treatment of chemo-refractory solid tumors.Citation5 T lymphocytes redirected with a clonal T cell receptor (TCR) anti-NY-ESO1 have demonstrated significant clinical responses, including remissions, in selected cases of synovial sarcomas.Citation6,Citation7 These findings constitute an important proof-of-concept on the potential efficacy of adoptive immunotherapy in sarcomas and brought new hopes and enthusiasm in the field. This first example is however restricted to the few selected cases that express the NY-ESO1 target antigen and is only applicable to patients with a precise HLA haplotype (e.g. HLA-A2). The exploration and development of alternative immunotherapy strategies, with a wider spectrum of applicability, are still needed for the majority of STS patients.
To this purpose, immunotherapy with CIK appears a promising option.
CIK are ex vivo expanded T lymphocytes, endowed with T-NK phenotype and intense MHC-independent antitumor ability reported against various types of solid and hematologic malignancies.Citation8-13 Recent clinical trials support their activity and an excellent safety profile in challenging settings such as lung, renal, liver, breast and gastrointestinal cancers.Citation14-16 A summary of the main clinical findings was also published in the International Registry on CIK Cells (IRCC).Citation17 Overall, a beneficial effect of CIK emerged in patients with hepatocellular carcinoma, renal cell carcinoma, non-small cell lung cancer (NSCLC), colorectal carcinoma, and breast cancer.Citation17
The intrinsic tumor killing ability of CIK is mainly mediated by the NKG2D receptor that recognizes, in MHC-independent manner, stress-inducible targets (MIC A/B, ULBPs) which are selectively expressed by transformed cells.Citation18,Citation19 The subset of CIK co-expressing CD3 and CD56 molecules (CD3+CD56+) is present at variable rates and is considered the most capable of antitumor activity. CIK with CD3+CD56− phenotype have a minor, but however positive, tumor-killing capacity.Citation20
Importantly, NKG2D ligands are not restricted to a specific tumor histotype since their expression has been described in various epithelial cancers and we recently confirmed this finding in various types of sarcomas.Citation11
We reported that patient-derived CIK are active against autologous STS, but observed that their function decreases at low effector/target (E/T) ratios, thus showing limitations in clinical perspective.
Adoptive immunotherapy with CIK might greatly benefit from the new redirection opportunities offered by the developing strategies with engineered tumor-specific receptors.Citation6,Citation7,Citation21,Citation22 In particular, CAR-based approaches showed impressive therapeutic potential in selected hematologic malignancies even if with relevant safety warnings.Citation23 CARs are constructed by fusing the single chain variable fragment (scFv) of a tumor surface antigen-specific monoclonal antibody with an intracellular TCR-derived signaling domain and costimulatory molecules.Citation24,Citation25
Most importantly, their clinical application is not restricted by HLA-haplotypes with the important implication that, if a meaningful tumor antigen is identified, this strategy may be in theory appropriate to all patients affected by tumors expressing that specific target. The effective translation of CAR-strategies to the field of solid tumors has been so far disappointing and it is still object of intense research.Citation22
The first evaluation of a CAR strategy in sarcoma patients, with anti-HER2-CAR T cells, provided initial proof of concept about its feasibility and limited evidence of activity.Citation26
CIK with their biologic features might provide an intriguing platform for CAR-strategies, with potential positive impact in the challenging translation to the field of solid tumors and favorable safety implications. CAR engineered CIK would generate bi-specific tumor killers, conjugating in the same effector the CAR-specificity with the intrinsic NKG2D-mediated antitumor capacity. Advantages may derive by enhanced efficacy against tumors with heterogeneous antigen expression and by the possible contrast to tumor clonal selection events. CIK have demonstrated a very favorable safety profile in the early clinical trials.Citation9,Citation27-29 Their limited lifespan and persistence in vivo requires multiple therapeutic infusions but might acquire a positive valence in terms of safety when considering CAR-CIK.Citation30
Initial feasibility of CAR engineering and preclinical activity of CAR-CIK were reported against selected hematologic malignancies,Citation31-34 boosting recent scientific interest and need for their investigation in solid tumor settings.Citation35,Citation36
A crucial issue in CAR strategies is the identification of a suitable target antigen. Ideally it should be selectively expressed by tumor cells and sustaining their growth. The CD44v6 may be an ideal target for immunotherapy as it is a tumor-promoting antigen, associated with the metastatic process and tumor initiating cells.Citation2,Citation37 CD44v6 is broadly expressed in hematologic malignancies and several solid tumors.Citation38 Few retrospective studies described CD44v6 expression in selected STS, reporting a possible correlation with the clinical aggressiveness and patients' prognosis.Citation1,Citation39
The significant preclinical antitumor efficacy of anti-CD44v6 CAR-T cells against acute myeloid leukemia and multiple myeloma was recently reported.Citation38
We aimed at defining and exploring a new anti-CD44v6 CAR-CIK adoptive immunotherapy within the challenging setting of high grade STS. In this model, CIK are engineered with a lentiviral vector encoding for anti-CD44v6-CAR with a CD28 signaling domain and HSV-TK inducible suicide switch. We exploited our experimental platform,Citation10-12,Citation40 based on patient-derived STS cell cultures, to assess the prevalence and intensity of CD44v6 within a wide range of STS histotypes. We assessed the efficacy of CAR-CIK from STS patients, including autologous essays, to provide reliable preclinical basis for clinical studies in this challenging field.
Results
Generation and characterization of CIK cells redirected with a CD44v6 CAR
CIK cells were generated from Peripheral Blood Mononuclear Cells (PBMCs) obtained from 12 patients, with diagnosis of Soft Tissue Sarcoma (STS), Supplemental table 1. We genetically engineered CIK to express the CD44v6-specific CAR by transducing PBMCs at day +3 of culture with the lentiviral vector LV-CD44v6.CAR28z. Three weeks after transduction we evaluated the expression of the CD44v6-specific CAR on CIK (CAR+.CIK) by flow cytometry: mean transduction efficiency, was 68%±3 (mean ± SEM) in bulk mature CIK (), the rate of the CD3+CD56+ subset was comparable between the CD44v6 CAR+/CD3+ () and the CD44v6 CAR−/CD3+ fractions (). The median ex vivo expansion of CAR+.CIK was 37 fold (range 11–645), such value is comparable to that observed with unmodified CIK (NTD.CIK) used as paired control (38 fold; range, 9–631, p = ns) (Supplemental table 1), ruling out any possible detrimental effect caused by lentiviral transduction or CD44v6 expression in CIK cells. We confirmed the functionality of the HSV-TK inducible suicide gene encoded within the LV-CD44v6.CAR28z vector. CAR+.CIK were efficiently ablated by the in vitro exposure to Ganciclovir (GCV, 10 µM for 72 h) (Supplemental Fig. 1).
Figure 1. Phenotype of antiCD44v6 CAR+.CIK cells. Mature CIK cells efficiently expressed anti-CD44v6CAR (A). The CD3+CD56+ subset of CIK cells was equally distributed between CAR+ (B) and CAR− (C) cells. CAR+.CIK were mostly effector memory (EM / EM RA), followed in order by naive and central memory phenotype (D). The phenotype was comparable with that of unmodified NTD.CIK (unpaired t test, P > 0.05). Abbreviations: Chimeric Antigen Receptor (CAR); Not Transduced (NTD); Effector Memory (EM: CD45RA−/CD62 L-); Effector Memory RA (EM RA: CD45RA+/CD62 L-); Central Memory (CM: CD45RA−/CD62 L+); NAÏVE: CD45RA+/CD62 L+.
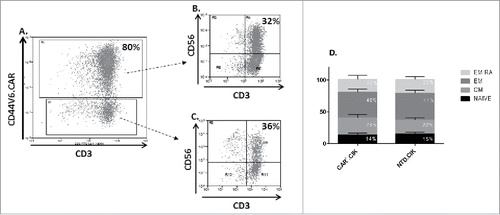
The phenotype of mature CAR+.CIK was comparable to that of paired controls with NTD CIK.
The majority of CAR+.CIK were CD8+ (67%±4, mean ± SEM) with a relevant subset co-expressing CD3 and CD56 molecules (CD3+CD56+) (37%±6, mean ± SEM).The median membrane expression of NKG2D receptor was (77%±4, mean ± SEM) and the effector memory (CD62 L+CD45RA−) was expectedly the most represented phenotype (40,6%±5, mean ± SEM) (). A complete phenotype description and comparison with NTD.CIK cells is summarized in supplemental table 1.
Expression of CD44v6 by soft tissue sarcomas
To explore the clinical relevance of CD44v6 as a target in STS, we analyzed its surface expression on 27 STS. Twenty-three out of 27 STS cell lines were directly generated from surgical biopsies obtained at our Center. CD44v6 was expressed at variable levels in 11 out of 27 (40.7%) STS, with a higher prevalence in UPS, liposarcoma and leiomiosarcoma, while lower prevalence was observed in GIST. The prevalence of CD44v6 expression in different STS histotypes is summarized in . Considering the relative fluorescence intensity (RFI) ratio, the expression of CD44v6 in STS could be classified as high (RFI >5, 27%), moderate (RFI >2 and <5, 46%) or low (RFI <2, 27%), . STS were confirmed to express variable levels of the principal known ligands recognized by the NKG2D receptor on CIK (MIC A/B, ULBP1, ULBP2/5/6, and ULBP3). MIC A/B was highly expressed only in two targets whereas less present or practically absent in the others. Analysis for ULBPs 1 was negligible, whereas ULBPs 2/5/6, and 3 displayed ubiquitous expression. HLA class-I expression was retained by all STS, .
Table 1. CD44v6 expression in STS.
Table 2. Phenotype of STS cell lines.
Antitumor activity of anti CD44v6 CAR+CIK against soft tissue sarcomas
Activity in vitro
We assessed the killing activity in vitro of CAR+.CIK, expanded from our patients, against 11 different STS. In 4/11 essays we could reproduce the autologous setting as PBMC and STS targets from the same patient were available (Supplemental Fig. 2), CIK from allogeneic patients were used in the 7 remaining cases.
CAR+.CIK efficiently killed STS and the killing activity was significantly higher if compared with unmodified NTD.CIK, especially at low effector/target ratios (n = 43; p<0.0001 and supplemental video 1). The STS killing by CAR+.CIK remained intense regardless the variable expression level (high, intermediate or low) of CD44v6 on tumor membrane, only a trend toward a better killing was observed against STS with high (RFI>5) expression of CD44v6 (). The rate of viable tumor cells continued to progressively decrease up to 72 hours after stopping the experiment and removal of CAR+.CIK (n = 2, supplemental Fig. 3A).
Figure 2. Sarcoma killing in vitro by antiCD44v6 CAR+.CIK Patient-derived antiCD44v6 CAR+.CIK (n = 12) efficiently killed STS (n = 11, autologous targets in 4/11). The specific killing by CAR+.CIK cells was significantly higher, especially at low E/T ratios, than that obtained with unmodified CIK generated from the same patients (A). Killing values (mean ± SEM) from 43 independent essays are reported, results were analyzed by two way ANOVA and Bonferroni post test analysis, statistical significance is reported as * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001. The killing activity remained intense regardless the expression level (RFI high, intermediate, low) of CD44v6 on targets (B). Representative flow-cytometry histograms for each RFI level are reported. The addition of a blocking antibody against CD44v6 reduced the killing ability of CAR+.CIK cells to levels comparable to NTD.CIK. Blocking NKG2D receptor significantly reduced the tumor killing activity of unmodified NTD.CIK but did not impair CAR+.CIK (n = 4) (C). CAR+.CIK were moderately capable of killing monocytes at (E/T 5:1), the effect sensibly decreased and ceased at lower E:T ratios (n = 3) (D). Abbreviations: CAR Chimeric Antigen Receptor; NTD, Not Transduced; STS, Soft Tissue Sarcoma.
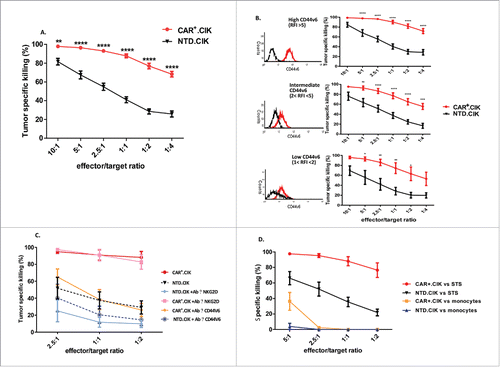
In selected control experiments (n = 4), the addition of anti-CD44v6 blocking antibody, directed against the same epitope recognized by the CAR (VFF-18), significantly reduced the killing ability of CAR+.CIK: (P ≤ 0.0001, ). The killing activity was not abrogated but reduced to levels comparable to unmodified NTD.CIK.
The addition of anti-NKG2D neutralizing antibody partially inhibited tumor killing of unmodified NTD.CIK cells but it did not interfere with the killing activity of CAR+.CIK, probably depending on the intense and homogenous membrane expression of CD44v6.
We assessed in vitro the killing activity of antiCD44v6 CAR+.CIK against monocytes as they were reported to express CD44v6 with potential risk of monocytopenia. CAR+.CIK were moderately active against monocytes at (E/T 5:1), their killing activity sensibly decreased at lower E:T ratios (n = 3) ().
Cytokine production
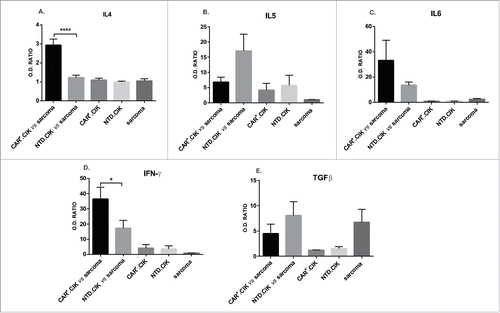
We explored the production of main Th1 and Th2 type cytokines by CAR+CIK upon recognition of CD44v6+ STS. A panel of 11 cytokines (IL-4, IL5, IL-6, IL-10, IL-12, IL-13, IL-17a, IFN-γ, TNF-α, GM-CSF and TGF-β) was determined. CAR+.CIK cells produced significant higher amounts of IL-4 (p < 0.0001) and IFN-γ (p < 0.05) compared with unmodified NTD.CIK (,). A positive trend was observed for IL6 (), while a negative trend was observed for IL5 and TGF-β (,). We did not observe any significant difference for the other cytokines analyzed. IFN-γ was the only cytokine with a progressive incremental trend (supplemental Fig. 4).
Figure 3. Cytokine production by CAR+.CIK. CAR+.CIK produced higher amounts of IL4, IFN-γ and IL6 compared with NTD.CIK. A trend toward lower production of IL5 and TGFβ was observed. Results are reported as optic density ratio (O.D. ratio) between samples and internal control (according to manufacturer instructions). Unpaired t test was adopted to compare CAR+.CIK with NTD.CIK. Significance is reported as * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001.
In vivo activity
We explored the antitumor activity of anti-CD44v6 CAR+.CIK intravenously infused, every 3–4 days, in NOD/SCID mice bearing subcutaneous STS xenograft generated from a patient-derived UPS.
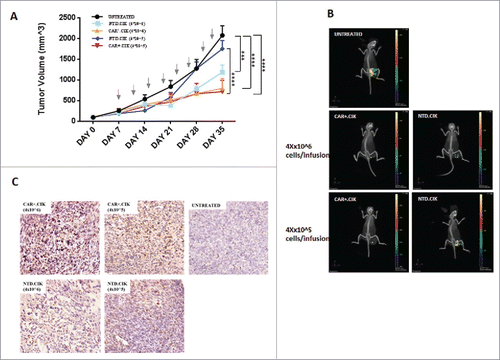
After 8 total infusions (dose 4 × 105) we observed a significant delay of tumor growth in mice (n = 6) treated with CAR+.CIK, compared with untreated controls (n = 6, p< 0.0001) or treated with equivalent doses of paired NTD.CIK cells (n = 6, p<0.0001) (). Comparable antitumor activity was obtained with 1 log-higher dose of CAR+.CIK (dose 4 × 106), however in this case the differences with NTD.CIK cells were less evident not reaching statistical significance (). In selected cases (n = 5) we could confirm that the therapeutic effect of CAR+CIK was maintained up to 10 days after stopping the infusions (Supplemental Fig. 3B). We did not observe any relevant toxicity or side effect in mice receiving CAR+.CIK at any dose. At the end of in vivo experiment, the antitumor activity of CAR+.CIK was confirmed by 3D analysis in one representative animal per group by assessing the tumor uptake of fluorescent glucose (probe XenoLight RediJect 2-DeoxyGlucosone) (). The tumor homing of CAR+CIK was confirmed by IHC in explanted tumors (), along with the expression of CD44v6 and NKG2D ligands (Supplemental Fig. 5).
Figure 4. In vivo anti-sarcoma activity of antiCD44v6 CAR+.CIK. CAR+.CIK were intravenously infused, every 3–4 days (total 8 infusions), in NOD/SCID mice bearing subcutaneous patient-derived UPS xenograft. Two different doses (4 × 105 and 4 × 106 per infusion) of CAR+.CIK were explored. We observed a significant delay of tumor growth in mice (n = 6) treated with CAR+.CIK cells (4 × 105), compared with untreated controls (n = 6, p< 0.0001) or treated with equivalent doses of paired NTD.CIK cells (n = 6, p< 0.0001). A similar antitumor activity was observed with 1 log-higher dose of CAR+.CIK (4 × 106). Arrows indicate CIK cell infusions. Results were analyzed by Two way Anova and correction for multiple comparisons test using Bonferroni method: Statistical significance is expressed as * P ≤ 0.05; ** P ≤ 0.01; *** P ≤ 0.001; **** P ≤ 0.0001 (A). In selected cases (1 representative mouse per group, n = 5) results were confirmed by a 3D imaging analysis based of fluorescent glucose uptake (B). Tumor homing of CAR+.CIK was confirmed by IHC in explanted tumors by staining with anti-human CD3 antibody (C). Abbreviations: CAR Chimeric Antigen Receptor; NTD, Not Transduced; NOD SCID mice, Non obese diabetic/severe combined immunodeficiency mice.
Discussion
In this study we reported for the first time the intense and safe anti-sarcoma activity of patient-derived CIK, genetically redirected with a CAR against CD44v6. Results were confirmed in vitro against multiple histotypes of STS, including autologous targets, and in an in vivo STS xenograft model.
CAR-T cell have been heralded as a major breakthrough in the field of adoptive immunotherapy. Impressive clinical results, admittedly along with important safety warnings, have been reported against hematologic malignancies, while their potentialities against solid tumors are still object of intense research. CAR-CIK cells may be an intriguing therapeutic alternative, especially in the challenging field of solid tumors including high grade STS. Promising preclinical data in this direction have been recently produced against hematologic malignancies, ovarian, nasopharyngeal and EGFR-positive solid tumors.Citation31,Citation32,Citation35,Citation36,Citation41-43
Biologic features of CIK may offer an advantageous experimental platform for CAR-engineered adoptive cell therapy. In particular the intense ex vivo expansibility, the intrinsic antitumor activity and even the limited persistence in vivo might be therapeutically exploited. We transduced CIK precursors in the early days of cultures, exploiting the subsequent massive expansion to generate relevant rates of CAR-engineered mature CIK. Lentiviral engineering did not impair the expansion potential or phenotype of CIK. Effector memory phenotype was the most represented within CAR-CIK, fully comparable to paired unmodified control CIK. The intense and cost-effective ex vivo expansibility may have a positive impact on the feasibility of this approach. Relevant doses of the final cell product may be obtained and cryopreserved, assuming the necessity of multiple infusions in consideration of the limited persistence in vivo of CIK.
This is an important difference with “conventional” CAR-T lymphocytes as they are mainly employed as single infusion, with subsequent capacity of in vivo proliferation and long-time persistence. The phenotype observed in our final cell product cannot exclude the hypothesis of selected CAR-CIK subsets capable of longer in vivo persistence, future dedicated studies with side by side CAR-T cell controls will be required to address this issue.
From the stand-point of antitumor efficacy, we previously reported that CIK were capable to kill STS but also showed that their killing activity sensibly decreased at low effector/target ratios, with possible limitations in clinical perspective. Such issue was overcome by CAR-CIK, whose intense killing activity was maintained at unfavorable, more clinical resembling, effector/target ratios. From these findings it is possible to imagine a therapeutic plan with multiple, low but highly effective, doses of CAR-CIK.
Compared with CAR-T lymphocytes, CAR-CIK already possess an intrinsic, CAR-independent MHC unrestricted, tumor-killing ability. We observed that blocking CD44v6 in tumor targets did not abrogate the killing activity of CAR-CIK, but it was instead reduced to values comparable with unmodified CIK. On the other hand, blocking NKG2D did not apparently diminish the efficacy of CAR-CIK, while it significantly inhibited unmodified CIK. This may be explained by the intense and homogenous expression of CD44v6 on target cells used in the essay, effectively eliminated by CAR-CIK. The bi-specific killing potential of CAR-CIK may limit tumor escape by clonal selection and be advantageous against tumors with heterogeneous expression of CD44v6. Similar biologic features, with intense transduction efficiency and bispecific killing properties were recently explored with an alternative CAR-strategy based on clinically applicable NK cell lines such as NK-92.Citation44 Phase I trials demonstrated their safety profile and initial evidence of antitumor activity.Citation45,Citation46 However, an important difference with the CAR-CIK platform is the allogeneic source of NK92, potentially triggering host immune-responses that might limit the repeatability and efficacy of multiple infusions.
Safety of CAR-based approaches is a central issue in the perspective of clinical translation. Important and even fatal events have been reported in some initial clinical studies with CAR-T lymphocytes against hematologic malignancies. Possible off-tumor recognition, dose and in vivo persistence are central aspects that may influence the safety profile of CAR-based strategies. Important pathogenetic roles may be played by peculiar, life-threatening immunological phenomena related to the activity of CAR-T cells, like cytokine release syndrome (CRS), macrophage activation syndrome or tumor lysis syndrome. Citation47,48We observed that CAR-CIK produce higher IFNγ compared with control CIK. This may indicate a stronger Th1 type response and favor additional innate and adaptive immune effectors, prompting Ag processing and HLA presentation. On the other hand the production of IL-4 may, at least in part, sustain a Th2 type polarization ad play a pro-tumoral role.Citation49 We did not observe an enhanced production of GM-CSF by CAR-CIK. Along with IFNγ, GM-CSF derived from CAR-T was reported to favorably shape tumor microenvironment through macrophage activation.Citation50 It is also true that high rates of IFNγ might in turn enhance the expression of PD-L1 at tumor level, with potential immune-suppressive effects but also providing speculative rational for synergism with checkpoint inhibitors. IL6 production also surged upon tumor engagement by CAR−CIK, warning about possible risks of cytokine release syndrome in clinical perspective. Dedicated studies, with immune-competent models, are warranted to explore a more complete cytokine panel comparing side by side the CRS risk of CAR+CIK with conventional CAR-T.Citation51
The limited in vivo persistence of CIK, previously discussed as a potential limitation of the antitumor activity, may be turned into positive safety implications. Multiple low-dose infusions of CAR-CIK may be planned to aim at disease control, but their withdrawal will allow the fast effective control of potential side effects. Also the theoretical risk of insertional mutagenesis, associated with the use viral vectors, is addressed by the limited lifespan of CAR−CIK. Furthermore the possible administration of ganciclovir will provide an additional safety measure to activate the inducible HSV-TK suicide switch.
The first clinical trial with an anti-CD44v6 mAb (bivatuzumab) directly conjugated with a potent chemotherapeutic agent (mertansine) raised concerns on potential skin toxicities.Citation52,Citation53 Previous experiments with primary keratinocytes, however, suggested a low level of CD44v6 expression, and relative resistance to CAR-T cell recognition, as compared to hematological tumor cells and monocytes.Citation38 Although in the present study we could not directly study keratinocyte recognition, we confirmed membrane expression of CD44v6 in monocytes, which were readily recognized by CAR-CIK, although with lower efficiency compared with sarcoma cells, suggesting a sufficiently wide therapeutic window also in the case of STS. Moreover, we previously reported that CD44v6 is indeed absent on tissue-macrophages, ruling out potential bystander damages to healthy tissues rich in monocyte-derived cells (e.g. macrophages; liver kupffer cells; microglia). We supported the relevance of the proposed approach by confirming the expression of CD44v6 in about 40% of STS screened. The distribution was not homogenous and we found that it was more frequently expressed by aggressive UPS, Liposarcoma, Fibrosarcoma and Leiomiosarcoma while less present in GIST (2/12).These findings, obtained by flow-cytometry, are consistent with a previous large retrospective immunohistochemistry analysis where CD44v6 was found in 57% of STS cases and associated with worse prognosis and higher risk of relapse.Citation1,Citation3 The expression of CD44v6 across several STS histotypes favorably compares with that of NY-ESO-1, that is a very relevant target for synovial sarcomasCitation6,Citation7 but only rarely expressed by other STS.
Our study was limited to STS but the approach described may be relevant also against other mesenchymal tumors like osteosarcoma. A recent meta-analysis and systematic review reported that CD44V6 over-expression is associated with poor survival and metastasis in osteosarcoma patients.Citation54
Overall our findings support the efficacy of antiCD44v6 CAR-CIK against currently high grade STS and lead basis for their dedicated exploration in clinical studies. CAR redirection of CIK may represent, in general, a valuable alternative or integration to adoptive immunotherapy strategies in the challenging field of solid tumors.
Materials and methods
Generation and characterization of antiCD44v6 CAR±.CIK
CIK cells were generated from peripheral blood mononuclear cells (PBMC) of patients with diagnosis of high grade STS. Patients released informed consent approved by the institutional review board according to the Declaration of Helsinki.
PBMC were isolated by density gradient centrifugation (Lymphosep, Aurogene) and seeded in cell culture flasks at a concentration of 2 × 106 cells/mL in RPMI-1640 medium (Gibco BRL), consisting of 10% FBS (Sigma), 100 U/mL penicillin, and 100 U/mL streptomycin (Gibco BRL). We added IFN-γ (Miltenyi Biotec; 1,000 U/mL) on day 0 and after 24 hours we activated PBMC using Anti-Biotin MACSiBead Particles loaded with CD2, CD3, and CD28 antibodies (Miltenyi Biotec) and human interleukin (IL)-2 IS (Miltenyi Biotec, 300 U/mL). On day +3, stimulated PBMC were transduced with a lentiviral vector encoding for anti-CD44v6-CAR containing a CH2CH3 spacer and a CD28 signaling domain (Casucci et al, Blood 2013), with MOI of 2 by overnight incubation. Non-transduced CIK were used as paired control. Cells were expanded over 3 weeks. Fresh medium and IL-2 (300 U/mL) were replaced every 2–3 days as needed and the cell concentration was maintained at 1.8 × 106 cells/mL.
Phenotype of CIK was analyzed by standard flow cytometric assays. The following monoclonal antibodies (mAb) were used: CD3–fluorescein isothiocyanate (FITC), CD8– phycoerythrin (PE), CD56–PE, CD314– allophycocyanin (APC, anti-NKG2D), CD45RA-FITC, CD45RO-PE Cy5, CD62 L-PE (Miltenyi Biotec); CAR expression was detected with a mAb specific for the IgG1/CH2CH3 spacer (The Jackson Laboratory).
STS cell lines
Patient-derived soft tissue sarcomas (STS) cell lines were generated from surgical specimens as previously described (Sangiolo et al., Can Res. 2014) and cultured in KO DMEM F12 (KO Out Dulbecco's Modified Eagle Medium, Gibco BRL) medium with 10% FBS, 25 mmol/L HEPES, 100 U/mL penicillin, and 100 U/mL streptomycin (Gibco BRL) in a humidified 5% CO2 incubator at 37°C. All tumor samples were derived from patient who had not previously received a chemotherapy treatment for their disease.
Patients released informed consent approved by the institutional review board according to the Declaration of Helsinki.
HT1080 cell line [American Type Culture Collection (ATCC)] used in this study was authenticated by genotype analysis with Cell_ID system (Promega) and comparing their profile with those published on the DMSZ database. We received DMR,Citation55 402.91Citation56 and NARA-HCitation55 cell lines as kind gifts from Spessotto P., Kawamoto T. and Akisue T., Aman P., respectively. All STS cell lines were grown in RPMI-1640 supplemented with 10% FBS (Sigma-Aldrich), 25 mmol/L HEPES, 100 U/mL penicillin, and 100 U/mL streptomycin (Gibco BRL) in a humidified 5% CO2 incubator at 37°C. Cell aliquots were stained with fluorescein isothiocyanate, PE, PE–Cyanin 7 (PC7), or APC-conjugated mouse mAbs against HLA-ABC (anti-HLA-ABC-FITC, BD Pharmingen) and CIK target antigens (anti-MIC A/B, BD Pharmingen; anti-ULBPs, R&D System, Space Import Export). Cells were stained for CD44v6 (CD44v6-PE e-Bioscience) and relative fluorescence intensity (RFI) of CD44v6 was calculated as follows: mean fluorescence intensity after mAb staining/mean fluorescence intensity after isotype-control staining.
Sarcoma killing activity of antiCD44v6 CAR±.CIK
The tumor-killing abilities of patient-derived antiCD44v6 CAR+.CIK and unmodified NTD.CIK were assessed in vitro against STS cell lines either by flow-cytometry or bioluminescent cell viability essay. In the first case target cells were stained with the vital dye CFSE (5,6-carboxyfluorescein diacetate succinimidyl ester; Molecular Probes) in accordance with the manufacturer's protocol. The immune-mediated killing was determined evaluating cell viability by flow cytometry (Cyan ADP, Beckman Coulter s.r.l.), after 72-hour incubation with expanded antiCD44v6 CAR+.CIK or unmodified NTD.CIK cells at various effectors/target ratios (10:1, 5:1, 2.5:1, 1:1, 1:2 and 1:4 for 72 hours in culture medium with 300 U/mL IL2 at 37°C, 5% CO2), according to the formula: experimental − spontaneous mortality/(100 − spontaneous mortality) × 100. Cell viability essays were performed at the same effector/target ration with CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
In selected blocking experiments, we pre-incubated CIK with 20 μg/mL of neutralizing NKG2D-specific mAb (Clone #149810, R&D Systems) or pre-incubated STS cells with 10 μg/mL of neutralizing CD44v6-specific mAb (bivatuzumab) that were maintained during the cytotoxicity assay against targets. Suicide gene functionality was analyzed after exposing antiCD44v6 CAR+.CIK for three days to ganciclovir 10 µM (Sigma Aldrich).
In selected assays (n = 2) at the end of experiment, the recovery of sarcoma cells was evaluated analyzing their growth rate 72 hours after the removal of CAR+.CIK.
Analysis of cytokine secretion
AntiCD44v6 CAR+.CIK and unmodified NTD.CIK were incubated with STS targets (E/T 1:1) for 72 h in CIK-culture medium described above. Thereafter, supernatants from the co-cultures and from wells with CIK or tumor cells alone were collected. Cytokine production was analyzed with multi-analyte EliSArray Kit (Human Th1 / Th2 / Th17 Cytokines Multi-Analyte ELISArray Kit, Qiagen). IL2 was excluded by the final analysis as already present in the CIK culture medium.
In vivo activity of patient-derived antiCD44v6 CAR±.CIK
Experiments were approved by internal review board. NOD/SCID (Charles River) female mice were subcutaneously injected with 3 × 106 primary cells of patient-derived pleomorphic sarcoma, resuspended in sterile PBS and BD Matrigel Basement Membrane Matrix (Becton Dickinson) 1:1. When tumors were approximatively 200 mm3, mice were infused twice-weekly with 4 × 106 or 4 × 105 mature antiCD44v6 CAR+.CIK or NTD.CIK cells resuspended in PBS (200 μL), for a total of 8 infusions. Mice injected with PBS only were used as control. Tumor growth was weekly monitored with a caliper and volume calculated according to the formula: V = 4/3 × π × (a/2)Citation2 × (b/2), where a is the length and b is the width diameter of the tumor.. Experiment ended and animals were euthanized 2 weeks after the last treatment or when tumor reached 2 cm in the main diameter.
Imaging by fluorescent glucose in vivo
At the end of in vivo treatments, a representative animal per cohort was intravenously infused with the fluorescent probe XenoLight RediJect 2-DeoxyGlucosone (DG)-750 (PerkinElmer) (5 nmol/mouse). Images based on the uptake of fluorescent glucose by tumors were acquired with Living Image ® Software and the IVIS ® Spectrum CT (Caliper Corporation, PerkinElmer Company).
Immunohistochemistry (IHC)
In selected cases (2 mice per group) we explored by IHC the presence of CAR+CIK in the explanted tumors along with the expression of NKG2D ligands (MIC A/B; ULBPs 2,5,6) and CD44v6.
Sections from formalin fixed, paraffin-embedded samples were cut into 3-μm thick sections. The tissue slides were treated according to standard immunohistochemistry procedures. In short, the slides were permeabilized in 0.1% Triton X-100 and 0.3% Tween 20 (Sigma-Aldrich) in TBS, treated for 30 min with 1% hydrogen peroxide to quench endogenous peroxidases. The slides were incubated with individual primary antibodies overnight at 4°C inside a moist chamber. After rinsing in PBS, a secondary antibody was added. Secondary HRP-conjugate antibodies (EnVision; DakoCytomation) were used for immunohistochemistry and the reaction was visualized with DAB chromogen (DakoCytomation Liquid DAB Substrate Chromogen System, Dako). The tissues were counterstained with Mayer hematoxylin (Bio-Optica), mounted on glass slides and visualized with a BX-60 microscope (Olympus) equipped with a color Qicam Fast 1394-digital CCD camera (12 bit; QImaging). The tissues were stained with the following primary antibodies: anti–CD3 (DAKO); anti-MIC A/B (clone #159207, R&D SYSTEM, BIOTECHNE BRAND); anti-ULBP 2/5/6 (R&D SYSTEM, BIOTECHNE BRAND); anti-CD44v6 (clone #SP37, Acris, an OriGene Company).
Statistical analysis
Descriptive data were calculated and reported as mean or median values, calculated as appropriate.
Phenotype and cytokine production between CAR+CIK and NTD CIK were compared by 2-tailed Student t tests. Comparison of antitumor activity among groups was calculated by two-way analysis of variance (ANOVA) and multiple comparison post-test analysis by Bonferroni method. Differences with a P value < 0.05 were considered statistically significant.Citation57
Conflict of interest disclosure
There are not potential conflicts of interest disclosed by the authors.
supp_data_1423167.zip
Download Zip (72 MB)Additional information
Funding
References
- Maula S, Huuhtanen RL, Blomqvist CP, Wiklund TA, Laurila P, Ristamäki R. The adhesion molecule CD44v6 is associated with a high risk for local recurrence in adult soft tissue sarcomas. Br J Cancer. 2001;84:244–52. doi:10.1054/bjoc.2000.1590. PMID:11161384.
- Manten-Horst E, Danen EH, Smit L, Snoek M, Le Poole IC, Van Muijen GN, Pals ST, Ruiter DJ. Expression of CD44 splice variants in human cutaneous melanoma and melanoma cell lines is related to tumor progression and metastatic potential. Int J Cancer 1995;64:182–8. doi:10.1002/ijc.2910640307. PMID:7542641.
- Kahara N, Ozaki T, Doi T, Nishida K, Kawai A, Shibahara M, et al. CD44 expression in soft tissue sarcomas. Virchows Arch. 2000;436:574–8. doi:10.1007/s004289900159. PMID:10917171.
- Andritsch E, Beishon M, Bielack S, Bonvalot S, Casali P, Crul M, Delgado Bolton R, Donati DM, Douis H, Haas R, et al. ECCO Essential Requirements for Quality Cancer Care: Soft Tissue Sarcoma in Adults and Bone Sarcoma. A critical review. Crit Rev Oncol Hematol. 2017;110:94–105. doi:10.1016/j.critrevonc.2016.12.002.
- Rosenberg SA. Cell transfer immunotherapy for metastatic solid cancer-what clinicians need to know. Nat Rev Clin Oncol. 2011;8:577–85. doi:10.1038/nrclinonc.2011.116. PMID:21808266.
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi:10.1200/JCO.2010.32.2537. PMID:21282551.
- Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21:1019–27. doi:10.1158/1078-0432.CCR-14-2708. PMID:25538264.
- Schmidt-Wolf IG, Lefterova P, Mehta BA, Fernandez LP, Huhn D, Blume KG, Weissman IL, Negrin RS. Phenotypic characterization and identification of effector cells involved in tumor cell recognition of cytokine-induced killer cells. Exp Hematol 1993;21:1673–9. PMID:7694868.
- Schmidt-Wolf IG, Finke S, Trojaneck B, Denkena A, Lefterova P, Schwella N, Heuft HG, Prange G, Korte M, Takeya M, et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br J Cancer 1999;81:1009–16. doi:10.1038/sj.bjc.6690800. PMID:10576658.
- Gammaitoni L, Giraudo L, Leuci V, Todorovic M, Mesiano G, Picciotto F, Pisacane A, Zaccagna A, Volpe MG, Gallo S, et al. Effective Activity of Cytokine Induced Killer Cells against Autologous Metastatic Melanoma including Cells with Stemness Features. Clin Cancer Res. 2013;19(16):4347–58. doi:10.1158/1078-0432.CCR-13-0061 PMID:23794732.
- Sangiolo D, Mesiano G, Gammaitoni L, Leuci V, Todorovic M, Giraudo L, Cammarata C, Dell'Aglio C, D'Ambrosio L, Pisacane A, et al. Cytokine-induced killer cells eradicate bone and soft-tissue sarcomas. Cancer Res. 2014;74:119–29. doi:10.1158/0008-5472.CAN-13-1559. PMID:24356422.
- Gammaitoni L, Giraudo L, Macagno M, Leuci V, Mesiano G, Rotolo R, Sassi F, Sanlorenzo M, Zaccagna A, Pisacane A, et al. Cytokine-Induced Killer Cells Kill Chemo-surviving Melanoma Cancer Stem Cells. Clin Cancer Res. 2017;23:2277–88. doi:10.1158/1078-0432.CCR-16-1524. PMID:27815354.
- Cappuzzello E, Tosi A, Zanovello P, Sommaggio R, Rosato A. Retargeting cytokine-induced killer cell activity by CD16 engagement with clinical-grade antibodies. Oncoimmunology. 2016;5:e1199311. doi:10.1080/2162402X.2016.1199311. PMID:27622068.
- Olioso P, Giancola R, Di Riti M, Contento A, Accorsi P, Iacone A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematol Oncol. 2009. doi:10.1002/hon.886. PMID:19294626.
- Giraudo L, Gammaitoni L, Cangemi M, Rotolo R, Aglietta M, Sangiolo D. Cytokine-induced killer cells as immunotherapy for solid tumors: current evidences and perspectives. Immunotherapy. 2015;7(9):999–1010. doi:10.2217/imt.15.61. PMID:26310715.
- Pan K, Guan XX, Li YQ, Zhao JJ, Li JJ, Qiu HJ, Weng DS, Wang QJ, Liu Q, Huang LX, et al. Clinical activity of adjuvant cytokine-induced killer cell immunotherapy in patients with post-mastectomy triple-negative breast cancer. Clin Cancer Res. 2014;20:3003–11. doi:10.1158/1078-0432.CCR-14-0082. PMID:24668644.
- Schmeel LC, Schmeel FC, Coch C, Schmidt-Wolf IG. Cytokine-induced killer (CIK) cells in cancer immunotherapy: report of the international registry on CIK cells (IRCC). J Cancer Res Clin Oncol. 2015;141(5):839–49. doi:10.1007/s00432-014-1864-3. PMID:25381063.
- Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 2001;14:123–33. doi:10.1016/S1074-7613(01)00095-4. PMID:11239445.
- Groh V, Rhinehart R, Secrist H, Bauer S, Grabstein KH, Spies T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc Natl Acad Sci U S A 1999;96:6879–84. doi:10.1073/pnas.96.12.6879. PMID:10359807.
- Sangiolo D, Martinuzzi E, Todorovic M, Vitaggio K, Vallario A, Jordaney N, Carnevale-Schianca F, Capaldi A, Geuna M, Casorzo L, et al. Alloreactivity and anti-tumor activity segregate within two distinct subsets of cytokine-induced killer (CIK) cells: implications for their infusion across major HLA barriers. Int Immunol. 2008;20:841–8. doi:10.1093/intimm/dxn042. PMID:18469328.
- Elia AR, Circosta P, Sangiolo D, Bonini C, Gammaitoni L, Mastaglio S, Genovese P, Geuna M, Avolio F, Inghirami G, et al. Cytokine-induced killer cells engineered with exogenous T-cell receptors directed against melanoma antigens: enhanced efficacy of effector cells endowed with a double mechanism of tumor recognition. Hum Gene Ther. 2015;26:220–31. doi:10.1089/hum.2014.112. PMID:25758764.
- Leuci V, Mesiano G, Gammaitoni L, Aglietta M, Sangiolo D. Genetically Redirected T Lymphocytes for Adoptive Immunotherapy of Solid Tumors. Curr Gene Ther. 2013.
- Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263:68–89. doi:10.1111/imr.12243. PMID:25510272.
- Dotti G, Savoldo B, Brenner M. Fifteen years of gene therapy based on chimeric antigen receptors: “are we nearly there yet?”. Hum Gene Ther. 2009;20:1229–39. doi:10.1089/hum.2009.142. PMID:19702437.
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 1993;90:720–4. doi:10.1073/pnas.90.2.720. PMID:8421711.
- Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–96. doi:10.1200/JCO.2014.58.0225. PMID:25800760.
- Leemhuis T, Wells S, Scheffold C, Edinger M, Negrin RS. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed Hodgkin disease and non-Hodgkin lymphoma. BiolBlood Marrow Transplant. 2005;11:181–7. doi:10.1016/j.bbmt.2004.11.019.
- Li R, Wang C, Liu L, Du C, Cao S, Yu J, et al. Autologous cytokine-induced killer cell immunotherapy in lung cancer: a phase II clinical study. Cancer Immunol Immunother. 2012;61:2125–33. doi:10.1007/s00262-012-1260-2. PMID:22581306.
- Olioso P, Giancola R, Di Riti M, Contento A, Accorsi P, Iacone A. Immunotherapy with cytokine induced killer cells in solid and hematopoietic tumours: a pilot clinical trial. Hematol Oncol. 2009;27:130–9. doi:10.1002/hon.886. PMID:19294626.
- Nishimura R, Baker J, Beilhack A, Zeiser R, Olson JA, Sega EI, Karimi M, Negrin RS. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood. 2008;112:2563–74. doi:10.1182/blood-2007-06-092817. PMID:18565854.
- Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, Biondi A, Biagi E, Bonnet D. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–605. doi:10.1038/leu.2014.62. PMID:24504024.
- Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, Maltese F, Galimberti S, Lopez AF, Biondi A, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401. doi:10.1111/bjh.12282. PMID:23432359.
- Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation”. Mol Ther. 2013;21:2268–77. doi:10.1038/mt.2013.192. PMID:23985696.
- Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genßler S, Rettinger E, Buchholz CJ, Pfeifer H, Schubert R, Ottmann OG, et al. CAR-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-ALL and enhance survival. Int J Cancer. 2016;139(8):1799–809. doi:10.1002/ijc.30217. PMID:27253354.
- Zuo S, Wen Y, Panha H, Dai G, Wang L, Ren X, Fu K. Modification of cytokine-induced killer cells with folate receptor alpha (FRα)-specific chimeric antigen receptors enhances their antitumor immunity toward FRα-positive ovarian cancers. Mol Immunol. 2017;85:293–304. doi:10.1016/j.molimm.2017.03.017. PMID:28360017.
- Guo X, Zheng H, Luo W, Zhang Q, Liu J, Yao K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci Rep. 2017;7:4859. doi:10.1038/s41598-017-04756-9. PMID:28687750.
- Zöller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–67. doi:10.1038/nrc3023. PMID:21390059.
- Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, Gentner B, Gullotta F, Ponzoni M, Bernardi M, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–72. doi:10.1182/blood-2013-04-493361. PMID:24016461.
- Kuryu M, Ozaki T, Nishida K, Shibahara M, Kawai A, Inoue H. Expression of CD44 variants in osteosarcoma. J Cancer Res Clin Oncol 1999;125:646–52. doi:10.1007/s004320050329. PMID:10541973.
- Gammaitoni L, Giraudo L, Macagno M, Leuci V, Mesiano G, Rotolo R, Sassi F, Sanlorenzo M, Zaccagna A, Pisacane A, et al. Cytokine-Induced Killer Cells Kill Chemo-surviving Melanoma Cancer Stem Cells. Clin Cancer Res. 2017;23(9):2277–88. doi:10.1158/1078-0432.CCR-16-1524. PMID:27815354.
- Marin V, Kakuda H, Dander E, Imai C, Campana D, Biondi A, D'Amico G. Enhancement of the anti-leukemic activity of cytokine induced killer cells with an anti-CD19 chimeric receptor delivering a 4-1BB-zeta activating signal. Exp Hematol. 2007;35:1388–97. doi:10.1016/j.exphem.2007.05.018. PMID:17656004.
- Ren X, Ma W, Lu H, Yuan L, An L, Wang X, Cheng G, Zuo S. Modification of cytokine-induced killer cells with chimeric antigen receptors (CARs) enhances antitumor immunity to epidermal growth factor receptor (EGFR)-positive malignancies. Cancer Immunol Immunother. 2015;64:1517–29. doi:10.1007/s00262-015-1757-6. PMID:26386966.
- Huang X, Park H, Greene J, Pao J, Mulvey E, Zhou SX, Albert CM, Moy F, Sachdev D, Yee D, et al. IGF1R- and ROR1-Specific CAR T Cells as a Potential Therapy for High Risk Sarcomas. PLoS One. 2015;10:e0133152. doi:10.1371/journal.pone.0133152. PMID:26173023.
- Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, Nowakowska P, Bönig H, Köhl U, Kloess S, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23:330–8. doi:10.1038/mt.2014.219. PMID:25373520.
- Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, et al. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–32. doi:10.1080/14653240802301872. PMID:18836917.
- Tonn T, Schwabe D, Klingemann HG, Becker S, Esser R, Koehl U, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–70. doi:10.1016/j.jcyt.2013.06.017. PMID:24094496.
- Gauthier J, Yakoub-Agha I. Chimeric antigen-receptor T-cell therapy for hematological malignancies and solid tumors: Clinical data to date, current limitations and perspectives. Curr Res Transl Med. 2017;65:93–102. doi:10.1016/j.retram.2017.08.003. PMID:28988742.
- Maus MV, Levine BL. Chimeric Antigen Receptor T-Cell Therapy for the Community Oncologist. Oncologist. 2016;21:608–17. doi:10.1634/theoncologist.2015-0421. PMID:27009942.
- Suzuki A, Leland P, Joshi BH, Puri RK. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine. 2015;75:79–88. doi:10.1016/j.cyto.2015.05.026. PMID:26088753.
- Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J Immunol. 2012;188:6389–98. doi:10.4049/jimmunol.1103019. PMID:22586039.
- Toi M, Bicknell R, Harris AL. Inhibition of colon and breast carcinoma cell growth by interleukin-4. Cancer Res 1992;52:275–9. PMID:1728401.
- Börjesson PK, Postema EJ, Roos JC, Colnot DR, Marres HA, van Schie MH, Stehle G, de Bree R, Snow GB, Oyen WJ, et al. Phase I therapy study with (186)Re-labeled humanized monoclonal antibody BIWA 4 (bivatuzumab) in patients with head and neck squamous cell carcinoma. Clin Cancer Res. 2003;9:3961. S-72S.
- Tijink BM, Buter J, de Bree R, Giaccone G, Lang MS, Staab A, et al. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin Cancer Res. 2006;12:6064–72. doi:10.1158/1078-0432.CCR-06-0910. PMID:17062682.
- Zhang Y, Ding C, Wang J, Sun G, Cao Y, Xu L, Zhou L, Chen X. Prognostic significance of CD44V6 expression in osteosarcoma: a meta-analysis. J Orthop Surg Res. 2015;10:187. doi:10.1186/s13018-015-0328-z. PMID:26697855.
- Irsan I, Akisue T, Hara H, Fujimoto T, Imabori M, Doita M, Kuroda R, Fujioka H, Kawamoto T, Yamamoto T, et al. Imatinib mesylate inhibits tumorigenicity of malignant fibrous histiocytoma cells in vivo. Anticancer Res. 2007;27:423–9. PMID:17352263.
- Germano G, Frapolli R, Simone M, Tavecchio M, Erba E, Pesce S, Pasqualini F, Grosso F, Sanfilippo R, Casali PG, et al. Antitumor and anti-inflammatory effects of trabectedin on human myxoid liposarcoma cells. Cancer Res. 2010;70:2235–44. doi:10.1158/0008-5472.CAN-09-2335. PMID:20215499.
- Fernández L, Metais JY, Escudero A, Vela M, Valentín J, Vallcorba I, Leivas A, Torres J, Valeri A, Patiño-García A, et al. MEMORY T CELLS EXPRESSING AN NKG2D-CAR EFFICIENTLY TARGET OSTEOSARCOMA CELLS. Clin Cancer Res. 2017;23(19):5824–35. doi:10.1158/1078-0432.CCR-17-0075.