
Abstract
DNA encoded adjuvants are well known for increasing the magnitude of cellular and/or humoral immune responses directed against vaccine antigens. DNA adjuvants can also tune immune responses directed against vaccine antigens to better protect against infection of the target organism. Two potent DNA adjuvants that have unique abilities to tune immune responses are the catalytic A1 domains of Cholera Toxin (CTA1) and Heat-Labile Enterotoxin (LTA1). Here, we have characterized the adjuvant activities of CTA1 and LTA1 using HIV and SIV genes as model antigens. Both of these adjuvants enhanced the magnitude of antigen-specific cellular immune responses on par with those induced by the well-characterized cytokine adjuvants IL-12 and GM-CSF. CTA1 and LTA1 preferentially enhanced cellular responses to the intracellular antigen SIVmac239-gag over those for the secreted HIVBaL-gp120 antigen. IL-12, GM-CSF and electroporation did the opposite suggesting differences in the mechanisms of actions of these diverse adjuvants. Combinations of CTA1 or LTA1 with IL-12 or GM-CSF generated additive and better balanced cellular responses to both of these antigens. Consistent with observations made with the holotoxin and the CTA1-DD adjuvant, CTA1 and LTA1 evoked mixed Th1/Th17 cellular immune responses. Together, these results show that CTA1 and LTA1 are potent DNA vaccine adjuvants that favor the intracellular antigen gag over the secreted antigen gp120 and evoke mixed Th1/Th17 responses against both of these antigens. The results also indicate that achieving a balanced immune response to multiple intracellular and extracellular antigens delivered via DNA vaccination may require combining adjuvants that have different and complementary mechanisms of action.
Introduction
For various known and unknown reasons, the promise of DNA vaccination has translated poorly from mice to primates. While all of the reasons for the loss of potency between mice and primates have not been fully elucidated, 2 reasons are believed to be most important - poor DNA uptake/expression and reduced immune-stimulatory signals at the inoculation site.Citation1-5 Ballistic particle bombardment with the gene gunCitation6 and in vivo electroporationCitation7 have dramatically enhanced DNA uptake generating substantially improved immunogenicity in primates.Citation2,5,7-21 However, delivery of DNA with either of these techniques is not ideal for large scale vaccination programs, particularly in underdeveloped countries. DNA expressed adjuvants, such as cytokinesCitation22-26 or chemokines,Citation27-29 have also been used to boost immunogenicity. Unfortunately, like DNA vaccination itself, many of these adjuvants did not translate well from mice to primates.Citation30-32 A notable exception is the Th1 polarizing cytokine, IL-12, that is perhaps the best DNA adjuvant tested to date in non-human primates and humans.Citation11,33-39 Unfortunately, IL-12 is not sufficiently potent to alleviate the need for delivery with electroporation.
Cholera Toxin (CT), heat-labile enterotoxin (LT) and their enzymatically active A1 domains have also shown great promise as DNA vaccine adjuvants.Citation6,19,21,40,41 CT and LT are closely related AB5 enterotoxins produced by Vibrio cholera and E. coli, respectively and are well known mucosal immunogens and adjuvants (reviewed in ref.Citation42). These toxins consist of catalytic A domains (separated into A1 and A2 subunits) anchored in rings of 5 identical B subunits.Citation43 Their enzymatic active sites reside within their A1 subunits, while the A2 subunits anchor the A1 subunits into the B pentamers.Citation43 The B pentamers engage gangliosides on cell membranes and facilitate the entry of the A subunits into lysosomes.Citation44 The A1 subunits then exploit host protein retention and degradation pathways to gain access to the cell cytoplasm (reviewed in ref.Citation45).
In the cytoplasm, the A1 subunits of CT and LT (CTA1 and LTA1, respectively) catalyze the transfer of ADP-ribose moieties from nicotinamide adenine dinucleotide (NAD) to stimulatory α-subunits of G proteins (Gsα).Citation46 The ADP-ribosylation of Gsα locks it into the GTP bound active state and subsequently causes the constitutive activation of adenylate cyclase leading to sustained high levels of cAMP within cells.Citation46 The ADP ribosyltransferase activities of CTA1 and LTA1 are responsible for their toxicity and for a large portion of their adjuvant effects.Citation41,47-53 Although Gsα is a well-established cellular target of both CTA1 and LTA1, other signaling proteins are known to be ADP-ribosylated by these toxins.Citation54,55 Whether Gsα is the principle target responsible for adjuvant activity is unknown.
Unfortunately, the CT and LT holotoxins cannot be used as mucosal adjuvants in humans since they are extremely toxic when administered at mucosal sites even at very small doses.Citation56 Several methods have been used to minimize toxicity including toxin-inactivating mutations,Citation51,57-59 immunizing at sites distant from the mucosa, such as the skin,Citation60-64 or by re-targeting the A1 subunits using plasmid DNACitation40,41 or other proteinsCitation47,48 absent the promiscuous native B subunit. CTA1, expressed as the native bacterial DNA sequence as a DNA adjuvant, enhanced both cellular and antibody responses to HIVBal-gp120 when delivered intramuscularly (i.m.).Citation41 Human codon-optimization enhanced the in vitro biological activity of CTA1 by 250-fold and enhanced antibody responses to SIV gag when administered by gene gun.Citation40 Cellular responses, however, were absent when delivered by the gene gun.Citation40
Much of the promise of DNA based adjuvants lies in their abilities to “tune” immune responses in directions better suited to protect vaccinees from target infectious agents. Enabling this outcome requires more in depth profiling of the immune responses induced by the selected adjuvants. Since CTA1 and LTA1 have proven to evoke potent antibody responses when administered via gene gun, we wanted to profile the antibody and T cell responses provided by these adjuvants when administered i.m. We also compared the immune profiles of CTA1 and LTA1 against IL-12 and GM-CSF and show that their adjuvant effects are on par with those of IL-12 and GM-CSF. More importantly, these enterotoxin subunit adjuvants had complementary effects when combined with either of these cytokine adjuvants.
In contrast to the cytokine adjuvants IL-12 and GM-CSF that preferentially enhanced cellular responses to the secreted antigen gp120 over the intracellular antigen gag, CTA1 and LTA1 preferentially enhanced cellular responses to gag over gp120. LTA1 and CTA1 also evoked a mixed Th1/Th17 cellular response consistent with what has been observed with the holotoxin adjuvants and the CTA1-DD adjuvant.Citation65-69 Together, the results of this study suggest that an optimal combination of DNA expressed adjuvants that enhance the magnitude of immune responses to vaccine antigens and tune the immune responses toward those best suited to combating targeted infectious agents may eliminate the need for suboptimal delivery techniques such as gene gun and electroporation for some vaccines, particularly when they are used as a prime for a recombinant subunit boost.
Results
One inoculation evokes near maximal magnitude cellular responses when CTA1 is used as an adjuvant
We first determined how many inoculations are necessary to evoke high-level cellular and/or antibody responses when a codon-optimized CTA1 is used as a DNA vaccine adjuvant. For this experiment, mice were immunized 1, 2 or 3 times with or without the CTA1 adjuvant on days 0, 14 and/or 28. Mice were inoculated with pHIV-gp120 and pSIV-gag plus either empty plasmid (as a no adjuvant control) or with pCTA1. Splenocytes from all mice were harvested on day 48 and stimulated with peptide pools encompassing HIVBaLgp120 or SIVmac239gag for IFN-γ ELISpot analysis. Day 48 correlates to 20 d post the final immunization for mice vaccinated 2 or 3 times and 20 d post the single immunization for mice vaccinated a single time.
As expected, the number of HIV gp120 and SIV gag IFN-γ ELISpots increased after each vaccination in those animals that did not receive an adjuvant (). By contrast, CTA1 adjuvanted mice reached high-magnitude cellular responses after a single inoculation and these cellular responses did not increase further with additional inoculations. SIV gag and HIV gp120 ELISpots were 7.3-fold and 5.1-fold higher, respectively than those of control mice that did not receive an adjuvant after a single inoculation (). Importantly, the number of gag-specific ELISpots from mice inoculated a single time with CTA1 was significantly higher (3.8-fold, p = 0.001) than the number of gag-specific ELISpots from mice inoculated 3 times in the absence of adjuvant (). Similarly, the number of gp120-specific ELISpots from mice inoculated a single time with CTA1 was higher than the number of gp120-specific ELISpots from mice inoculated 3 times in the absence of adjuvant (); however, the difference between these responses did not reach statistical significance.
Figure 1. CTA1 evokes near maximal cellular immune responses after a single vaccination: Groups of 6 BALB/c mice were immunized 1, 2 or 3 times with or without the CTA1 adjuvant on days 0, 14 and/or 28 (i.e., mice vaccinated twice were vaccinated on days 14 and 28 and mice vaccinated once were vaccinated on day 28). Mice were inoculated with 30 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 25 μg of empty plasmid as a no adjuvant control or 25 μg of pCTA1. ELISpot analysis was performed on day 48. Day 48 correlates to 20 d post the final immunization for mice vaccinated 2 or 3 times and 20 d post the single immunization for mice vaccinated a single time. (A) Splenocytes were assayed on day 48 by IFN-γ ELISpots with peptide pools encompassing SIVmac239gag or HIVBaLgp120. (B) Anti-gp120 reciprocal half maximal IgG titers were determined on day 48 sera by ELISA. The error bars represent the standard errors of the means. The P values (compared to the matching no adjuvant group) were calculated with a Students T Test using SigmaPlot v11 software. NS stands for not significantly different from the unadjuvanted control. The results shown are from a single experiment performed.
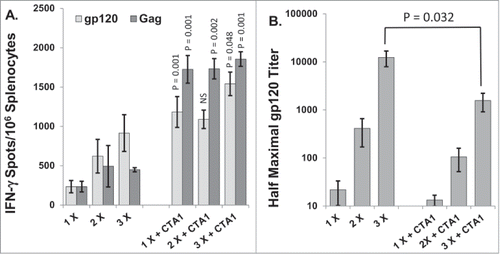
In contrast to the cellular responses, the codon optimized CTA1 did not enhance antibody responses to HIV gp120 (). The antibody titers to gp120 increased with subsequent immunizations whether CTA1 was included or not, however, mice that received CTA1 had gp120 titers that were significantly lower (8-fold) than those of unadjuvanted controls after 3 inoculations. The gag titers for most mice in this study did not rise above background levels (data not shown). Subsequent experiments focus comparison on the cellular responses as little to no differences were found between the antibody responses when we used CTA1 or LTA1 delivered i.m.
Dose response of CTA1
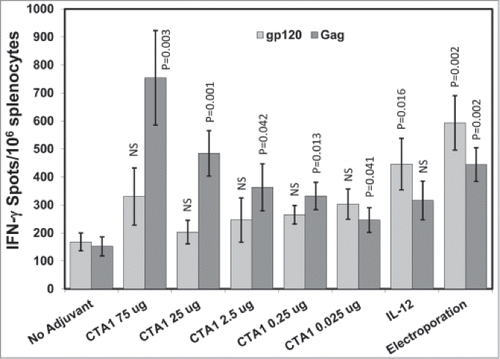
To determine the optimal dose range of the codon optimized CTA1, mice were immunized i.m. once on day 0 with pHIV-gp120 and pSIV-gag plus either an empty plasmid, pIRES-IL-12 or different doses of pCTA1. Enough empty plasmid was included to keep the total DNA delivered in each vaccination to 70 μg. An additional group that received unadjuvanted DNA delivered i.m. with electroporation was included as a comparator since delivery by electroporation has been shown to dramatically increase antigen expression and ensuing immune responses.Citation7,13-16,70-75 Splenocytes were harvested on day 14 and stimulated with HIVBaLgp120 or SIVmac239gag peptide pools for IFN-γ ELISpot assays.
shows that CTA1 significantly enhanced the anti-gag ELISpot responses over the unadjuvanted controls at all doses tested. Increases in the anti-gp120 ELISpot responses were also observed but did not reach statistical significance. These observations are similar to those from the previous study and demonstrate that CTA1 preferentially enhances cellular responses to the intracellular expressed antigen gag over those to the secreted antigen gp120. IL-12 did the opposite by significantly increasing the ELISpot responses to gp120 but not significantly enhancing the responses to gag (). Strikingly, CTA1 induced anti-gag ELISpot responses that were equal or greater in magnitude than those evoked by electroporation at the 25 and 75 μg doses (). We can conclude that CTA1 has a very wide effective dose range (over 3 orders of magnitude) and that it has the unusual ability to preferentially enhance responses to the intracellular antigen gag over the secreted antigen gp120.
Figure 2. CTA1 has a wide dose range: Groups of 6 BALB/c mice were immunized i.m. 1 time on day 0 with 30 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 25 μg of empty plasmid or 25 μg of pIRES-mIL-12 as a comparator adjuvant or decreasing doses of pCTA1. Enough empty plasmid was included to keep the total DNA delivered in each vaccination to at least 70 μg. An additional group had the vaccine DNA delivered i.m. with electroporation. Day 14 splenocytes were stimulated with SIVmac239gag or HIVBaLgp120 peptide pools for IFN-γ ELISpot assays. The error bars represent the standard errors of the means. The P values (compared to the no adjuvant group) were calculated with a Students T Test using SigmaPlot v12 software. NS stands for not significantly different from the unadjuvanted control. The results shown are from a single experiment performed.
CTA1 has complementary adjuvant effects with IL-12 and GM-CSF
IL-12 is a Th1 polarizing cytokine with direct effects on T cellsCitation76-81 and is one of the most potent DNA vaccine adjuvants ever tested in mice and primates.Citation11,33-39 GM-CSF is another well-studied and potent DNA vaccine adjuvant.Citation82-85 Since CTA1 likely has a different mechanism of DNA adjuvant activity than IL-12 or GM-CSF, we speculated that their adjuvant effects would be complementary (i.e., additive or synergistic). To test these hypotheses, mice were immunized i.m. 1 time on day 0 with pHIV-gp120 and pSIV-gag plus either empty plasmid, pCTA1, pIRES-IL-12, pGM-CSF or combinations of pCTA1 and pIL-12 or pGM-CSF. Day 14 splenocytes were harvested and assessed by IFN-γ ELISpot assays to HIVBalgp120 or SIVmac239gag.
CTA1, IL-12 and GM-CSF all increased ELISpot responses to gp120 and gag (). CTA1 again preferentially enhanced responses to gag (4.9-fold) over gp120 (2-fold). By contrast, IL-12 again preferentially enhanced responses to gp120 (4.6-fold) over gag (1.4-fold). Similarly to IL-12, GM-CSF preferentially enhanced responses to gp120 (6.9-fold) over gag (2.5-fold). As predicted, the total gp120 + gag ELISpot response in the CTA1 + IL-12 group (1865 spots) was approximately additive when compared to groups receiving either adjuvant alone (1033 total spots for CTA1 and 985 total spots for the IL-12 group). The difference in the combined ELISpot magnitudes between the IL-12 alone group and the CTA1 + IL-12 group was significant (p = .036), but the difference between CTA1 alone group and the CTA1 + IL-12 group did not reach statistical significance.
Figure 3. CTA1 has complementary adjuvant effects with IL-12 and GM-CSF: Groups of 6 BALB/c mice were immunized i.m. 1 time on day 0 with 30 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 40 μg of empty plasmid, 40 μg of pCTA1, 40 μg of pIL-12, 40 μg of pGM-CSF or 20 μg of pCTA1 + 20 μg of pIL-12 or 20 μg of pCTA1 + 20 μg of pGM-CSF. Day 14 splenocytes were stimulated with SIVmac239gag or HIVBalgp120 peptide pools for IFN-γ ELISpot assays. The error bars represent the standard errors of the means. The P values (compared to the no adjuvant group) were calculated with a Students T Test using SigmaPlot v12 software. NS stands for not significantly different from the unadjuvanted control. The results shown are from a single experiment of 3 performed.
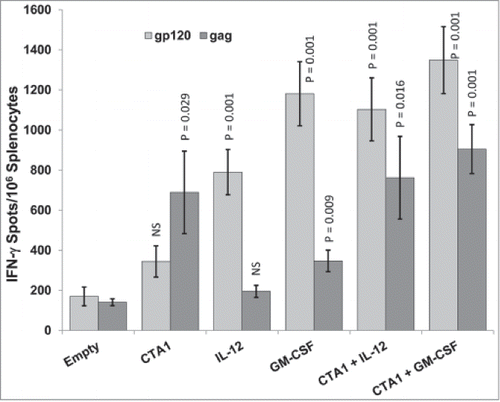
CTA1 + GM-CSF had a similar additive effect as CTA1 + IL-12 (). The total gp120 + gag ELISpot response in the CTA1 + GM-CSF group (2254 spots) was approximately additive when compared to groups receiving either adjuvant alone (1033 total spots for CTA1 and 1528 total spots for the GM-CSF group). The difference in the combined ELISpot magnitudes between the CTA1 alone group and the CTA1 + GM-CSF group was significant (p = 0.007) as was the difference between the GM-CSF alone group and the CTA1 + GM-CSF group (p = 0.039). The adjuvant combinations also yielded better balanced cellular responses to both antigens as compared to the adjuvants used alone ().
LTA1 and CTA1 have similar adjuvant effects
CT and LT are highly homologous enterotoxins with the majority of their differences lying in the targeting domains of their respective B subunits.Citation86 Subtle differences have been intermittently observed between immune responses when the holotoxins are used either as antigens or adjuvants.Citation87 Since there are only 26 amino acid differences between the 196 amino acid A1 domains of our CTA1 and LTA1 DNA adjuvants (87% homology), we wanted to confirm that they would have similar DNA vaccine adjuvant effects and that LTA1 would also have additive effects with IL-12.
In this experiment, mice were immunized i.m. on days 0 and 21 with pHIV-gp120 and pSIV-gag plus either an empty plasmid, pIL-12, pLTA1, or a combination pLTA1 and pIL-12. Day 35 splenocytes were stimulated with HIV-BaLgp120 or SIVmac239gag peptide pools for IFN-γ ELISpot assays. Consistent with the previous experiments using CTA1, LTA1 preferentially enhanced responses to gag (8.8-fold) over gp120 (3.6-fold) (). Again, IL-12 did the opposite by preferentially improving responses to gp120 (2.7-fold) over gag (1-fold) (). Similar to when CTA1 was combined with IL-12, the LTA1 + IL-12 group had total gp120 + gag ELISpot responses (2432) approximately additive when compared to the LTA1 alone (1377 total spots) and the IL-12 alone (502 total spots) groups. The differences in the total gp120 + gag ELISpot responses between the LTA1 + IL-12 group and the LTA1 or IL-12 alone group were also statistically significant (p = 0.032 for the LTA1 alone group compared to the LTA1 + IL-12 group and p = 0.001 for the IL-12 alone group compared to the LTA1 + IL-12 group).
Figure 4. Complementary adjuvant effects of LTA1 with IL-12: Groups of 5 BALB/c mice were immunized i.m. on days 0 and 21 with 25 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 30 μg of empty plasmid, 30 μg of pLTA1, 30 μg of pIL-12 or 15 μg of pLTA1 + 15 μg of pIL-12. Day 35 splenocytes were stimulated with SIVmac239gag or HIV-BaLgp120 peptide pools for IFN-γ ELISpot assays and for supernatant cytokine analysis. (A) Splenocytes assayed by IFN-γ ELISpots. (B) Supernatants from peptide stimulated splenocytes plated at 8 × 105 cells/well analyzed by IFN-γ ELISA. (C) Supernatants from peptide stimulated splenocytes plated at 8 × 105 cells/well analyzed by Granzyme B ELISA. The error bars represent the standard errors of the means. The P values in panel (A) (compared to the no adjuvant group) were calculated with a Students T Test using SigmaPlot v12 software. The P values in panels (B and C)(compared to the no adjuvant group) were calculated with a Mann-Whitney Rank Sum-test using SigmaPlot v12 software. NS stands for not significantly different from the unadjuvanted control. The results shown are froma single experiment of 2 performed.
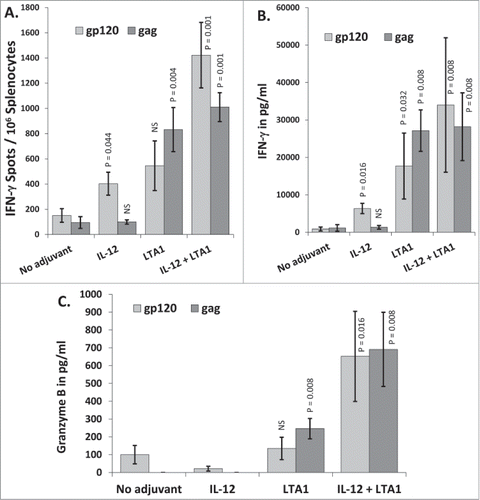
Similar to the ELISpot results, LTA1 preferentially enhanced IFN-γ secretion in response to gag peptides (23.7-fold) over gp120 peptides (20-fold) whereas IL-12 did the opposite by preferentially enhancing IFN-γ secretion in response to gp120 (7.2-fold) over gp120 (1.2-fold) (). Strikingly, the animals that received LTA1 secreted significantly more IFN-γ when calculated on a per cell basis (3.26 pg/spot) when compared to both the unadjuvanted group (0.82 pg/spot, p = 0.016), and the IL-12 adjuvanted group (1.53 pg/spot, p = 0.008). The LTA1 + IL-12 group also had strong IFN-γ secretion in response to both gp120 and gag () with an average secretion of 2.56 pg/spot.
Granzyme B is a Cytotoxic T cell (CTL) effector molecule that directly participates in the killing of target cells.Citation88–90 The release of Granzyme B by peptide stimulated splenocytes is often used as a measure of the killing capacity of the evoked cellular immune responses. shows that LTA1 significantly enhanced Granzyme B secretion from gag peptide stimulated splenocytes 3.8-fold over the unadjuvanted group, but did not enhance Granzyme B secretion from gp120 peptide stimulated splenocytes affirming the preferential boosting of gag specific cellular immune responses by the A1 subunit adjuvants. IL-12, on the other hand, did not boost Granzyme B secretion for either antigen. The LTA1 + IL-12 combination yielded enhanced combined total Granzyme B secretion (gp120 + gag) from splenocytes stimulated by peptides that was balanced between both antigens and was significantly higher than the unadjuvanted group (p = 0.008), the IL-12 group (p = 0.008) and the LTA1 group (p = 0.032) ().
Together, these data imply that the main benefit of the CTA1/LTA1 + IL-12 or CTA1 + GM-CSF combinations is that these combinations significantly enhance the combined gp120 + gag responses over the responses evoked by the individual adjuvants. This represents mostly an additive effect rather than a synergistic effect. In other words, when CTA1 or LTA1 (that preferentially enhance responses to gag) are combined with IL-12 or GM-CSF (that preferentially enhance responses to gp120) the result is an enhancement to both antigens and the total response (the additive total for gag and gp120) is significantly enhanced compared to the individual adjuvants alone.
LTA1 evokes mixed Th1/Th17 antigen-specific T cell responses
The dramatically enhanced secretion of IFN-γ induced by the LTA1 adjuvant prompted us to profile the cytokines and chemokines induced by the adjuvant more thoroughly. Mice were immunized i.m. on days 0 and 21 with pHIV-gp120 and pSIV-gag plus either an empty plasmid, pLTA1 or pIL-12. Day 35 splenocytes were stimulated with HIVbal-gp120 or SIV mac239-gag peptide pools for IFN-γ ELISpot assays and supernatants from replicate cultures were harvested, pooled within groups and analyzed by Cytokine Bead Array (CBA) for the concentrations of 34 different cytokines and chemokines (listed in the Materials and Methods).
As expected, the LTA1 and IL-12 adjuvants evoked IFN-γ ELISpot responses that were consistent with the previous study (). Immunization with IL-12 enhanced the gp120 IFN-γ ELISpot response 3.8-fold but did not enhance the gag response as compared to the unadjuvanted controls (). LTA1 enhanced gag IFN-γ ELISpots 5.1-fold and anti-gp120 ELISpots 3.5-fold compared to the no adjuvant controls (). Peptide stimulated splenocytes from LTA1 adjuvanted mice again secreted more IFN-γ per ELISpot than those from unadjuvanted controls () (0.19 pg/spot for LTA1 mice verses 0.084 pg/spot for unadjuvanted mice). Examining the production of other cytokines revealed that LTA1 induced the secretion of more IL-2, IL-3, IL-6, RANTES, GM-CSF, TNF-α and MIP-1β compared to unadjuvanted or IL-12 adjuvanted mice (). The concentrations of these cytokines and chemokines from LTA1 adjuvanted mice were also higher for gag than for gp120 further highlighting the preferential enhancement of gag responses over gp120 responses by this adjuvant. The concentrations of the remaining 24 cytokines and chemokines were either undetectable or were not different between groups (data not shown). LTA1 also dramatically enhanced the secretion of IL-17 (both the IL-17A and IL-17F isoforms) from gp120 and gag peptide stimulated splenocytes indicating that LTA1 evokes a mixed Th1/Th17 response (). Although LTA1 evoked IL-17 secretion from both gag and gp120 peptide stimulated splenocytes, again the IL-17 concentrations were higher for gag than for gp120. We further verified that antigen-specific T cells from CTA1 immunized mice also presented this same mixed Th1/Th17 profile (data not shown).
Figure 5. LTA1 evokes IL-17 secretion: Groups of 5 BALB/c mice were immunized i.m. on days 0 and 21 with 25 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 30 μg of empty plasmid, 30 μg of pLTA1 or 30 μg of pIL-12. Day 35 splenocytes were stimulated with SIVmac239gag or HIV-BaLgp120 peptide pools for IFN-γ ELISpot assays. Supernatants from peptide stimulated splenocytes plated a 1 × 105 cells/well were pooled within groups and analyzed by CBA. (A) Splenocytes assayed by IFN-γ ELISpots. (B) Supernatants from peptide stimulated splenocytes analyzed by multi-analyte CBA. C. Pooled supernatants from peptide stimulated splenocytes analyzed for IL-17 concentration by multi-analyte CBA. The error bars represent the standard errors of the means. The P values (compared to the no adjuvant group) were calculated with a Students T Test using SigmaPlot v12 software. NS stands for not significantly different from the unadjuvanted control. The results shown are from a single experiment of 2 performed.
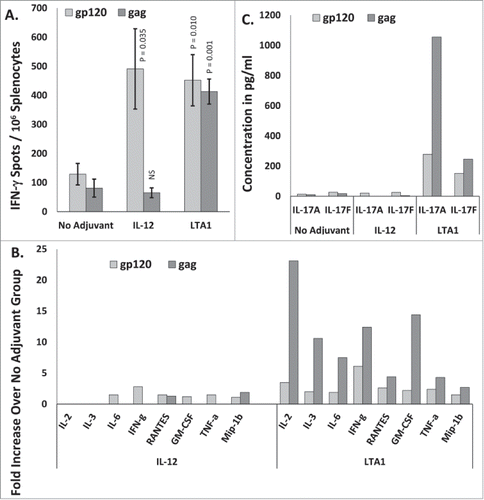
CTA1 and LTA1 preferentially enhance cellular responses to gag over gp120
The studies above indicate that CTA1 and LTA1 behave similarly as DNA adjuvants and that both adjuvants preferentially enhance cellular responses to gag over gp120. We next determined if the preferential enhancement of cellular responses to gag over gp120 by these adjuvants was retained between different studies performed at different times using different reagents. A total of 9 individual mouse studies were performed using CTA1, 6 were performed using LTA1 and 9 were performed using IL-12. These studies were performed over a period of 4 y and often used different batches of DNA and/or peptides.
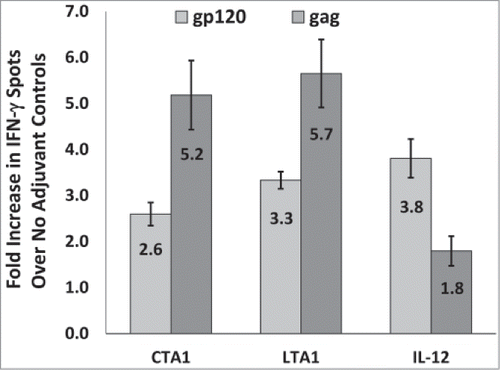
shows that CTA1 and LTA1 evoked similar average fold enhancements in the gp120 and gag specific IFN-γ ELISpot responses between these studies. In fact, there were no statistically significant differences in the fold-enhancements in the gp120 (p = 0.96) or gag (p = 0.72) responses evoked by CTA1 and LTA1. The average fold-enhancements for gag were also 2-times higher for CTA1 and 1.7-times higher for LTA1 than the corresponding fold-enhancements for gp120 (). These data further demonstrate the preferential enhancing effects of CTA1 and LTA1 for gag over gp120. also shows that IL-12 did the opposite by preferentially enhancing IFN-γ ELISpot responses to gp120 over those to gag. In this regard, the average fold-enhancements for gp120 were 2.1-times higher for IL-12 than the corresponding fold-enhancements for gag.
Figure 6. CTA1 and LTA1 preferentially enhance cellular responses to gag over gp120: The results from 9 independent mouse studies for CTA1, 6 independent studies for LTA1 and 9 independent studies for IL-12 were compared. Each study had groups of 5 BALB/c mice immunized i.m. on days 0 and 14 or days 0 and 21 with 25 μg of pSIV-gag and 15 μg of pHIV-gp120 plus either 25 μg of empty plasmid, pLTA1 or pIL-12. Day 28 or day 35 splenocytes were stimulated with SIVmac239gag or HIV-BaLgp120 peptide pools for IFN-γ ELISpot assays. For each study, the average gp120 and gag-specific IFN-γ ELISpot responses were determined for the no adjuvant groups and the CTA1, LTA1 or IL-12 adjuvanted groups. Within each study, the average fold increases for the gp120 (light gray bars) and gag (dark gray bars)-specific ELISpot responses were calculated by dividing the mean gp120 and gag-specific ELISpot responses from the adjuvanted groups by the corresponding mean gp120 and gag-specific ELISpot responses from the unadjuvanted groups. The error bars represent the standard errors of the means. The P values (provided in the text) were calculated with a Students T test using SigmaPlot v12 software.
Discussion
The reduced immunogenicity of DNA vaccines in primates necessitates the development of improved delivery methods and/or adjuvants. Most DNA vaccine adjuvants tested to date have been cytokines or chemokines.Citation22-26 Unfortunately, most of these adjuvants have not translated well from mice to primates.Citation30-32,91 We and others have exploited the inherent adjuvant activities of CT and LT for use as plasmid DNA expressed adjuvants.Citation6,19,21,40,41 We initially found that a non-optimized CTA1 (with the natural bacterial DNA sequence) is an effective DNA vaccine adjuvant for HIV-gp120 when delivered i.m.Citation41 CT, LT and their A1 subunits are also potent adjuvants for a variety of DNA encoded antigens when delivered by gene gun.Citation19,21,40,92,93 In our hands however, gene gun delivery improved antibody responses to gag but did not evoke measurable cellular responses.Citation40 Since in our hands gene gun delivery yielded suboptimal cellular immune responses and since there is not currently a clear clinical path for gene gun delivery, we focused our efforts on i.m. delivery of our vaccines and adjuvants.
The codon optimized CTA1 proved to be approximately 250-fold more active than the non-optimized CTA1 in vitro.Citation40 This optimized CTA1 dramatically increased cellular responses to SIVmac239gag, and to a lesser extent to HIVBaL gp120 (), but did not improve the magnitude of the anti-gp120 antibody response (). Similar results were observed with an optimized LTA1 (). This apparent discrepancy from our previous report where we used a non-optimized CTA1Citation41 is most likely an artifact of the experimental design. Here, the mice were sacrificed 20 d post the final immunization which may have been too short for antibody responses to fully develop since it took 26 weeks for the antibody response to reach its maximum after immunization with the native CTA1 sequence.Citation41 Also, when delivered by gene gun, CTA1 dramatically enhanced antibody responses to SIV-gag (over 2 orders of magnitude) but had little to no enhancing effects on antibody responses to HIV-gp120.Citation40
The CTA1 and LTA1 adjuvants are closely related enzymes with likely identical activities and targets.Citation54 Our data indicate that these enzymes also behave indistinguishably from each other as DNA adjuvants. For this reason, we believe that CTA1 and LTA1 can be thought of interchangeably as DNA adjuvants. We have chosen LTA1 as a lead only because there is a general perception that CTA1 is more toxic than LTA1. The preferential enhancing effects that CTA1 and LTA1 have for the intracellular antigen gag over the secreted antigen gp120 may be due to their ability to facilitate the release of intracellular antigens into the extracellular milieu. In this regard, we found that CTA1 and LTA1 increased the release of an intracellular retained reporter protein into the extracellular milieu by approximately 2 orders of magnitude (manuscript in preparation). This may facilitate the transfer of intracellular expressed antigens from transfected muscle cells to APCs. This ability of CTA1 and LTA1 may have also caused or contributed to the reduced antibody responses to gp120 observed in . In this regard, it is plausible that increased antibody responses to gag may have decreased the antibody responses to gp120 (i.e., antigen competition). We are currently studying the mechanism(s) of the adjuvant effects of CTA1 and LTA1 in greater detail. The results of these studies should shed more light on the mechanism(s) of action of these adjuvants and may better answer the questions of why CTA1 and LTA1 preferentially enhance responses to gag over gp120 and why CTA1 reduced antibody responses to gp120.
Interestingly, the results of this study, where CTA1 boosted cellular responses but not antibody responses are the converse of those of our previous study where CTA1 was delivered by gene gun.Citation40 In that study, CTA1 boosted antibody responses but not cellular responses. However, in that study CTA1 dramatically enhanced antibody responses to gag, but did not significantly enhance antibody responses to gp120.Citation40 This is consistent with the preferential enhancing effects that CTA1 and LTA1 had for gag over gp120 in the current studies. The results of that study may also be explainable by the ability of CTA1 and LTA1 to cause the release of normally intracellular retained proteins into the extracellular milieu.
We do not believe that our failure to detect cellular responses when the adjuvants were delivered by gene gun represents a failure of the adjuvants, but rather a failure of the DNA delivery. In this vein, others have consistently measured robust cellular responses after gene gun immunization, even in the absence of adjuvants.Citation6,19,94-101 We, however, failed to measure cellular responses with gene gun delivery even when IL-12 was used as the adjuvant (data not shown). Our inability to evoke cellular responses with the gene gun likely stems from undetermined differences in formulation and/or delivery parameters. Differences in the local tissue environments between the epidermis (where DNA is delivered by gene gun) and muscle (where DNA is delivered by i.m. inoculation) also likely impacts how well the vaccines evoke humoral immune responses. For instance the epidermis is rich in Langerhans cells which may be efficient at priming humoral responses, whereas muscle tissue has a much smaller population of antigen presenting cells (APCs).
The results of the current study also indicate that a single immunization with CTA1 provides maximal, or near maximal, cellular immune responses that are not improved by further boosting (). The antibody response, on the other hand, did benefit from additional boosts (). The adjuvant effects of combinations of CTA1 with IL-12 or GM-CSF were approximately additive () principally because the adjuvants showed an effective preference for either the gag or gp120 when used alone that combined to give more balanced responses when the adjuvants were used together. The propensity for CTA1 and LTA1 to preferentially enhance cellular responses to the intracellular SIVmac239gag antigen () may be a unique feature of these adjuvants since using no adjuvant, adjuvanting with IL-12 or GM-CSF, or delivery of DNA with electroporation all favored cellular responses to secreted HIVBaLgp120 (). We are currently evaluating whether this observation can be generalized to other intracellular antigens. If so, then CTA1 and LTA1 may be ideal adjuvants for therapeutic vaccines for chronic viral infections and cancer where the majority of targeted antigens are intracellular or surface bound.
To date, no single adjuvant has been identified that can alleviate the need for delivery by electroporation. IL-12 has been promising in primates,Citation33,38,39,98,102 including humans,Citation39 but is still not sufficiently potent to negate the need for electroporation. Combining adjuvants may provide additive or synergistic effects that may eclipse those provided by the best individual adjuvants or delivery regimens. Our results with combinations of CTA1 or LTA1 with IL-12 or GM-CSF support this possibility (). With the right combination of adjuvants, it may be possible to eliminate the need for electroporation and potentially reduce the cost, and increase the deployability of some DNA vaccines, particularly those dependent solely on cellular responses. One caveat is that none of the adjuvants or combinations that we have tested have substantially boosted humoral responses when delivered i.m. without electroporation. For this reason, vaccines that require a humoral component for protection may require an enhanced delivery regimen such as electroporation or a heterologous boost with recombinant protein. We are currently exploring the effects of CTA1 and LTA1 delivered by electroporation.
Perhaps the most interesting finding of this study is that CTA1 and LTA1 evoked mixed Th1/Th17 cellular responses ( and data not shown). This may also be a unique feature of these bacterial toxin A1 subunit adjuvants since we have not detected the secretion of IL-17 from mice immunized with DNA alone or co-formulated with IL-12 or GM-CSF ( and data not shown). Th17 cells are a T helper CD4+ subsetCitation103-105 that can secrete IL-17ACitation104,105 and IL-17F,Citation103 as well as IL-21Citation106,107 and IL-22.Citation108,109 Th17 cells generally orchestrate the eradication of extracellular bacteria and fungi by activating neutrophils through production of IL-17A and IL-17F (reviewed in ref.Citation110). Like Th1 cells, Th17 cells also promote B-cell class switching to opsonizing IgG antibodies.Citation111 Th17 cells have also been found to play roles in combating certain viruses and cancers and have been found to play important roles in several autoimmune disorders.Citation112,113 Th17 cells can also acquire the ability to co-express IFN-γ.Citation114 A number of recent studies have demonstrated that the CT and LT holotoxins evoke Th17 responses when delivered to mucosal sites.Citation65-68 Additionally, the novel CTA1-DD adjuvant that retargets the A1 subunit of CT to B cells also stimulates mixed Th1/Th17 responsesCitation69. The results of that studyCitation69 along with our results here indicate that this Th17 skewing activity resides in the A1 subunits of these toxins, and occurs whether delivered mucosally of systemically. This suggests that CTA1 and LTA1 might be ideal adjuvants for anti-bacterial and anti-fungal vaccines (reviewed in ref.Citation115). Interestingly, the CTA1-DD adjuvant targets B cellsCitation47,116,117 whereas i.m. delivered CTA1 or LTA1 almost exclusively targets muscle cells, yet both adjuvants evoke similar mixed Th1/Th17 responses. This indicates that the cell types targeted by CTA1-based adjuvants may be less important than the biological effects that these enzymes have on the targeted cells.
Numerous DNA-based adjuvants, including CTA1 and LTA1 have been discovered and studied, almost always individually. Given the cacophony of cytokines and chemokines that are induced upon infection, we believe that no single DNA adjuvant will solve the shortcomings of DNA vaccines. In this regard, during natural infections, the innate immune system senses multiple pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs) released by host cells in response to infection. It is the integration of signals from these multiple PAMPs and DAMPs that fully triggers innate and adaptive immune responses. Single DNA adjuvants can at best only mimic one of the many signals triggered by natural infections. We therefore propose that combining adjuvants with different and potentially complimentary mechanisms of action will yield improved adaptive immune responses. The data generated under this study support this hypothesis and provide incentive to continue exploring new adjuvant combinations for unique and useful immunological outcomes. Additionally, our data show that different adjuvants can enhance immune responses to different antigens, suggesting that a single adjuvant may not be appropriate for every antigen.
Although the studies presented in this manuscript provide new insights into improving DNA vaccine performance, there are limitations to these studies. For instance, we focused on delivering our vaccines and adjuvants as naked DNA by i.m. injection, while most vaccines that are in clinical trials or are nearing clinical trials are delivered using electroporation. We have also evaluated CTA1 and LTA1 and combinations of CTA1 and LTA1 with IL-12 and other adjuvants delivered by electroporation. We found similar enhancements in the cellular immune responses when these adjuvants were delivered using electroporation yielding dramatically higher cellular responses than those achieved by delivery with electroporation without adjuvants or by i.m. delivery with the same adjuvant combinations (manuscript in preparation).
Another limitation of these studies is that they do not elucidate the mechanism(s) of action of the CTA1 and LTA1 adjuvants. However, we have determined that their adjuvant effects are a result of their enzymatic activities and that their adjuvant effects cannot be mimicked by the elevation of cAMP levels within transfected muscle cells (manuscript in preparation). Therefore, Gsα may not be the primary enzymatic target that mediates the adjuvant activity of CTA1 and LTA1. Together, our results indicate that CTA1 and LTA1 are unique DNA adjuvants that are worthy of further study, particularly in combinations with other adjuvants.
Materials and Methods
DNA vaccine construction
The pMAX-PRO DNA vaccine vector was created by replacing the yellow fluorescent protein (YFP) gene from pMAX-FP-Yellow (Amaxa Corp., Cologne, Germany) with a multiple cloning site using the Nhe-I and Bss-HII sites. The DNA sequences of CTA1 and LTA1 (minus their bacterial secretion signals), mouse GM-CSF, SIVmac239-gag and HIVBaL-gp120 were codon-optimized for expression in human and mouse cells by GeneArt (Resensburg, Germany). The optimized sequences that were provided in the GeneArt shuttle vector were sub-cloned into pMAX-PRO using the 5' Kpn-1 and 3'; Xho-1 sites. The plasmids pMAX-PRO-CTA1, pMAX-PRO-LTA1, pMAX-PRO-SIVmac239-gag, pMAX-PRO-HIVBaL-gp120 and pMAX-PRO-mGM-CSF are referred to as pCTA1, pLTA1, pGM-CSF, pSIV-gag and pHIV-gp120 respectively in the text of this manuscript.
In early studies, we used an IL-12 expressing vector that expressed the mouse IL-12 p35 subunit from a CMV promoter and the P40 subunit from an Internal Ribosome Entry Site (IRES). This plasmid (pMAX-PRO-mIL-12) was created by excising the mIL-12 expression cassette from pUMVC3-mIL-12 (Aldeveron, Fargo, ND) and sub-cloning it into the pMAX-PRO backbone. The expression of mIL-12 from pMAX-PRO-mIL-12 was equivalent to that of pUMVC3-mIL-12 (data not shown). The pMAX-PRO-mIL-12 plasmid is referred to as pIRES-IL-12 in the text of this manuscript. For later studies, a dual promoter plasmid (PBS-mIL-12) expressing the p35 and p40 subunits of mIL-12 was used. This plasmid was described previously.Citation33,37 PBS-mIL-12 plasmid is referred to as pIL-12 in the text of this manuscript.
Vaccination procedures
DNA vaccinations given i.m. without electroporation were given in the quadriceps muscle(s). The mice were anesthetized with a Ketamine-based anesthetic before vaccination. Hair was removed from the areas over the quadriceps muscle(s) using clippers. The animals were inoculated in both quadriceps muscles using a 27 gauge needle and an insulin syringe. A total injection volume of 100 μl (50 μl/muscle) was injected. For mice receiving electroporation, the vaccines were also delivered into the quadriceps muscles. Within one minute after the inoculation, the muscles were subjected to electroporation with the following parameters: 6 pulses of 100 V lasting 50 ms each with 200 ms intervals between pulses. Electroporation was carried out using an 830 electroporation generator (BTX Harvard Apparatus, Holliston, MA) and a 2 needle gene probe with a 5 mm gap.
Animal housing and handling
For the studies in , mice were housed in the Advanced BioScience Laboratories (ABL) Animal Facility in Rockville, MD. The facility is AAALAC-International Accredited, USDA Registered, and has a Category 1 Assurance from the Office of Laboratory Animal Welfare (Assurance # A3467-01). The Animal Research Facility complies with USDA regulations pertaining to animal care (USDA registration #51-R-0059) and with the “PHS Policy on Humane Care and Use of Laboratory Animals” The facility also complies with all policies of the “Guide for the Care and Use of Laboratory Animals” (National Academy of Sciences, 1996). ABL also performed the IFN-γ ELISpot assays shown in .
For the remaining studies, performed by Profectus, the mice were housed at the vivarium of the Department of Comparative Medicine of New York Medical College. This facility is AAALAC International accredited, USDA Registered and has an Assurance from the Office of Laboratory Animal Welfare (formerly OPRR) OLAW Assurance #A3362-1. The Comparative Medicine Department also complies with all the policies promulgated in the “Guide for the Care and Use of Laboratory Animals” (National Academy of Sciences, 1996).
Humoral responses to SIVmac239gag
Solid-phase ELISAs were used to determine SIV-gag-specific antibody (Ab) titers in the sera. Briefly, 96 well high-binding microtiter plates (Nunc, Rochester, NY) were coated with 100 μl of 10 μg/ml of SIVmac-251-rp27-gag (Immunodiagnostics, Woburn, Massachusetts) in PBS overnight at 4°C. Plates were washed 3 times with Tris Buffered Saline (TBS) and then blocked with 300 μl of Blotto (5% w/v non-fat dried milk in TBS) at room temperature for 30 minutes. Serially diluted sera were added to the wells and incubated at room temperature for one hour, then washed 3 times with TBS. Horseradish peroxidase-conjugated goat anti-mouse IgG (Kirkegaard & Perry, Gaithersburg, MD) diluted 1:1000 in Blotto was added (100 μl/well) and incubated at room temperature for one hour. The plates were washed 3 times with TBS before adding SureBlue TMB peroxidase substrate (Kirkegaard & Perry) (100 μl/well) and incubating for 3–5 minutes. The reactions were stopped by adding 50 μl/well of 1N H2SO4. Absorbance was read at 450 nm using a Beckman Coulter AD 200 Plate Reader (Brea, CA). Half-maximal serum binding titers were calculated using SigmaPlot v11 software.
Humoral responses to HIVBaLgp120
Capture ELISAs were used to determine HIVBaL-gp120-specific Ab titers in the sera of mice. Briefly, 96 well high-binding microtiter plates (Nunc, Rochester, NY) were coated with 200 ng/ml of the sheep capture antibody D7324 (Aalto Scientific, Carlsbad, California)Citation118,119 (100 μl/well) overnight at 4°C. After three washes with TBS, 1 μg/ml of Balgp120 (purified recombinant) in PBS (100 μl/well) was added for one hour at 37°C. Plates were washed 3 times with TBS, then blocked with (300 μl/well) Blotto at room temperature for 30 minutes. Serially diluted sera were added to the wells and incubated at room temperature for one hour, then washed 3 times with TBS. Horseradish peroxidase-conjugated goat anti-mouse IgG (Kirkegaard & Perry, Gaithersburg, MD) diluted 1:1000 in Blotto was added (100 μl/well) and incubated at room temperature for one hour. The plates were washed 3 times with TBS before adding SureBlue TMB peroxidase substrate (100 μl/well) and incubating for 3–5 minutes. The reactions were stopped by adding (50 μl/well) of 1 N H2SO4. Absorbance was read at 450 nm using a Beckman Coulter AD 200 plate reader. Half-maximal serum binding titers were calculated using SigmaPlot v11 software.
IFN-γ ELISpot Assays
Ninty six-well flat-bottom ELISpot plates (Millipore, Bedford, MA) were coated overnight at 4 C with an anti-mouse IFN-γ monoclonal antibody (BD-PharMingen, San Jose, CA) at a concentration of 10 μg/mL after which the plates were washed 3 times, then blocked for 2 hours with PBS containing 5% heat-inactivated FBS. Mouse splenocytes were resuspended in complete RPMI 1640 medium containing either medium alone, 50 mg/mL PHA-M (Sigma), or peptide pools (15 mers overlapping by 11 amino acids; 1 μM each final peptide concentration) spanning HIV BaLgp120 or SIVmac239-gag. Input cell numbers were 4 × 105 cells/well, and were assayed in duplicate wells. Cells were incubated for 22–24 hours at 37°C and then removed from the ELISpot plates by first washing with de-ionized water, then washing 6 times with PBS containing 0.25% Tween-20 and then an additional 3 times with PBS. Plates were treated with a biotinylated anti-mouse IFN-γ detection antibody (0.5 μg/well; BD-Pharmingen) and incubated at room temperature for 2 hours. ELISpot plates were washed 10 times with PBS containing 0.25% Tween-20 and treated with 100 μL per well of streptavidin-horseradish peroxidase conjugate (BD-PharMingen) diluted 1:100 and incubated an additional 1 hour at room temperature. Unbound conjugate was removed by rinsing the plate 10 times with PBS containing 0.25% Tween-20. Chromogenic substrate (100 μL/well; AEC chromagen; BD-Pharmingen) was then added for 3–5 minutes before being rinsed away with water, after which the plates were air-dried and the resulting spots counted using an Immunospot Reader (CTL Inc., Cleveland, OH). Peptide-specific ELISpot responses were considered positive if the response (minus media background) was >3-fold above the media response and ≥50 Spot Forming Cells (SFC)/106 cells. The ELISpot assays shown in were performed by ABL. The ELISpot assays shown in were performed by Profectus.
Cytokine ELISAs and Cytokine Bead Array (CBA)
Splenocytes were resuspended in complete RPMI 1640 medium containing either medium alone, 50 μg/mL PHA-M (Sigma), or peptide pools (15 mers overlapping by 11 amino acids; 1 μM each final peptide concentration) spanning HIVBaLgp120 or SIVmac239gag, and plated in 96-well flat-bottom tissue culture plates (Becton Dickenson). Input cell numbers were 8 × 105 cells/well for the study shown in or 1 × 105 cells/well for the study shown in . Cells were incubated for 24 hours at 37°C and then supernatants were removed for cytokine ELISAs or CBA. IFN-γ and Granzyme B Ready-SET-Go!® ELISA kits were purchased from Affymetrix/eBioscience (San Diego, CA) and were performed according to the manufacturer's instructions. CBA was performed by Affymetrix/Panomics (Santa Clara, CA) using their Luminex-based platform (ProcartaPlex™ Multiplex). This service included sample handling, performance of the assays and data analysis. The concentrations of the following cytokines or chemokines were determined in the supernatants: IL-1α, IL1-β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, IL13, IL-15, IL-17A, IL-17F, IL-21, IL-23, EOTAXIN, G-CSF, GM-CSF, IFN-γ, IP-10, KC, LIX, MCP-1, MCP-3, MIP-1α, MIP-1β, MIP-2, M-CSF, sRANKL, RANTES, TGF-β, TNF-α and VEGF. The concentrations of the cytokines and chemokines not shown in the figures were either below the limits of detection or were not significantly different between the groups.
Statistical methods
The error bars in the figures are the standard errors of the means (SEMs). The SEMs were calculated by dividing the standard deviations of the means (STDEVs) (determined using MicroSoft Excel software) by the square root of the number of animals in the group. To make comparisons between groups in a study, the medians of the immune measures for each group were calculated and then evaluated for normality to determine if the distribution required parametric or non-parametric statistical tests for analysis. When a parametric test was required, a student t-test comparison was used. When a non-parametric statistical test was required, a Wilcoxon rank-sum test (2-group comparison) was used. Tests were performed using SigmaPlot v11 or v12 software. A p value ≤0.05 indicates that there is a significant difference in the immune measure between groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank ABL for assistance with the mouse studies shown in .
Funding
This work was supported in part by the SBIR Grants 1R43AI074334-01 and 2R44AI074334-02 awarded to Timothy R. Fouts.
References
- Abdulhaqq SA, Weiner DB. DNA vaccines: developing new strategies to enhance immune responses. Immunol Res 2008; 42:219-32; PMID:19066740; http://dx.doi.org/10.1007/s12026-008-8076-3
- Babiuk S, Baca-Estrada ME, Foldvari M, Storms M, Rabussay D, Widera G, Babiuk LA. Electroporation improves the efficacy of DNA vaccines in large animals. Vaccine 2002; 20:3399-408; PMID:12213410; http://dx.doi.org/10.1016/S0264-410X(02)00269-4
- Coban C, Kobiyama K, Aoshi T, Takeshita F, Horii T, Akira S, Ishii KJ. Novel Strategies to Improve DNA Vaccine Immunogenicity. Curr Gene Ther 2011; 11; PMID:22023477
- Liu MA. DNA vaccines: an historical perspective and view to the future. Immunol Rev 2011; 239:62-84; PMID:21198665; http://dx.doi.org/10.1111/j.1600-065X.2010.00980.x
- Wang S, Zhang C, Zhang L, Li J, Huang Z, Lu S. The relative immunogenicity of DNA vaccines delivered by the intramuscular needle injection, electroporation and gene gun methods. Vaccine 2008; 26:2100-10; PMID:18378365; http://dx.doi.org/10.1016/j.vaccine.2008.02.033
- Fynan EF, Webster RG, Fuller DH, Haynes JR, Santoro JC, Robinson HL. DNA vaccines: protective immunizations by parenteral, mucosal, and gene-gun inoculations. Proc Natl Acad Sci U S A 1993; 90:11478-82; PMID:8265577; http://dx.doi.org/10.1073/pnas.90.24.11478
- Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol 1998; 16:867-70; PMID:9743122; http://dx.doi.org/10.1038/nbt0998-867
- Babiuk S, van Drunen Littel-van den Hurk S, Babiuk LA. Delivery of DNA vaccines using electroporation. Methods Mol Med 2006; 127:73-82; PMID:16988447
- Heller LC, Ugen K, Heller R. Electroporation for targeted gene transfer. Expert Opin Drug Deliv 2005; 2:255-68; PMID:16296752; http://dx.doi.org/10.1517/17425247.2.2.255
- Laddy DJ, Yan J, Khan AS, Andersen H, Cohn A, Greenhouse J, Lewis M, Manischewitz J, King LR, Golding H, et al. Electroporation of synthetic DNA antigens offers protection in nonhuman primates challenged with highly pathogenic avian influenza virus. J Virol 2009; 83:4624-30; PMID:19211745; http://dx.doi.org/10.1128/JVI.02335-08
- Luckay A, Sidhu MK, Kjeken R, Megati S, Chong SY, Roopchand V, Garcia-Hand D, Abdullah R, Braun R, Montefiori DC, et al. Effect of plasmid DNA vaccine design and in vivo electroporation on the resulting vaccine-specific immune responses in rhesus macaques. J Virol 2007; 81:5257-69; PMID:17329330; http://dx.doi.org/10.1128/JVI.00055-07
- Mathiesen I. Electropermeabilization of skeletal muscle enhances gene transfer in vivo. Gene Ther 1999; 6:508-14; PMID:10476210; http://dx.doi.org/10.1038/sj.gt.3300847
- Otten G, Schaefer M, Doe B, Liu H, Srivastava I, zur Megede J, O'Hagan D, Donnelly J, Widera G, et al. Enhancement of DNA vaccine potency in rhesus macaques by electroporation. Vaccine 2004; 22:2489-93; PMID:15193413; http://dx.doi.org/10.1016/j.vaccine.2003.11.073
- Scheerlinck JP, Karlis J, Tjelle TE, Presidente PJ, Mathiesen I, Newton SE. In vivo electroporation improves immune responses to DNA vaccination in sheep. Vaccine 2004; 22:1820-5; PMID:15068866; http://dx.doi.org/10.1016/j.vaccine.2003.09.053
- Selby M, Goldbeck C, Pertile T, Walsh R, Ulmer J. Enhancement of DNA vaccine potency by electroporation in vivo. J Biotechnol 2000; 83:147-52; PMID:11000470; http://dx.doi.org/10.1016/S0168-1656(00)00308-4
- Widera G, Austin M, Rabussay D, Goldbeck C, Barnett SW, Chen M, Leung L, Otten GR, Thudium K, Selby MJ, et al. Increased DNA vaccine delivery and immunogenicity by electroporation in vivo. J Immunol 2000; 164:4635-40; PMID:10779767; http://dx.doi.org/10.4049/jimmunol.164.9.4635
- Feltquate DM, Heaney S, Webster RG, Robinson HL. Different T helper cell types and antibody isotypes generated by saline and gene gun DNA immunization. J Immunol 1997; 158:2278-84; PMID:9036975
- Weiss R, Scheiblhofer S, Freund J, Ferreira F, Livey I, Thalhamer J. Gene gun bombardment with gold particles displays a particular Th2-promoting signal that over-rules the Th1-inducing effect of immunostimulatory CpG motifs in DNA vaccines. Vaccine 2002; 20:3148-54; PMID:12163266; http://dx.doi.org/10.1016/S0264-410X(02)00250-5
- Arrington J, Braun RP, Dong L, Fuller DH, Macklin MD, Umlauf SW, Wagner SJ, Wu MS, Payne LG, Haynes JR. Plasmid vectors encoding cholera toxin or the heat-labile enterotoxin from Escherichia coli are strong adjuvants for DNA vaccines. J Virol 2002; 76:4536-46; PMID:11932419; http://dx.doi.org/10.1128/JVI.76.9.4536-4546.2002
- Babiuk LA, van Drunen Littel-van den H, Babiuk SL. Immunization of animals: from DNA to the dinner plate. Vet Immunol Immunopathol 1999; 72:189-202; PMID:10614509; http://dx.doi.org/10.1016/S0165-2427(99)00132-4
- Haynes JR, Arrington J, Dong L, Braun RP, Payne LG. Potent protective cellular immune responses generated by a DNA vaccine encoding HSV-2 ICP27 and the E. coli heat labile enterotoxin. Vaccine 2006; 24:5016-26; PMID:16621198; http://dx.doi.org/10.1016/j.vaccine.2006.03.046
- Chow YH, Huang WL, Chi WK, Chu YD, Tao MH. Improvement of hepatitis B virus DNA vaccines by plasmids coexpressing hepatitis B surface antigen and interleukin-2. J Virol 1997; 71:169-78; PMID:8985336
- Jang HS, Cho TH, Jang YS, Choung PH. Administration of multiple cytokine genes with anti-tumor activity inhibits both tumor incidence and tumor growth. Yonsei Med J 1999; 40:355-62; PMID:10487139; http://dx.doi.org/10.3349/ymj.1999.40.4.355
- Kim JJ, Trivedi NN, Nottingham LK, Morrison L, Tsai A, Hu Y, Mahalingam S, Dang K, Ahn L, Doyle NK, et al. Modulation of amplitude and direction of in vivo immune responses by co- administration of cytokine gene expression cassettes with DNA immunogens. Eur J Immunol 1998; 28:1089-103; PMID:9541605; http://dx.doi.org/10.1002/(SICI)1521-4141(199803)28:03%3c1089::AID-IMMU1089%3e3.0.CO;2-L
- Kim JJ, Yang JS, Montaner L, Lee DJ, Chalian AA, Weiner DB. Coimmunization with IFN-gamma or IL-2, but not IL-13 or IL-4 cDNA can enhance Th1-type DNA vaccine-induced immune responses in vivo. J Interferon Cytokine Res 2000; 20:311-9; PMID:10762079; http://dx.doi.org/10.1089/10799900050023906
- Sin JI, Kim JJ, Arnold RL, Shroff KE, McCallus D, Pachuk C, McElhiney SP, Wolf MW, Pompa-de BruinSJ, Higgins TJ, et al. IL-12 gene as a DNA vaccine adjuvant in a herpes mouse model: IL-12 enhances Th1-type CD4+ T cell-mediated protective immunity against herpes simplex virus-2 challenge. J Immunol 1999; 162:2912-21; PMID:10072541
- Lu Y, Xin KQ, Hamajima K, Tsuji T, Aoki I, Yang J, Sasaki S, Fukushima J, Yoshimura T, Toda S, et al. Macrophage inflammatory protein-1alpha (MIP-1alpha) expression plasmid enhances DNA vaccine-induced immune response against HIV-1. Clin Exp Immunol 1999; 115:335-41; PMID:9933462; http://dx.doi.org/10.1046/j.1365-2249.1999.00793.x
- Xin KQ, Lu Y, Hamajima K, Fukushima J, Yang J, Inamura K, Okuda K. Immunization of RANTES expression plasmid with a DNA vaccine enhances HIV-1-specific immunity. Clin Immunol 1999; 92:90-6; PMID:10413656; http://dx.doi.org/10.1006/clim.1999.4730
- Youssef S, Maor G, Wildbaum G, Grabie N, Gour-Lavie A, Karin N. C-C chemokine-encoding DNA vaccines enhance breakdown of tolerance to their gene products and treat ongoing adjuvant arthritis. J Clin Invest 2000; 106:361-71; PMID:10930439; http://dx.doi.org/10.1172/JCI9109
- Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol 2005; 175:633-9; PMID:16002657; http://dx.doi.org/10.4049/jimmunol.175.2.633
- Forde GM. Rapid-response vaccines–does DNA offer a solution? Nat Biotechnol 2005; 23:1059-62; PMID:16151391; http://dx.doi.org/10.1038/nbt0905-1059
- Lu S, Wang S, Grimes-Serrano JM. Current progress of DNA vaccine studies in humans. Expert Rev Vaccines 2008; 7:175-91; PMID:18324888; http://dx.doi.org/10.1586/14760584.7.2.175
- Chong SY, Egan MA, Kutzler MA, Megati S, Masood A, Roopchard V, Garcia-Hand D, Montefiori DC, Quiroz J, Rosati M, et al. Comparative ability of plasmid IL-12 and IL-15 to enhance cellular and humoral immune responses elicited by a SIVgag plasmid DNA vaccine and alter disease progression following SHIV(89.6P) challenge in rhesus macaques. Vaccine 2007; 25:4967-82; PMID:17335943; http://dx.doi.org/10.1016/j.vaccine.2006.11.070
- Egan MA, Charini WA, Kuroda MJ, Schmitz JE, Racz P, Tenner-Racz K, Manson K, Wyand M, Lifton MA, Nickerson CE, et al. Simian immunodeficiency virus (SIV) gag DNA-vaccinated rhesus monkeys develop secondary cytotoxic T-lymphocyte responses and control viral replication after pathogenic SIV infection. J Virol 2000; 74:7485-95; PMID:10906202; http://dx.doi.org/10.1128/JVI.74.16.7485-7495.2000
- Egan MA, Chong SY, Megati S, Montefiori DC, Rose NF, Boyer JD, Sidhu MK, Quiroz J, Rosati M, Schadeck EB, et al. Priming with plasmid DNAs expressing interleukin-12 and simian immunodeficiency virus gag enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res Hum Retroviruses 2005; 21:629-43; PMID:16060834; http://dx.doi.org/10.1089/aid.2005.21.629
- Egan MA, Chong S-Y, Megati S, Montefiori DC, Rose NF, Sidhu M, Quiroz J, Schadeck EB, Pavlakis G, Weiner DB, et al. Priming with plasmid DNAs expressing IL-12 and SIV Gag protein enhances the immunogenicity and efficacy of an experimental AIDS vaccine based on recombinant vesicular stomatitis virus. AIDS Res Hum Retroviruses 2005; 7:629-643; PMID:16060834; http://dx.doi.org/10.1089/aid.2005.21.629
- Egan MA, Megati S, Roopchand V, Garcia-Hand D, Luckay A, Chong SY, Rosati M, Sackitey S, Weiner DB, Felber BK, et al. Rational design of a plasmid DNA vaccine capable of eliciting cell-mediated immune responses to multiple HIV antigens in mice. Vaccine 2006; 24:4510-23; PMID:16140439; http://dx.doi.org/10.1016/j.vaccine.2005.08.024
- Schadeck EB, Sidhu M, Egan MA, Chong SY, Piacente P, Masood A, Garcia-Hand D, Cappello S, Roopchand V, Megati S, et al. A dose sparing effect by plasmid encoded IL-12 adjuvant on a SIVgag-plasmid DNA vaccine in rhesus macaques. Vaccine 2006; 24:4677-87; PMID:16288822; http://dx.doi.org/10.1016/j.vaccine.2005.10.035
- Kalams S. Shocking results from HVTN 080. HVTN Conference. Seattle, WA: HIV Vaccine Trials Network, 2010.
- Bagley KC, Lewis GK, Fouts TR. The adjuvant activity of the catalytic A1 domain of cholera toxin for retroviral antigens delivered by GeneGun. Clin Vaccine Immunol 18, 922-930 (2011).
- Bagley KC, Shata MT, Onyabe DY, DeVico AL, Fouts TR, Lewis GK, Hone DM. Immunogenicity of DNA vaccines that direct the coincident expression of the 120 kDa glycoprotein of human immunodeficiency virus and the catalytic domain of cholera toxin. Vaccine 2003; 21:3335-41; PMID:12804865; http://dx.doi.org/10.1016/S0264-410X(03)00038-0
- Lycke N. The mechanism of cholera toxin adjuvanticity. Res Immunol 1997; 148:504-20; PMID:9588829; http://dx.doi.org/10.1016/S0923-2494(98)80144-2
- Ohtomo N, Muraoka T, Tashiro A, Zinnaka Y, Amako K. Size and structure of the cholera toxin molecule and its subunits. J Infect Dis 1976; 133 Suppl:31-40; PMID:815449; http://dx.doi.org/10.1093/infdis/133.Supplement_1.S31
- Heyningen SV. Cholera toxin: interaction of subunits with ganglioside GM1. Science 1974; 183:656-7; PMID:4810267; http://dx.doi.org/10.1126/science.183.4125.656
- Hazes B, Read RJ. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997; 36:11051-4; PMID:9333321; http://dx.doi.org/10.1021/bi971383p
- Cassel D, Selinger Z. Mechanism of adenylate cyclase activation by cholera toxin: inhibition of GTP hydrolysis at the regulatory site. Proc Natl Acad Sci U S A 1977; 74:3307-11; PMID:198781; http://dx.doi.org/10.1073/pnas.74.8.3307
- Agren L, Lowenadler B, Lycke N. A novel concept in mucosal adjuvanticity: the CTA1-DD adjuvant is a B cell-targeted fusion protein that incorporates the enzymatically active cholera toxin A1 subunit. Immunology and cell biology 1998; 76:280-7; PMID:9682972; http://dx.doi.org/10.1046/j.1440-1711.1998.00750.x
- Agren L, Sverremark E, Ekman L, Schon K, Lowenadler B, Fernandez C, Lycke N. The ADP-ribosylating CTA1-DD adjuvant enhances T cell-dependent and independent responses by direct action on B cells involving anti- apoptotic Bcl-2- and germinal center-promoting effects. J Immunol 2000; 164:6276-86; PMID:10843681; http://dx.doi.org/10.4049/jimmunol.164.12.6276
- Bagley KC, Abdelwahab SF, Tuskan RG, Fouts TR, Lewis GK. Cholera Toxin and Heat-Labile Enterotoxin Activate Human Monocyte- Derived Dendritic Cells and Dominantly Inhibit Cytokine Production through a Cyclic AMP-Dependent Pathway. Infect Immun 2002; 70:5533-9; PMID:12228279; http://dx.doi.org/10.1128/IAI.70.10.5533-5539.2002
- Bagley KC, Abdelwahab SF, Tuskan RG, Lewis GK. An enzymatically active a domain is required for cholera-like enterotoxins to induce a long-lived blockade on the induction of oral tolerance: new method for screening mucosal adjuvants. Infect Immun 2003; 71:6850-6; PMID:14638772; http://dx.doi.org/10.1128/IAI.71.12.6850-6856.2003
- Giuliani MM, Del Giudice G, Giannelli V, Dougan G, Douce G, Rappuoli R, Pizza M. Mucosal adjuvanticity and immunogenicity of LTR72, a novel mutant of Escherichia coli heat-labile enterotoxin with partial knockout of ADP-ribosyltransferase activity. J Exp Med 1998; 187:1123-32; PMID:9529328; http://dx.doi.org/10.1084/jem.187.7.1123
- Lycke N, Tsuji T, Holmgren J. The adjuvant effect of Vibrio cholerae and Escherichia coli heat-labile enterotoxins is linked to their ADP-ribosyltransferase activity. Eur J Immunol 1992; 22:2277-81; PMID:1381311; http://dx.doi.org/10.1002/eji.1830220915
- Ryan EJ, McNeela E, Pizza M, Rappuoli R, O'Neill L, Mills KH. Modulation of innate and acquired immune responses by Escherichia coli heat-labile toxin: distinct pro- and anti-inflammatory effects of the nontoxic AB complex and the enzyme activity. J Immunol 2000; 165:5750-9; PMID:11067933; http://dx.doi.org/10.4049/jimmunol.165.10.5750
- De Haan L, Hirst TR. Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review). Mol Membr Biol 2004; 21:77-92; PMID:15204437; http://dx.doi.org/10.1080/09687680410001663267
- Morinaga N, Kaihou Y, Vitale N, Moss J, Noda M. Involvement of ADP-ribosylation factor 1 in cholera toxin-induced morphological changes of Chinese hamster ovary cells. J Biol Chem 2001; 276:22838-43; PMID:11279243; http://dx.doi.org/10.1074/jbc.M101184200
- Holmgren J. Actions of cholera toxin and the prevention and treatment of cholera. Nature 1981; 292:413-17; PMID:7019725; http://dx.doi.org/10.1038/292413a0
- Douce G, Turcotte C, Cropley I, Roberts M, Pizza M, Domenghini M, Rappuoli R, Dougan G. Mutants of Escherichia coli heat-labile toxin lacking ADP- ribosyltransferase activity act as nontoxic, mucosal adjuvants. Proc Natl Acad Sci U S A 1995; 92:1644-8; PMID:7878032; http://dx.doi.org/10.1073/pnas.92.5.1644
- Yamamoto S, Takeda Y, Yamamoto M, Kurazono H, Imaoka K, Fujihashi K, Noda M, Kiyono H, McGhee JR. Mutants in the ADP-ribosyltransferase cleft of cholera toxin lack diarrheagenicity but retain adjuvanticity. J Exp Med 1997; 185:1203-10; PMID:9104807; http://dx.doi.org/10.1084/jem.185.7.1203
- Pizza M, Giuliani MM, Fontana MR, Douce G, Dougan G, Rappuoli R. LTK63 and LTR72, two mucosal adjuvants ready for clinical trials. Int J Med Microbiol 2000; 290:455-61; PMID:11111926; http://dx.doi.org/10.1016/S1438-4221(00)80064-8
- Glenn GM, Flyer DC, Ellingsworth LR, Frech SA, Frerichs DM, Seid RC, Yu J. Transcutaneous immunization with heat-labile enterotoxin: development of a needle-free vaccine patch. Expert Rev Vaccines 2007; 6:809-19; PMID:17931160; http://dx.doi.org/10.1586/14760584.6.5.809
- Glenn GM, Rao M, Matyas GR, Alving CR. Skin immunization made possible by cholera toxin. Nature 1998; 391:851; PMID:9495336; http://dx.doi.org/10.1038/36014
- Glenn GM, Scharton-Kersten T, Vassell R, Mallett CP, Hale TL, Alving CR. Transcutaneous immunization with cholera toxin protects mice against lethal mucosal toxin challenge. J Immunol 1998; 161:3211-4; PMID:9759833
- Glenn GM, Scharton-Kersten T, Vassell R, Matyas GR, Alving CR. Transcutaneous immunization with bacterial ADP-ribosylating exotoxins as antigens and adjuvants. Infect Immun 1999; 67:1100-6; PMID:10024549
- Glenn GM, Taylor DN, Li X, Frankel S, Montemarano A, Alving CR. Transcutaneous immunization: a human vaccine delivery strategy using a patch. Nat Med 2000; 6:1403-6; PMID:11100128; http://dx.doi.org/10.1038/82225
- Brereton CF, Sutton CE, Ross PJ, Iwakura Y, Pizza M, Rappuoli R, Lavelle EC, Mills KH. Escherichia coli heat-labile enterotoxin promotes protective Th17 responses against infection by driving innate IL-1 and IL-23 production. J Immunol 2011; 186:5896-906; PMID:21490151; http://dx.doi.org/10.4049/jimmunol.1003789
- Datta SK, Sabet M, Nguyen KP, Valdez PA, Gonzalez-Navajas JM, Islam S, Mihajlov I, Fierer J, Insel PA, Webster NJ, et al. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc Natl Acad Sci U S A 2010; 107:10638-43; PMID:20479237; http://dx.doi.org/10.1073/pnas.1002348107
- Lee JB, Jang JE, Song MK, Chang J. Intranasal delivery of cholera toxin induces th17-dominated T-cell response to bystander antigens. PLoS One 2009; 4:e5190; PMID:19360100; http://dx.doi.org/10.1371/journal.pone.0005190
- Norton EB, Lawson LB, Mahdi Z, Freytag LC, Clements JD. The A subunit of Escherichia coli heat-labile enterotoxin functions as a mucosal adjuvant and promotes IgG2a, IgA, and Th17 responses to vaccine antigens. Infect Immun 2012; 80:2426-35; PMID:22526674; http://dx.doi.org/10.1128/IAI.00181-12
- Nedrud JG, Bagheri N, Schon K, Xin W, Bergroth H, Eliasson DG, Lycke NY. Subcomponent vaccine based on CTA1-DD adjuvant with incorporated UreB class II peptides stimulates protective Helicobacter pylori immunity. PLoS One 2013; 8:e83321
- Gehl J, Mir LM. Determination of optimal parameters for in vivo gene transfer by electroporation, using a rapid in vivo test for cell permeabilization. Biochem Biophys Res Commun 1999; 261:377-80; PMID:10425193; http://dx.doi.org/10.1006/bbrc.1999.1014
- Bachy M, Boudet F, Bureau M, Girerd-Chambaz Y, Wils P, Scherman D, Meric C. Electric pulses increase the immunogenicity of an influenza DNA vaccine injected intramuscularly in the mouse. Vaccine 2001; 19:1688-93; PMID:11166892; http://dx.doi.org/10.1016/S0264-410X(00)00406-0
- Buchan S, Gronevik E, Mathiesen I, King CA, Stevenson FK, Rice J. Electroporation as a “prime/boost” strategy for naked DNA vaccination against a tumor antigen. J Immunol 2005; 174:6292-8; PMID:15879128; http://dx.doi.org/10.4049/jimmunol.174.10.6292
- Tollefsen S, Vordermeier M, Olsen I, Storset AK, Reitan LJ, Clifford D, Lowrie DB, Wiker HG, Huygen K, Hewinson G, et al. DNA injection in combination with electroporation: a novel method for vaccination of farmed ruminants. Scand J Immunol 2003; 57:229-38; PMID:12641651; http://dx.doi.org/10.1046/j.1365-3083.2003.01218.x
- Zhao YG, Peng B, Deng H, Chen G, Yang F, Shao M, Lu H, Li Y, Peng J, Xu L, et al. Anti-HBV immune responses in rhesus macaques elicited by electroporation mediated DNA vaccination. Vaccine 2006; 24:897-903; PMID:16253404; http://dx.doi.org/10.1016/j.vaccine.2005.08.093
- Zucchelli S, Capone S, Fattori E, Folgori A, Di Marco A, Casimiro D, Simon AJ, Laufer R, La Monica N, Cortese R, et al. Enhancing B- and T-cell immune response to a hepatitis C virus E2 DNA vaccine by intramuscular electrical gene transfer. J Virol 2000; 74:11598-607; PMID:11090158; http://dx.doi.org/10.1128/JVI.74.24.11598-11607.2000
- Belladonna ML, Renauld JC, Bianchi R, Vacca C, Fallarino F, Orabona C, Fioretti MC, Grohmann U, Puccetti P. IL-23 and IL-12 have overlapping, but distinct, effects on murine dendritic cells. J Immunol 2002; 168:5448-54; PMID:12023338; http://dx.doi.org/10.4049/jimmunol.168.11.5448
- Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets 2009; 8:40-52; PMID:19275692; http://dx.doi.org/10.2174/187152809787582507
- Grohmann U, Belladonna ML, Vacca C, Bianchi R, Fallarino F, Orabona C, Fioretti MC, Puccetti P. Positive regulatory role of IL-12 in macrophages and modulation by IFN-gamma. J Immunol 2001; 167:221-7; PMID:11418652; http://dx.doi.org/10.4049/jimmunol.167.1.221
- Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ. IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 2004; 202:96-105; PMID:15546388; http://dx.doi.org/10.1111/j.0105-2896.2004.00214.x
- Novelli F, Casanova JL. The role of IL-12, IL-23 and IFN-gamma in immunity to viruses. Cytokine Growth Factor Rev 2004; 15:367-77; PMID:15450252; http://dx.doi.org/10.1016/j.cytogfr.2004.03.009
- Ryffel B. Interleukin-12: role of interferon-gamma in IL-12 adverse effects. Clin Immunol Immunopathol 1997; 83:18-20; PMID:9073529; http://dx.doi.org/10.1006/clin.1996.4306
- Barouch DH, Santra S, Tenner-Racz K, Racz P, Kuroda MJ, Schmitz JE, Jackson SS, Lifton MA, Freed DC, Perry HC, et al. Potent CD4+ T cell responses elicited by a bicistronic HIV-1 DNA vaccine expressing gp120 and GM-CSF. J Immunol 2002; 168:562-8; PMID:11777947; http://dx.doi.org/10.4049/jimmunol.168.2.562
- Iwasaki A, Stiernholm BJ, Chan AK, Berinstein NL, Barber BH. Enhanced CTL responses mediated by plasmid DNA immunogens encoding costimulatory molecules and cytokines. J Immunol 1997; 158:4591-601; PMID:9144471
- Kim JJ, Simbiri KA, Sin JI, Dang K, Oh J, Dentchev T, Lee D, Nottingham LK, Chalian AA, McCallus D, et al. Cytokine molecular adjuvants modulate immune responses induced by DNA vaccine constructs for HIV-1 and SIV. J Interferon Cytokine Res 1999; 19:77-84; PMID:10048771; http://dx.doi.org/10.1089/107999099314441
- Lai L, Vodros D, Kozlowski PA, Montefiori DC, Wilson RL, Akerstrom VL, Chennareddi L, Yu T, Kannanganat S, Ofielu L, et al. GM-CSF DNA: an adjuvant for higher avidity IgG, rectal IgA, and increased protection against the acute phase of a SHIV-89.6P challenge by a DNA/MVA immunodeficiency virus vaccine. Virology 2007; 369:153-67; PMID:17698160; http://dx.doi.org/10.1016/j.virol.2007.07.017
- Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect Immun 1988; 56:1748-53; PMID:3290106
- Freytag LC, Clements JD. Mucosal adjuvants. Vaccine 2005; 23:1804-13; PMID:15734046; http://dx.doi.org/10.1016/j.vaccine.2004.11.010
- Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev 2010; 235:105-16; PMID:20536558
- Lord SJ, Rajotte RV, Korbutt GS, Bleackley RC. Granzyme B: a natural born killer. Immunol Rev 2003; 193:31-8; PMID:12752668; http://dx.doi.org/10.1034/j.1600-065X.2003.00044.x
- Shresta S, Heusel JW, Macivor DM, Wesselschmidt RL, Russell JH, Ley TJ. Granzyme B plays a critical role in cytotoxic lymphocyte-induced apoptosis. Immunol Rev 1995; 146:211-21; PMID:7493755; http://dx.doi.org/10.1111/j.1600-065X.1995.tb00690.x
- Liu MA. DNA vaccines: a review. J Intern Med 2003; 253:402-10; PMID:12653868; http://dx.doi.org/10.1046/j.1365-2796.2003.01140.x
- Jones S, Evans K, McElwaine-Johnn H, Sharpe M, Oxford J, Lambkin-Williams R, Mant T, Nolan A, Zambon M, Ellis J, et al. DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccine 2009; 27:2506-12; PMID:19368793; http://dx.doi.org/10.1016/j.vaccine.2009.02.061
- Sharpe M, Lynch D, Topham S, Major D, Wood J, Loudon P. Protection of mice from H5N1 influenza challenge by prophylactic DNA vaccination using particle mediated epidermal delivery. Vaccine 2007; 25:6392-8; PMID:17640779; http://dx.doi.org/10.1016/j.vaccine.2007.06.009
- Fuller DH, Rajakumar P, Che JW, Narendran A, Nyaundi J, Michael H, Yager EJ, Stagnar C, Wahlberg B, Taber R, et al. Therapeutic DNA vaccine induces broad T cell responses in the gut and sustained protection from viral rebound and AIDS in SIV-infected rhesus macaques. PLoS One 2012; 7:e33715; PMID:22442716; http://dx.doi.org/10.1371/journal.pone.0033715
- Yager EJ, Dean HJ, Fuller DH. Prospects for developing an effective particle-mediated DNA vaccine against influenza. Expert Rev Vaccines 2009; 8:1205-20; PMID:19722894; http://dx.doi.org/10.1586/erv.09.82
- Yager EJ, Stagnar C, Gopalakrishnan R, Fuller JT, Fuller DH. Optimizing particle-mediated epidermal delivery of an influenza DNA vaccine in ferrets. Methods Mol Biol 2013; 940:223-37; PMID:23104347
- Boyer JD, Robinson TM, Kutzler MA, Vansant G, Hokey DA, Kumar S, Parkinson R, Wu L, Sidhu MK, Pavlakis GN, et al. Protection against simian/human immunodeficiency virus (SHIV) 89.6P in macaques after coimmunization with SHIV antigen and IL-15 plasmid. Proc Natl Acad Sci U S A 2007; 104:18648-53; PMID:18000037; http://dx.doi.org/10.1073/pnas.0709198104
- Halwani R, Boyer JD, Yassine-Diab B, Haddad EK, Robinson TM, Kumar S, Parkinson R, Wu L, Sidhu MK, Phillipson-Weiner R, et al. Therapeutic vaccination with simian immunodeficiency virus (SIV)-DNA + IL-12 or IL-15 induces distinct CD8 memory subsets in SIV-infected macaques. J Immunol 2008; 180:7969-79; PMID:18523260; http://dx.doi.org/10.4049/jimmunol.180.12.7969
- Hung CF, Monie A, Alvarez RD, Wu TC. DNA vaccines for cervical cancer: from bench to bedside. Exp Mol Med 2007; 39:679-89; PMID:18160838; http://dx.doi.org/10.1038/emm.2007.74
- Trimble CL, Peng S, Kos F, Gravitt P, Viscidi R, Sugar E, Pardoll D, Wu TC. A phase I trial of a human papillomavirus DNA vaccine for HPV16+ cervical intraepithelial neoplasia 2/3. Clin Cancer Res 2009; 15:361-7; PMID:19118066; http://dx.doi.org/10.1158/1078-0432.CCR-08-1725
- Zeng Q, Peng S, Monie A, Yang M, Pang X, Hung CF, Wu TC. Control of cervicovaginal HPV-16 E7-expressing tumors by the combination of therapeutic HPV vaccination and vascular disrupting agents. Hum Gene Ther 2011; 22:809-19; PMID:21128743; http://dx.doi.org/10.1089/hum.2010.071
- Gherardi MM, Ramirez JC, Esteban M. Interleukin-12 (IL-12) enhancement of the cellular immune response against human immunodeficiency virus type 1 env antigen in a DNA prime/vaccinia virus boost vaccine regimen is time and dose dependent: suppressive effects of IL-12 boost are mediated by nitric oxide. J Virol 2000; 74:6278-86; PMID:10864637; http://dx.doi.org/10.1128/JVI.74.14.6278-6286.2000
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005; 201:233-40; PMID:15657292; http://dx.doi.org/10.1084/jem.20041257
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123-32; PMID:16200070; http://dx.doi.org/10.1038/ni1254
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, et al. A distinct lineage of CD4T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133-41; PMID:16200068; http://dx.doi.org/10.1038/ni1261
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007; 448:480-3; PMID:17581589; http://dx.doi.org/10.1038/nature05969
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007; 448:484-7; PMID:17581588; http://dx.doi.org/10.1038/nature05970
- Chung Y, Yang X, Chang SH, Ma L, Tian Q, Dong C. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res 2006; 16:902-7; PMID:17088898; http://dx.doi.org/10.1038/sj.cr.7310106
- Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 2006; 203:2271-9; PMID:16982811; http://dx.doi.org/10.1084/jem.20061308
- Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev 2013; 252:89-103; PMID:23405897; http://dx.doi.org/10.1111/imr.12035
- Mitsdoerffer M, Lee Y, Jager A, Kim HJ, Korn T, Kolls JK, Cantor H, Bettelli E, Kuchroo VK. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc Natl Acad Sci U S A 2010; 107:14292-7; PMID:20660725; http://dx.doi.org/10.1073/pnas.1009234107
- Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev 2008; 223:87-113; PMID:18613831; http://dx.doi.org/10.1111/j.1600-065X.2008.00628.x
- Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol 2013; 182:10-20; PMID:23159950; http://dx.doi.org/10.1016/j.ajpath.2012.08.041
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol 2011; 12:255-63; PMID:21278737; http://dx.doi.org/10.1038/ni.1993
- Kumar P, Chen K, Kolls JK. Th17 cell based vaccines in mucosal immunity. Curr Opin Immunol 2013; 25:373-80; PMID:23669353; http://dx.doi.org/10.1016/j.coi.2013.03.011
- Agren L, Norin M, Lycke N, Lowenadler B. Hydrophobicity engineering of cholera toxin A1 subunit in the strong adjuvant fusion protein CTA1-DD. Protein Eng 1999; 12:173-8; PMID:10195289; http://dx.doi.org/10.1093/protein/12.2.173
- Lycke N. From toxin to adjuvant: the rational design of a vaccine adjuvant vector, CTA1-DD/ISCOM. Cell Microbiol 2004; 6:23-32; PMID:14678328; http://dx.doi.org/10.1046/j.1462-5822.2003.00338.x
- Moore JP, Jarrett RF. Sensitive ELISA for the gp120 and gp160 surface glycoproteins of HIV-1. AIDS Res Hum Retroviruses 1988; 4:369-79; PMID:2848557; http://dx.doi.org/10.1089/aid.1988.4.369
- Moore JP, McKeating JA, Jones IM, Stephens PE, Clements G, Thomson S, Weiss RA. Characterisation of recombinant gp120 and gp160 from HIV-1: binding to monoclonal antibodies and sCD4. AIDS 1990; 4:307-15; PMID:2190605; http://dx.doi.org/10.1097/00002030-199004000-00004