
ABSTRACT
Engineering dendritic cells (DCs) to treat cancer is a long sought-after goal for cell-based immunotherapies. In this review, we focus on the experience with CMN-001, formally AGS-003, a DC-based immunotherapy, employing autologous DC electroporated with autologous tumor RNA to treat subjects with metastatic renal cell carcinoma (mRCC). We will review the early clinical development of CMN-001 up to and including deployment in a multicenter phase 3 study and provide a rationale to continue the development of CMN-001 in an ongoing randomized phase 2 study. The synergy between CMN-001 and everolimus observed in the phase 3 study provides an opportunity to design a phase 2b study building on the mechanism of action of CMN-001 and underlying immune and clinical outcomes revealed in the earlier studies. The design of the phase 2b study combines CMN-001 with first-line checkpoint inhibition therapy and second line lenvatinib/everolimus in poor-risk mRCC subjects.
Rationale for CMN-001 product development
Dendritic cells (DCs) are antigen-presenting cells with the ability to prime T cells, driving long-term immunity against infectious agents and tumor cells. Enabling DC to prime anti-tumor responses is a long-standing goal of cell-based immunotherapies to treat cancer.Citation1,Citation2 This is predicated on the fact that DC can be loaded with tumor antigens in a variety of forms, including tumor lysates,Citation3 tumor proteins, peptides, and transduction with viral vectors encoding tumor antigensCitation4 and tumor antigens encoded in the form of DNA or RNA.Citation5–9 Clinical trials employing antigen-loaded DC have been shown to be safe and provide positive clinical benefit, albeit only in a small proportion of subjects.Citation10–13 There are many potential factors that can impact DC-based therapy. These include immune suppression in the tumor microenvironment, reduction of tumor antigen expression, the presence of regulatory T cells, and strong checkpoint signaling,Citation14,Citation15 all designed to dampen the induced immune response. This has led investigators to look for ways to improve the overall efficacy of DC therapy to attain durable clinical responses.
In this review, we focus on the experience with CMN-001, formally AGS-003, a DC-based immunotherapy, employing autologous DC electroporated with autologous amplified tumor RNA to treat patients with metastatic renal cell carcinoma (mRCC). CMN-001 is specifically designed to elicit an adaptive T-cell response against the antigens present in the patient’s own tumor tissue. We will review early clinical development of CMN-001 through its deployment in a large multicenter phase 3 study, the ADAPT study (Autologous Dendritic Cell Immunotherapy Plus Standard Treatment of Advanced Renal Cell Carcinoma),Citation16 with a specific focus on lessons learned during the execution of the ADAPT study leading to the design of a new clinical study. The new study design is reflective of the changing landscape of therapies now available to treat mRCC in the first and second lines, and is based on a retrospective data analysis of subjects who received CMN-001.
Mechanism of action of CMN-001
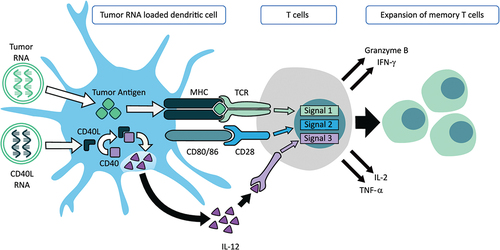
The fundamental mechanism of action of CMN-001 is via the induction of an adaptive T cell response specifically directed to the subjects’ tumor antigens encoded in the amplified tumor RNA. This mechanism of action involves three critical signals essential for T-cell activation (). Signal 1, the translation of electroporated amplified tumor RNA, processing, and presentation of tumor antigens via human leukocyte antigen (HLA) molecules to T cells, and subsequent activation and expansion of memory cytotoxic T lymphocytes (CTL). Signal 2, CMN-001 expresses CD80 and CD86 and delivers costimulatory signals to the T cell through the CD28 receptor on the T cell. The addition of CD40L RNA ensures a critical third signal required for cytotoxic T cells and DC secretion of IL-12 for the expansion and function of antigen-specific memory CTL.Citation17–19 CD40L binds to CD40 within the DC and delivers the necessary signal for IL-12 secretion. Therefore, CMN-001 incorporates all three signals in a single cellular product: antigen presentation, co-stimulation, and IL-12 secretion enabling an adaptive T cell response. Once activated, CTL will expand into memory T cells and secrete cytokines such as IFN-γ, TNF-a, and IL-2. These CTL can kill tumor cells bearing antigens for which they have been primed through the release of granzyme B. All three components help overcome the immunosuppressive environment present in the tumor microenvironment, bypassing the need for additional CD4+ T cell help.Citation20 Preclinical work has validated this mechanism of action by showing that CMN-001 is capable of priming memory CTL that retains a high avidity for antigen-specific targets.Citation21 Moreover, the potency of CMN-001 and the ability of DC to secrete IL-12 correlated with the induction of memory CTL to secrete multiple cytokines and the killing of cells expressing the correct antigen target.Citation22 In the following sections, we will present data from the reported clinical studies where monitoring T cell responses in subjects receiving CMN-001 showed a correlation between the number of induced memory CTL and clinical outcomes, thus validating the mechanism of action of CMN-001 modeled in vitro with applicable data from the clinical setting.
Figure 1. CMN-001 mechanism of action. Signal 1, mature DC expresses human leukocyte antigen (HLA) (a–c) surface molecules. In the CMN-001 manufacturing process, electroporation of autologous DC with amplified autologous tumor RNA enables the surface presentation of autologous tumor antigens in an appropriate HLA context. Signal 2, mature DCs express CD80/86 costimulatory molecules on the cell surface and deliver a costimulatory signal through the CD28 receptor present on T cells. Signal 3, the induction of IL-12 secretion from DC is achieved by cell surface ligation of CD40 by interaction with CD4+ helper T cells, which express CD40L on their cell surface. The efficiency of CD40/CD40L ligation is improved by performing it ex vivo in the CMN-001 manufacturing process. Electroporation of the DC with CD40L RNA allows for ectopic expression of CD40L with subsequent ligation with CD40 within the DC. Signaling via the formation of CD40/CD40L complexes within the DC stimulates IL-12 release from the DC, thus bypassing the requirement for a chance CD4+ helper T cell encounter in vivo and providing IL-12 for the expansion of cytotoxic T cells.
CMN-001 manufacturing
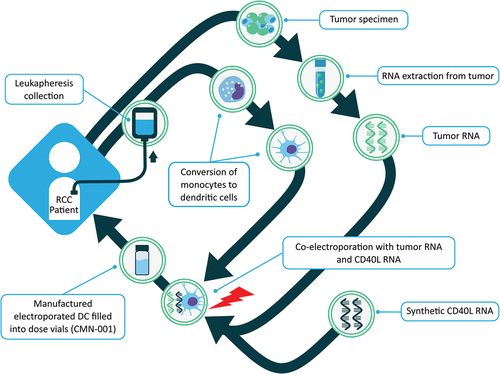
CMN-001 is manufactured at a centralized good manufacturing process (GMP) compliant facility (CoImmune Inc., Durham, NC), as has been previously published.Citation16,Citation23 An overview of the key steps in the manufacturing process for producing CMN-001 is outlined in . Briefly, CMN-001 is manufactured using two cellular components obtained from the subject. The first is a tumor specimen collected during a nephrectomy or biopsy. Tumor tissue confirmed to have predominantly clear cell histology was used to generate amplified tumor RNA.Citation24,Citation25 Autologous total tumor RNA is extracted, and polyadenylated messenger RNA is amplified using reverse transcriptase polymerase chain reaction and in vitro transcription to generate amplified tumor RNACitation25 for electroporation into the subjects’ DC. This approach captures all the tumor-specific neoantigens that have escaped central tolerance without the need to identify them. The second component is monocytes, isolated from peripheral blood mononuclear cells (PBMC) by elutriation obtained from subjects by leukapheresis. The monocytes are converted to immature DC by the addition of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4. Immature DC are matured using TNF-α, IFN-γ, and PGE2. These mature DC are then co-electroporated with autologous amplified tumor RNA and synthetic CD40L RNA using a post-maturation electroporation protocol as described previously.Citation16,Citation18,Citation23,Citation24 CD40L RNA was manufactured using in vitro transcription and a post-transcriptional capping method.Citation26 The resulting CMN-001 product is formulated as a single dose, and vials are frozen for cryogenic storage until the time of administration. This process results in multiple doses being produced for a subject in a single manufacturing run. Thawed samples of the final product were assessed for sterility, mycoplasma, endotoxin, and viability prior to release for clinical use.
Figure 2. CMN-001 manufacturing process. Two cellular components are collected from an mRCC patient. The first is a tumor specimen collected from subjects during nephrectomy or biopsy, and autologous total tumor RNA is extracted and amplified. The second component is monocytes, which are DC precursors isolated from the leukocytes by elutriation from a leukapheresis collection. GM-CSF and IL-4 are used initially to convert the monocytes into immature DC, which are then matured using TNF-α, IFN-γ, and PGE2. These mature DCs are then co-electroporated with amplified tumor RNA and synthetic CD40L RNA. The resulting RNA-electroporated mature DC is formulated as a single dose and vialed for cryogenic storage.
Results of clinical studies
Clinical development of CMN-001
The clinical experience with CMN-001 spans over two decades with CMN-001 (formally AGS-003) first employed as a monotherapy in a single-arm study. This study enrolled 22 mRCC subjects with an overall response rate (ORR) of 36% with seven subjects having stable disease (SD) and one with a partial response (PR). The median overall survival (OS) was 15.7 months and the median progression free survival (PFS) was 5.5 months (). During the study, the landscape for first-line therapy for subjects with mRCC changed from monotherapy treatment with IFN-α or IL-2 to approval of tyrosine kinase inhibitors (TKI), such as sorafenib and sunitinib, to treat mRCC as first-line therapy.Citation27–29 This prompted the design of a second-phase 2b study to include CMN-001 with sunitinib as a combination therapy. This study was initiated in January 2006, and results on 21 subjects have been published.Citation23 The study reported an ORR of 62% with five PR and eight SD. The median OS was 30.2 months, and the median PFS was 11.2 months (). At the time of this study, this reported OS was twice as long as what had been reported in a similar population of newly diagnosed, intermediate-and poor-risk RCC subjects receiving only sunitinib.Citation30 Strikingly, two subjects treated with CMN-001 in combination with sunitinib had durable, prolonged PRs greater than 8 years after diagnosis.
Table 1. Summary of clinical experience with CMN-001 in metastatic renal cell cancer.
Based on these encouraging clinical results, a phase 3 study was undertaken to assess the clinical activity of CMN-001 in combination with sunitinib compared to sunitinib alone as SOC treatment. The ADAPT study is one of the largest DC-based phase 3 studies, which enrolled 462 subjects randomized 2:1 to receive either CMN-001 on the combination arm with sunitinib or on the SOC treatment arm receiving sunitinib alone. CMN-001 was administered after at least one cycle of sunitinib and then five doses for induction, irrespective of progression. On the combination arm, after completion of induction, booster dosing continued until two or more progression events occurred within the first 48 weeks. The study provided for the inclusion of second-line therapy initiation if discontinuation of sunitinib was indicated for either toxicity or disease progression. Subjects who received CMN-001 continued with CMN-001 dosing through second-line therapy. The study allowed, at the discretion of the treating physician, the use of second-line therapies including everolimus, axitinib, pazopanib, cabozantinib, bevacizumab, nivolumab, IL-2, and pembrolizumab. All agents approved for use in second line.Citation31–35 The immunogenicity of CMN-001 was confirmed through the immune monitoring analysis of immunological responses in treated subjects. However, based on an interim analysis conducted by the data safety monitoring committee, the study was discontinued for futility, concluding that the study was unlikely to demonstrate a statistically significant improvement in OS in the combination arm based on the prospectively defined statistical analysis plan. Full study results have been publishedCitation16 and reported a median OS of 27.7 months and 32.4 months and median PFS of 6.0 months and 7.2 months for the CMN-001 combination arm and SOC arms, respectively (). While CMN-001 did not improve OS in subjects treated with combination therapy, the induced immune response correlated with OS and confirmed the immunogenicity of CMN-001. Moreover, the study identified two potential predictive survival biomarkers, IL-12 produced by the DC and higher numbers of T regulatory cells present in mRCC subjects.
Furthermore, no significant safety issues were attributable to the use of CMN-001, and no deaths or discontinuations were related to the administration of CMN-001. In the ADAPT study, few serious adverse events were attributed to CMN-001. Overall, CMN-001 was well tolerated, with only 6 out of 268 subjects (2.0%) on ADAPT study reporting Grade III/IV/V Treatment-Emergent Adverse Events (TEAEs) in the subjects who received at least one dose of CMN-001.Citation16 The majority of CMN-001-related adverse events were general injection site reactions, including erythema, pain, pruritus, and swelling. CMN-001 was successfully manufactured and delivered across multiple cancer centers for >94% of enrolled subjects. In addition to the ADAPT trial, another large phase 3 study deploying autologous DC (DCvax-L) to treat glioblastoma has been reported.Citation36 These studies taken together show the feasibility of manufacturing and delivery of DC-based cell therapies for deployment in large clinical studies. All subjects enrolled in the clinical studies administering CMN-001 NCT00272649, NCT00678119, NCT01582672, and NCT04203901 provided written informed consent prior to participation, and the trial was approved by each institutional review board in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines of the International Conference on Harmonization.
Retrospective analysis of clinical data from subjects receiving CMN-001
A retrospective analysis of the ADAPT study clinical data revealed a surprising survival benefit for subjects who were treated with everolimus as a subsequent second-line therapy on the combination arm. None of the other second-line therapies showed synergy with CMN-001. A deeper analysis of the subject data revealed an interesting observation. When the 60 subjects who received everolimus in combination with CMN-001 were compared to the 31 subjects who received everolimus after sunitinib, the median OS was 23.0 months and 13.6 months, respectively, with a hazard ratio (HR) of 0.76 (95% CI, 0.45–1.31) ().Citation37 Further analysis of this subset of subjects who received everolimus in second-line therapy after progression revealed two interesting trends. First, if subjects received everolimus during the treatment phase of the study (n = 49), which included all randomized subjects who received at least one or more doses of CMN 001 (n = 38) or at least one or more doses of sunitinib as SOC (n = 11), median OS was 19.3 months and 17.8 months, respectively, with an HR of 0.89 (95% CI, 0.42–2.10) (). Moreover, if subjects received everolimus during the follow-up phase of the study (n = 42), which included all randomized subjects who received at least one or more doses of CMN-001 (n = 22) or at least one or more doses of sunitinib as SOC treatment (n = 20), median OS was 25.8 months and 13.4 months, respectively, with an HR of 0.51 (95% CI, 0.23–1.11) (). This increase in clinical benefit for subjects receiving everolimus during the follow-up period of the study only manifests itself if subjects are administered CMN-001. There is no survival advantage for subjects who received SOC and subsequent everolimus during either the treatment period or into the follow-up period of the study, OS of 17.8 months and 13.4 months, respectively (HR 0.96; 95% CI, 0.40–2.32) (). However, if subjects received CMN-001 were then given everolimus during the follow-up period of the study there was a clinical benefit compared to subjects receiving CMN-001 and everolimus in the treatment phase of the study. Overall survival increased from 19.3 months to 25.8 months (HR 0.60; 95% CI, 0.31–1.14) ().
Figure 3. Kaplan–Meier survival curves for subjects treated with everolimus in second line. (a) A comparison of survival curves between ADAPT subjects receiving everolimus in second-line therapy in the SOC arm during follow-up (blue line) (n = 20, 30% censored subjects) or in treatment (orange line) (n = 11, 27% censored subjects). Median OS was 17.8 months and 13.4 months for follow-up and treatment, respectively (HR 0.96, CI, 0.40–2.32). (b) A comparison of survival curves between ADAPT subjects receiving everolimus in second-line therapy in the combination arm, during follow-up (blue line) (n = 22, 45% censored) or in treatment (orange line) (n = 38, 31.5% censored). Median OS was 25.7 months and 19.3 months for follow-up and treatment, respectively (HR 0.60, 95% CI, 0.31–1.14). Statistical analysis was performed with GraphPad Prism v9.4 software and hazard ratios were calculated using log-rank test.
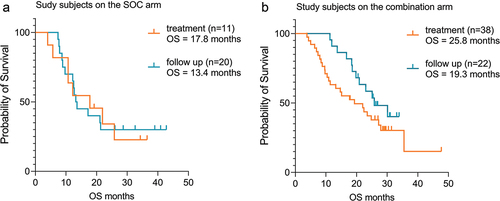
Table 2. Subjects treated with everolimus during the Phase 3 ADAPT study.
A further stratifying of subjects by the number of Heng risk factorsCitation38 revealed a stepwise improvement in clinical benefit for subjects with a greater number of risk factors. As was reported for all subjects with one or more risk factors, there was no clinical benefit between the arms with an HR of 1.00 (95% CI, 0.76–1.37).Citation16 However, if subjects were grouped by increasing number of risk factors a clinical benefit could be observed. In subjects with two or more risk factors the HR was 0.87 (95% CI, 0.63–1.18), with three or more risk factors the HR was 0.80 (95% CI, 0.48–1.33) and with four more risk factors the HR was 0.66 (95% CI, 0.26–1.69) (). This revealed a 33% lower incidence in death for subjects with four or more risk factors who have received CMN-001. Overall, those subjects with the greatest number of risk factors saw the lowest incidence of disease. The data suggests that poor-risk subjects may preferentially benefit from combination therapy of CMN-001 with everolimus due to reduced responses to SOC for this subject population.
Table 3. Hazard ratio between CMN-001 arm and SOC arm by risk factors.
Results of laboratory studies
Immunogenicity of CMN-001 in mRCC subjects
In addition to reporting clinical outcomes, the phase 2 and phase 3 studies included a detailed immune response monitoring platform to evaluate T cell responses after treatment with CMN-001 in combination therapy. The immune response monitoring platform employed multicolor flow cytometry to measure increases in the numbers of multifunctional memory CTL that produce the cytokines IFN-γ, TNF-α, and IL-2 with concurrent cytolytic function. In the phase 2b study 10 of 14 subjects had increases in multifunctional memory CTL post-CMN-001 treatment. Importantly, increases in absolute numbers of multifunctional memory CTL (post-treatment versus pre-treatment) demonstrated a statistically significant correlation with OS.Citation23 Furthermore, when the memory T cell response was measured in subjects receiving CMN-001 on the ADAPT phase 3 study, the induced increase of multifunctional memory CTL correlated with OS in subjects who received at least five doses of CMN-001.Citation16 Taken together, these data confirmed the mechanism of action of CMN-001 previously reported in preclinical studies to be operating in subjects receiving CMN-001.Citation21 Furthermore, we reported that the amount of IL-12 produced by CMN-001 product correlated with both the magnitude of the induced memory CTL response and clinical benefit after five and seven doses of CMN-001.Citation16
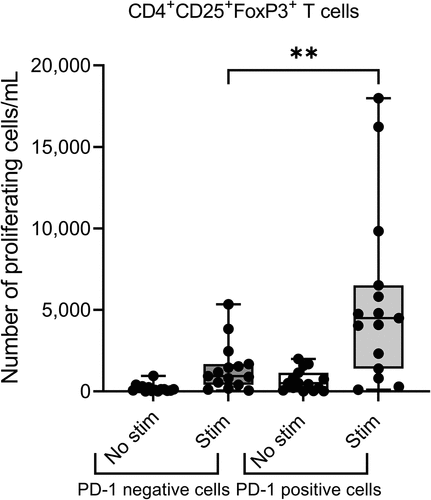
Moreover, a strong positive correlation between OS and CMN-001-treated subjects on the ADAPT study who had a greater percentage of T regulatory cells at baseline and at any subsequent time point measured, predicted better clinical outcomes. There was no correlation with OS and T regulatory cell percentages in the SOC group, at any time point analyzed. In fact, subjects not treated with CMN-001 who had high percentages of T regulatory cells performed worse.Citation16 This is in direct contrast with previously reported studies where higher percentages of T regulatory cells are considered a poor prognostic indicator for mRCC.Citation39 This posed an interesting question of why a higher frequency of T regulatory cells in the peripheral blood would be a biological indicator of increased efficacy with CMN-001 administration. Further analysis of the T regulatory population of cells in CMN-001-treated subjects revealed an interesting observation that may shed light on the underlying mechanism. When PBMC isolated from mRCC subjects were stimulated with CMN-001 in vitro, we identified increases in the numbers of proliferating CD4+CD25+FoxP3+ T cells upon stimulation with CMN-001. This population was distinct from non-proliferating CD4+CD25+FoxP3+ T regulatory cells based on the expression of PD-1. The proliferating CD4+CD25+FoxP3+ T cells were PD-1 positive (). Again, this contrasts with published work showing CD4+CD25+FoxP3+ T regulatory cells are PD-1 negative and proliferate poorly.Citation40 Our data suggest that the CMN-001 may be stimulating a population of early activated CD4 effector T cells as has been reported elsewhere,Citation40–42 and the detection of increased frequencies of CD4+CD25+Foxp3 + T cells in the peripheral blood may not be true T regulatory cells but represent a population of proliferating CD4+CD25+Foxp3+ effector T cells that can be stimulated in vivo by CMN-001 administration. However, it remains to be tested to see if these cells have true regulatory T cell activity.
Figure 4. CMN-001 expansion of proliferating CD4+CD25+FoxP3+ T cells in vitro. PBMC collected from mRCC subjects (n = 15) enrolled in the ADAPT study were co-cultured in vitro with autologous CMN-001 product (Stim) or left in culture (No stim) for 6 days, and the expansion of FoxP3+CD4+ T cells was determined. Intracellular staining detected FoxP3+ CD25+CD4+ T cells on the PD-1 positive and negative cell populations. The number of proliferating cells was determined by Ki67 expression for each population of cells. White bars are unstimulated cells, and gray bars are PBMCs stimulated with CMN-001. Statistical significance was determined by paired T-test ** p value < .05 using GraphPad Prism v9.4 software.
Everolimus modulation of TGF-β secretion by B cells
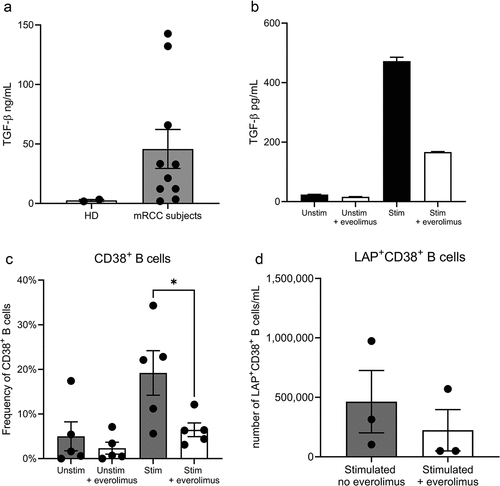
The unexpected clinical benefit of everolimus treatment in combination with CMN-001 could be due to modulating the immunosuppressive nature of mRCC. One potential immune suppressive target to modulate is the high concentration of TGF-β in the plasma of mRCC subjects.Citation43,Citation44 We measured the concentration of TGF-β in the plasma of a cohort of eight randomly selected ADAPT subjects. We detected high concentrations of TGF-β (ranging from 12 to 143 ng/mL) as compared to plasma collected from two individual healthy donors (1.9 and 3.4 ng/mL) (). One potential source of TGF-β is regulatory B cellsCitation45 whose production is regulated through the mTOR signaling pathway.Citation46 This prompted us to investigate the impact everolimus may have on B cell TFG-β production. PBMC from mRCC subjects was stimulated to activate B cells to secrete TGF-β. When B cells were stimulated in the presence of everolimus, there was a decrease in the concentration of TGF-β secreted by stimulated B cells (). Stimulation also resulted in the expansion of CD38+ B cells ranging from 5.6% to 34.3% of the CD19+ B cell population, which was reversed when stimulated B cells were treated with everolimus (). TGF-β exists as a latent form inside B cells complexed with the latency-associated peptide (LAP).Citation47 Within the B cell population, we could detect CD19+CD38+ B cells containing LAP, and treatment of stimulated B cells with everolimus resulted in a decrease in the numbers of CD19+CD38+LAP+ B cells (). Our ability to detect TGF-β in activated CD38+ B cells from mRCC subjects may reflect the regulatory state of B cells in mRCC subjects. TGF-β does not exist in an active form initially but must be cleaved from the latent protein for activation, suggesting a link between TGF-β in the plasma and CD19+CD38+LAP+ B cells as the source of TFG-β.Citation48
Figure 5. Everolimus reduces TGF-β secretion and the frequency of CD38+ B cells. (a) TGF-β was measured by ELISA in plasma collected from mRCC subjects (n = 8) (gray box) or healthy donors (HD n = 2) (white box). (b) PBMC from mRCC subjects were stimulated (Stim) with the combination of reagents described in the Material and Method section or left unstimulated (Unstim) in the presence of everolimus (+everolimus) to induce TGF-β secretion. TGF-β concentrations in the supernatant were measured after 6 days by ELISA. (c) The frequency of CD38+ B cells was determined by flow cytometry by gating on the viable CD19+ B cells in groups unstimulated or stimulated with everolimus (white bars) or without everolimus (gray bars). Data is from five individual mRCC subject PBMC samples. (d) the number of LAP+CD38+ B cells was determined by flow cytometry in cultures stimulated without everolimus (gray bar) or with everolimus (white bar). Data are representative of three independent experiments from four individual mRCC subject PBMC samples. Statistical significance was determined by paired T-test * p value < .05 using GraphPad Prism v9.4 software.
In vitro modeling of CMN-001 and lenvatinib plus everolimus
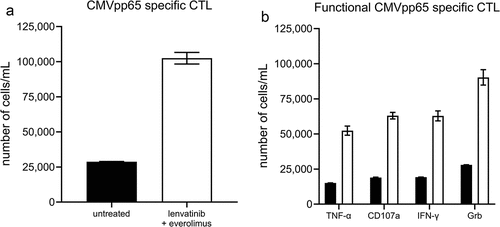
One outstanding question in selecting combination therapy is to understand the potential interactions between agents with different mechanisms of action. By targeting TKI and mTOR pathways with the combination of lenvatinib plus everolimus may impact the generation of T cell memory responses.Citation49,Citation50 The induction of memory T cells is the primary mechanism of action for CMN-001. To address this question, we assessed the combination of lenvatinib plus everolimus on the ability of CMN-001 to stimulate antigen-specific CTL in vitro. Healthy donors with known reactivity to the CMV antigen pp65 were selected to generate CMN-001 like DC expressing the CMVpp65 antigen. CMVpp65 expressing DC stimulated the expansion of functional recall CMV-specific CTL in cocultures of PBMC and DC expressing the CMVpp65 antigen in the presence of lenvatinib plus everolimus (). Furthermore, an increase in the number of functional CMV-specific memory CTL secreting IFN–γ, TNF–α, and expressing the lytic markers CD107a and Grb after restimulation with CMV antigen targets is shown in . This in vitro observation corroborates our immune monitoring data, which shows that we can detect memory CTL in subjects who received CMN-001 in combination with everolimus in the ADAPT study.Citation16 This observation agrees with another report showing that everolimus may be a positive regulator of antiviral memory T cells in mice and nonhuman primates treated with rapamycin.Citation51
Figure 6. Expansion of CMVpp65-specific CTL after coculture with CMVpp65 antigen-loaded DC and lenvatinib plus everolimus. (a) The number of CMVpp65-specific CTL after 10 days coculture with CMVpp65 antigen-loadedantigen loaded DC (untreated, black bar) or cocultured with the combination of lenvatinib plus everolimus (white bar) was determined by flow cytometry using Trucount tubes to report the number of CMVpp65-specific CTL cells/mL. (b) Day 10 CTL were then restimulated with CMVpp65 antigen-loaded DC for an additional 4 hrs to measure cytokine production and Grb expression without the addition of lenvatinib plus everolimus (black bars) or with lenvatinib plus everolimus (white bars). The number of functional CMVpp65-specific CTL expressing TNF-α, CD107a, IFN-γ, or Grb after 4-hr stimulation was measured by flow cytometry using Trucount tubes to report the number of cells/mL. The data shown are from one of four representative donors with similar results.
State of the art of DC therapy and future plans for CMN-001 deployment in mRCC
There are currently over 470 clinical trials either actively recruiting or completed employing DC as therapy for the treatment of many different cancers (as of April 17, 2023 clinicaltrial.gov). These DC-based therapies cover a wide range of methodologies to induce antitumor responses. The first trials using autologous DC pulsed with antigens from B cell lymphoma subjects was carried out over 30 years ago with some subjects seeing clinical benefit.Citation52 The loading of DCs can be achieved via ex vivo methods with recombinant tumor-associated antigens (TAAs), or transection with TAA encoding plasmids or by mRNA, or tumor DC fusions.Citation12 Earlier work has shown that DC can be loaded with total tumor RNA, having the advantage of allowing the DC to select the most likely immunodominant epitopes.Citation53 CMN-001 embodies this model of loading DC with tumor antigens in the form of RNA to allow the immunogenic tumor epitopes from the patient to be presented to the immune system. As the number of treatment options for cancer therapy keeps expanding, there is a need to choose appropriate combination therapies to provide the best opportunity for clinical benefit. DC is a long sought-after immunotherapy to induce cell-mediated antitumor responses. However, there is limited data on how to design clinical studies to leverage multiple mechanisms of action of different treatment combinations.
One approach is to leverage DC-based therapy in combination with other cancer therapies such as chemotherapy, TKI/mTOR inhibitors, and checkpoint inhibitors (CPI),Citation54 all SOC treatments for cancer. Recent trials in non-small cell lung cancer (NSCLC) and ovarian cancer have shown promising clinical results when DC loaded with tumor lysate are combined with chemotherapy.Citation55,Citation56 Other approaches have modified DC with RNA to deliver antigens of choice.Citation57 For example, DC loaded with RNA encoding WT1 antigen has been used to treat acute myeloid leukemia with improved OS rates and induced WT1-specific T cells.Citation58 DC loaded with Gp100 and tyrosinase RNA plus chemotherapy used to treat melanoma subjects showed antigen-specific immune responses but no improvement in clinical outcomes.Citation59 DC has also been used in the adjuvant setting to prevent relapse in high-risk prostate cancer subjects.Citation60 DC therapy has been combined with anti-CD40 agonistic antibodies for the treatment of pancreatic cancer.Citation61 Interestingly when DC loaded with non-mutated antigens derived from blood vessels surrounding the tumor were used in combination with chemotherapy objective clinical response were seen in 6 of 13 melanoma subjects.Citation62
More recently, DC therapy has been combined with CPI using DC loaded with tumor antigens encoded by RNA or tumor lysate leading to responses in melanoma subjects.Citation3,Citation6,Citation9,Citation63 Moreover, DC therapy has been employed to treat hepatocellular carcinomaCitation13 ovarian cancer and pancreatic cancer,Citation61,Citation64 with tumor lysate-loaded DC in combination with CPI.Citation65,Citation66 Peptide loading of DC with neoantigens plus anti-PD-1 therapy resulted in complete remission in a patient with metastatic gastric cancer.Citation67 These studies along with others in renal cell carcinoma have shown that there is a place for DC-based cell therapy in the era of checkpoint blockade.Citation68,Citation69
Design of the phase 2b randomized study of CMN-001 in combination therapy
Given our experience with CMN-001 in combination with SOC treatments, we have designed a new phase 2b study. The phase 2b study (CMN-001-1) was designed to affirm the observed synergy between CMN-001 and everolimus. The study is actively recruiting since its opening in July 2020 and plans to enroll 90 subjects (NCT04203901). The study design () includes three important protocol modifications based on retrospective observations from the ADAPT study. The first being the study will enroll primarily poor-risk subjects ≥ 3 risk factors with up to 50% intermediate-risk subjects (1–2 risk factors) with mRCC based on the Heng risk model.Citation70 The subjects must be eligible for nephrectomy or biopsy to provide a tumor specimen to generate tumor RNA. The study design is focused on leveraging a population of poor-risk patients who are in the most need of treatment for their disease. Median survival for poor-risk subjects with two or more risk factors is 7 monthsCitation71 and treatment options are limited, and current studies are confounded in this population due to small numbers of subjects. Therefore, CMN-001-1 provides an opportunity to combine and sequence therapies with different mechanisms of action in a population of patients that has few alternatives.
Figure 7. CMN-001-1 study schema. This study is a two-arm trial with the only difference between the arms being the administration of CMN-001 in the combination arm versus SOC alone (Control arm). The study will recruit primarily poor-risk subjects with up to 50% intermediate-risk subjects with metastatic RCC who are eligible for nephrectomy or biopsy (n = 90). Subjects will be randomized 1:1 either to the combination arm, CMN-001 plus SOC or the SOC alone arm. Subjects will receive CPI (checkpoint inhibition therapy) in first line and after first progression will proceed to second line with lenvatinib plus everolimus. First-line CPI therapy will be the combination of ipilimumab and nivolumab. Subjects on the combination arm will continue to receive CMN-001 through a second progression. Any subject not eligible for second-line therapy with lenvatinib plus everolimus will be replaced. The primary end point is OS with secondary end points for PFS, RR, and safety.
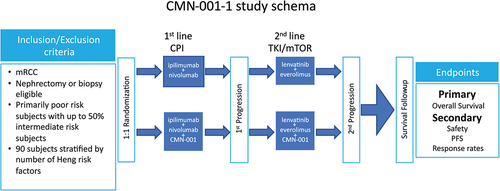
Second, the first-line therapy initiates with CPI therapy, ipilimumab plus nivolumab then moves to lenvatinib plus everolimus as second-line therapy post the first-line failure. Given the movement of CPI to first-line in mRCC, the new study incorporates CPI with CMN-001 during the induction phase of the study. This treatment option was not available during the ADAPT study. CPI was only adminstered as a second line option. By blocking the inhibitory pathway linked between PD-L1 on the tumor cell and PD-1 on activated T cells, CPI blockade may invoke a more robust memory T cell response primed by CMN-001. Ipilimumab plus nivolumab is administered at 3-week intervals for four administrations followed by nivolumab administration every 4 weeks until progression. CMN-001 dosing initiates during CPI therapy and is administered as one dose every 3 weeks for three doses (Induction phase), followed by maintenance doses, one dose every 4 weeks for seven doses (Maintenance phase), followed by booster doses, 1 dose every 12 weeks (Booster phase). Third, if discontinuation of first-line CPI therapy is indicated, either for intolerable toxicity or due to disease progression, second-line therapy can be initiated with lenvatinib plus everolimus and continue CMN-001 dosing in combination with lenvatinib plus everolimus. By fixing the second-line therapy during the maintenance phase and through progression, we eliminated any confounding results by the addition of other treatments. As subjects progress, they will continue to receive CMN-001 in combination with lenvatinib plus everolimus.
The primary objective is to compare OS in subjects treated with CMN-001 in combination to subjects treated with standard treatment alone, CPI in the first- and second-line Lenvatinib plus everolimus starting from the date of randomization. Secondary objectives include safety assessments, PFS, and tumor responses between the study arms. Anticipated adverse reactions to CMN-001 are administration site conditions (injection site events), rash, and fatigue. Injection site events are expected to resolve without sequelae. Manageable grade 3–4 immune-related adverse events associated with the ipilimumab/nivolumab combination were reported for the CheckMate 920 study.Citation72 Adverse events reported for the lenvatinib/everolimus combination were grade 3–4 toxicities, including hypertriglyceridemia, proteinuria, diarrhea, and fatigue.Citation73 Based on the safety profile for CMN-001, with few serious grade 3–5 adverse reactions and no discontinuations due to CMN-001, our expectation for the study is for little or no additional toxicity and a safety profile in accordance with that expected for the administration of CPI therapy and second-line lenvatinib plus everolimus.
Another aspect of the study design incorporates an immune monitoring platform to measure subjects’ immune responses to the product. We will continue to monitor increases in memory CTL after CMN-001 administration and how the magnitude of the induced immune response relates to clinical outcomes. Furthermore, we will explore the relationship between induced Foxp3+ CD4+ T cells and clinical outcomes. By tracking the frequency of T regulatory cells in the peripheral blood, we will affirm the observation that the frequency of T regulatory cells in subjects is a positive correlate for subjects treated with CMN-001.
The choice of first-line therapy with CPI is predicated on the evolving treatment paradigm for mRCC.Citation68 The use of CPI therapy is now considered first-line therapy with TKI and or mTOR being deployed in second line as reported in the CHECKMATE 214 study.Citation74 This design leverages the available treatment options for treating mRCC as it continues to evolve to include the combination of checkpoint inhibition as first-line therapy.Citation75 The rationale for relegating TKI/mTOR therapy to second-line post-CPI therapy was shown in two retrospective analyses of subjects receiving second-line TKI therapy post-first-line CPI. The first study reported a PFS of 16.6 and 4.0 months,Citation76 and the second study reported a PFS of 8 and 5 monthsCitation77 for intermediate- and poor-risk subjects, respectively. The ADAPT study allowed the use of CPI in second line but only as a single agent.Citation16 When CPI was deployed in second line after CMN-001 administration, no clinical benefit was detected. This new study design will allow the deployment of CMN-001 with CPI therapy in first line which was not possible in the ADAPT study. We hope to leverage check point inhibition to correct the immune suppressive tumor microenvironment allowing CMN-001 to induce a more robust antitumor immune response without the impact of the PD-1/PD-1 L axis of inhibition.
Selection of the combination of lenvatinib plus everolimus as second-line therapy in the ongoing phase 2 study is based on the following lines of evidence. The antitumor mechanism of action of everolimus is targeting the mTOR signaling pathway, which results in a block in protein synthesis and loss of tumor cell proliferation.Citation78 Addition of an mTOR inhibitor to combination therapy can leverage the dysregulation of the mTOR pathways in mRCC, particularly in poor-risk subjects.Citation79,Citation80 However, everolimus is not used in the first-line settingCitation35 but is approved for the treatment of mRCC following the failure of >1 or more prior TKI therapies based on results from the RECORD-1 registration study.Citation81 Furthermore, given the poor response rates with single-agent mTOR inhibitors, combination studies of lenvatinib plus everolimus showed an improvement in PFS of 14.6 over 5.5 months with everolimus alone leading to approval in the USA and Europe of the combination of lenvatinib plus everolimus for subjects with mRCC following one prior anti-angiogenic therapy.Citation82,Citation83
Another interest to us is to understand how an immune suppressant would provide clinical benefit to subjects receiving CMN-001, a DC therapy designed to induce memory T cell responses.Citation19,Citation22 Even though everolimus is an approved therapy for the treatment of mRCC,Citation73 it, along with other mTOR inhibitors, has traditionally been used as an immune suppressant to prolong organ transplant survival, particularly kidney graft survival.Citation84,Citation85 Our data may reveal a unique mechanism of action for everolimus in combination therapy based on the ability to modulate B cell secretion of TGF-β into the plasma of mRCC subjects. TGF-β secreted by B cells is a mechanism to maintain the function of regulatory T cells in models of transplant tolerance and in renal cell carcinoma playing a role in regulating T cell activation.Citation45,Citation86,Citation87 Moreover, TGF-β-induced regulatory T cells are efficient in suppressing autoimmune responses to self-antigens and disease progression.Citation88 TGF-β does not initially exist in an active form and must be cleaved from the latent protein for activation. The latent protein is produced by B cells, suggesting a link between TGF-β in the plasma and activated B cells as a mediator to deliver the TFG-β to T cells.Citation48 We have shown that activated CD38+ B cells present in the peripheral blood of mRCC subjects contain latent TGF-β. It has been reported that CD38+ B cells are a source of regulatory B cells which suppress autoimmune inflammatory responses and CD4 T cell differentiation.Citation89 Our ability to detect latent TGF-β in activated CD38+ B cells from mRCC subjects may reflect the regulatory state of B cells in mRCC subjects. Everolimus inhibits B cell proliferation and Ig secretion,Citation90 and mTOR inhibitors block antiviral B cell responses but increase numbers of memory CD4+ T cells differentiating to Th1 T cellsCitation91 promoting T cell memory if administered in low doses.Citation92 This mechanism may serve as a potential explanation of the enhanced clinical benefit seen in mRCC subjects who received CMN-001 and everolimus in second-line therapy. We propose a novel mechanism whereby T cell responses induced by CMN-001 administration are augmented by everolimus treatment, which reduces the frequency of LAP+/CD38+ B cells secreting TGF-β. As a result, the combination of CMN-001 and everolimus leads to expansion of DC induced memory T cells.
In summary, lessons learned from the ADAPT study are reflective of the changing landscape of therapies now available to treat mRCC in first and second line. The design of the ongoing follow-up randomized phase 2b study builds on the mechanism of action of CMN-001 and underlying correlates with clinical outcomes. By evaluating CMN-001 plus standard treatment for mRCC, we have an opportunity to combine and sequence therapies with different mechanisms of action. This new study design leverages three distinct mechanisms of action. First to allow CPI to block the suppressive tumor microenvironment, second CMN-001 to induce antigen-specific anti-tumor CTL, activation of CD4 T helper cells versus T regulatory cells, and third everolimus regulates systemic immune suppression allowing for the expansion of the induced T cell response. Furthermore, by focusing on poor-risk subjects, this provides an opportunity to evaluate clinical benefits for a patient population with limited treatment options. Moreover, by assessing the immunogenicity of CMN-001, we uncovered two potential mechanisms by which CMN-001 may alter the immunosuppressive environment of mRCC subjects. The first is the presence of CD4+ Foxp3+ T cells that may not be true regulatory T cells but are CD4 T helper cells activated as a consequence of CMN-001 administration. Second, the in vitro observation that everolimus may block TGF-b secretion by B cells. These two observations will be an area for future study in subjects receiving CMN-001 in combination therapy.
Author contributions
Joe Horvatinovich, Alicia Gamble, Marcus Norris, Mark DeBenedette, and Charles A. Nicolette designed research studies, analyzed data and contributed to writing the manuscript. Alicia Gamble, Joe Horvatinovich and Marcus Norris conducted experiments and acquired data.
Supplemental Material
Download PDF (202.7 KB)Acknowledgments
We would like to thank all the patients and their families and caregivers for their participation in the clinical studies. Additionally, we thank all the clinical study investigators for their contributions, administration, and execution of the listed clinical studies.
Disclosure statement
Mark DeBenedette, Alicia Gamble, Marcus Norris, Joe Horvatinovich, and Charles A. Nicolette are all full-time employees of CoImmune Inc. and have stock options in the company.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website at https://doi.org/10.1080/21645515.2023.2220629.
Additional information
Funding
References
- Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized dendritic cell vaccines-recent breakthroughs and encouraging clinical results. Front Immunol. 2019;10:766. doi:10.3389/fimmu.2019.00766.
- Gardner A, de Mingo Pulido Á, Ruffell B. Dendritic cells and their role in immunotherapy. Front Immunol. 2020;11:924. doi:10.3389/fimmu.2020.00924.
- Chick RC, Faries MB, Hale DF, Kemp Bohan PM, Hickerson AT, Vreeland TJ, Myers JW, Cindass JL, Brown TA, Hyngstrom J, et al. Multi-institutional, prospective, randomized, double-blind, placebo-controlled phase IIb trial of the tumor lysate, particle-loaded, dendritic cell (TLPLDC) vaccine to prevent recurrence in high-risk melanoma patients: a subgroup analysis. Cancer Med. 2021;10:4302–14. doi:10.1002/cam4.3969.
- Baldin AV, Savvateeva LV, Bazhin AV, Zamyatnin AA Jr. Dendritic cells in anticancer vaccination: rationale for ex vivo loading or in vivo targeting. Cancers. 2020;12:12. doi:10.3390/cancers12030590.
- Chung DJ, Sharma S, Rangesa M, DeWolf S, Elhanati Y, Perica K, Young JW. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. 2022;6:1547–58. doi:10.1182/bloodadvances.2021005941.
- De Keersmaecker B, Claerhout S, Carrasco J, Bar I, Corthals J, Wilgenhof S, Neyns B, Thielemans K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: link between T-cell activation and clinical responses in advanced melanoma. J ImmunoTher Cancer. 2020;8:e000329. doi:10.1136/jitc-2019-000329.
- Dörrie J, Schaft N, Schuler G, Schuler-Thurner B. Therapeutic cancer vaccination with ex vivo RNA-transfected dendritic cells-an update. Pharmaceutics. 2020;12:12. doi:10.3390/pharmaceutics12020092.
- Cozzi S, Najafi M, Gomar M, Ciammella P, Iotti C, Iaccarino C, Dominici M, Pavesi G, Chiavelli C, Kazemian A, et al. Delayed effect of dendritic cells vaccination on survival in glioblastoma: a systematic review and meta-analysis. Curr Oncol. 2022;29:881–91. doi:10.3390/curroncol29020075.
- Jansen Y, Kruse V, Corthals J, Schats K, van Dam PJ, Seremet T, Heirman C, Brochez L, Kockx M, Thielemans K, et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol Immunother. 2020;69:2589–98. doi:10.1007/s00262-020-02618-4.
- Filin IY, Kitaeva KV, Rutland CS, Rizvanov AA, Solovyeva VV, Solovyeva VV. Recent advances in experimental dendritic cell vaccines for cancer. Front Oncol. 2021;11:730824. doi:10.3389/fonc.2021.730824.
- Batich KA, Mitchell DA, Healy P, Herndon JE 2nd, Sampson JH. Once, twice, three times a finding: reproducibility of dendritic cell vaccine trials targeting cytomegalovirus in Glioblastoma. Clin Cancer Res. 2020;26:5297–303. doi:10.1158/1078-0432.CCR-20-1082.
- Laureano RS, Sprooten J, Vanmeerbeerk I, Borras DM, Govaerts J, Naulaerts S, Berneman ZN, Beuselinck B, Bol KF, Borst J, et al. Trial watch: dendritic cell (DC)-based immunotherapy for cancer. Oncoimmunology. 2022;11:2096363. doi:10.1080/2162402X.2022.2096363.
- Jeng LB, Liao LY, Shih FY, Teng CF. Dendritic-cell-vaccine-based immunotherapy for hepatocellular carcinoma: clinical trials and recent preclinical studies. Cancers. 2022;14:14. doi:10.3390/cancers14184380.
- Grenier JM, Yeung ST, Khanna KM. Combination immunotherapy: taking cancer vaccines to the next level. Front Immunol. 2018;9:610. doi:10.3389/fimmu.2018.00610.
- Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348:74–80. doi:10.1126/science.aaa6204.
- Figlin RA, Tannir NM, Uzzo RG, Tykodi SS, Chen DYT, Master V, Kapoor A, Vaena D, Lowrance W, Bratslavsky G, et al. Results of the ADAPT phase 3 study of rocapuldencel-T in combination with sunitinib as first-line therapy in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2020;26:2327–36. doi:10.1158/1078-0432.CCR-19-2427.
- Coates PT, Colvin BL, Hackstein H, Thomson AW. Manipulation of dendritic cells as an approach to improved outcomes in transplantation. Expert Rev Mol Med. 2002;4:1–21. doi:10.1017/S1462399402004283.
- Tcherepanova IY, Adams MD, Feng X, Hinohara A, Horvatinovich J, Calderhead D, Healey D, Nicolette CA. Ectopic expression of a truncated CD40L protein from synthetic post-transcriptionally capped RNA in dendritic cells induces high levels of IL-12 secretion. BMC Mol Biol. 2008;9:90. doi:10.1186/1471-2199-9-90.
- Calderhead DM, De Benedette MA, Ketteringham H, Gamble AH, Horvatinovich JM, Tcherepanova IY, Nicolette CA, Healey DG. Cytokine maturation followed by CD40L mRNA electroporation results in a clinically relevant dendritic cell product capable of inducing a potent proinflammatory CTL response. J Immunother. 2008;31:731–41. doi:10.1097/CJI.0b013e318183db02.
- Cesana GC, De Raffele G, Cohen S, Moroziewicz D, Mitcham J, Stoutenburg J, Cheung K, Hesdorffer C, Kim-Schulze S, Kaufman HL. Characterization of CD4+CD25+ regulatory T cells in patients treated with high-dose interleukin-2 for metastatic melanoma or renal cell carcinoma. J Clin Oncol. 2006;24:1169–77. doi:10.1200/JCO.2005.03.6830.
- De Benedette MA, Calderhead DM, Ketteringham H, Gamble AH, Horvatinovich JM, Tcherepanova IY, Nicolette CA, Healey DG. Priming of a novel subset of CD28+ rapidly expanding high-avidity effector memory CTL by post maturation electroporation-CD40L dendritic cells is IL-12 dependent. J Immunol. 2008;181:5296–305. doi:10.4049/jimmunol.181.8.5296.
- De Benedette MA, Calderhead DM, Tcherepanova IY, Nicolette CA, Healey DG. Potency of mature CD40L RNA electroporated dendritic cells correlates with IL-12 secretion by tracking multifunctional CD8(+)/CD28(+) cytotoxic T-cell responses in vitro. J Immunother. 2011;34:45–57. doi:10.1097/CJI.0b013e3181fb651a.
- Amin A, Dudek AZ, Logan TF, Lance RS, Holzbeierlein JM, Knox JJ, Master VA, Pal SK, Miller WH, Karsh LI, et al. Survival with AGS-003, an autologous dendritic cell-based immunotherapy, in combination with sunitinib in unfavorable risk patients with advanced renal cell carcinoma (RCC): phase 2 study results. J ImmunoTher Cancer. 2015;3:14. doi:10.1186/s40425-015-0055-3.
- Harris J, Monesmith T, Ubben A, Norris M, Freedman JH, Tcherepanova I. An improved RNA amplification procedure results in increased yield of autologous RNA transfected dendritic cell-based vaccine. Biochim Biophys Acta. 2005;1724:127–36. doi:10.1016/j.bbagen.2005.03.013.
- Slagter-Jager JG, Raney A, Lewis WE, De Benedette MA, Nicolette CA, Tcherepanova IY. Evaluation of RNA amplification methods to improve DC immunotherapy antigen presentation and immune response. Mol Ther Nucleic Acids. 2013;2:e91. doi:10.1038/mtna.2013.18.
- Tcherepanova I, Harris J, Starr A, Cleveland J, Ketteringham H, Calderhead D, Horvatinovich J, Healey D, Nicolette CA. Multiplex RT-PCR amplification of HIV genes to create a completely autologous DC-based immunotherapy for the treatment of HIV infection. PLos One. 2008;3:e1489. doi:10.1371/journal.pone.0001489.
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–24. doi:10.1056/NEJMoa065044.
- Escudier B. Sunitinib for the management of advanced renal cell carcinoma. Expert Rev Anticancer Ther. 2010;10:305–17. doi:10.1586/era.10.26.
- Escudier B. Sorafenib for the management of advanced renal cell carcinoma. Expert Rev Anticancer Ther. 2011;11:825–36. doi:10.1586/era.11.55.
- Ko JJ, Choueiri TK, Rini BI, Lee JL, Kroeger N, Srinivas S, Harshman LC, Knox JJ, Bjarnason GA, MacKenzie MJ, et al. First-, second-, third-line therapy for mRCC: benchmarks for trial design from the IMDC. Br J Cancer. 2014;110:1917–22. doi:10.1038/bjc.2014.25.
- Escudier B, Cosaert J, Pisa P. Bevacizumab: direct anti-VEGF therapy in renal cell carcinoma. Expert Rev Anticancer Ther. 2008;8:1545–57. doi:10.1586/14737140.8.10.1545.
- Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi:10.1056/NEJMoa066838.
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–56. doi:10.1016/S0140-6736(08)61039-9.
- Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–8. doi:10.1200/JCO.2009.23.9764.
- Lalani AA, McGregor BA, Albiges L, Choueiri TK, Motzer R, Powles T, Wood C, Bex A. Systemic treatment of metastatic clear cell renal cell carcinoma in 2018: current paradigms, use of immunotherapy, and future directions. Eur Urol. 2018;75:100–10. doi:10.1016/j.eururo.2018.10.010.
- Liau LM, Ashkan K, Brem S, Campian JL, Trusheim JE, Iwamoto FM, Tran DD, Ansstas G, Cobbs CS, Heth JA, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2023;9(1):112–21. doi:10.1001/jamaoncol.2022.5370.
- Master VA, Uzzo RG, Bratlavsky G, Karam JA. Autologous dendritic vaccine therapy in metastatic kidney cancer: the ADAPT trial and beyond. Eur Urol Focus. 2022;8:651–3. doi:10.1016/j.euf.2022.04.003.
- Heng DY, Xie W, Regan MM, Warren MA, Golshayan AR, Sahi C, Eigl BJ, Ruether JD, Cheng T, North S, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor–targeted agents: results from a large, multicenter study. J Clin Oncol. 2009;27:5794–9. doi:10.1200/JCO.2008.21.4809.
- Liotta F, Gacci M, Frosali F, Querci V, Vittori G, Lapini A, Santarlasci V, Serni S, Cosmi L, Maggi L, et al. Frequency of regulatory T cells in peripheral blood and in tumour-infiltrating lymphocytes correlates with poor prognosis in renal cell carcinoma. BJU Int. 2010;107:1500–6. doi:10.1111/j.1464-410X.2010.09555.x.
- Raimondi G, Shufesky WJ, Tokita D, Morelli AE, Thomson AW. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J Immunol. 2006;176:2808–16. doi:10.4049/jimmunol.176.5.2808.
- Asma G, Amal G, Raja M, Amine D, Mohammed C, Amel BA. Comparison of circulating and intratumoral regulatory T cells in patients with renal cell carcinoma. Tumour Biol. 2015;36:3727–34. doi:10.1007/s13277-014-3012-8.
- Li J, Ridgway W, Fathman CG, Tse HY, Shaw MK. High cell surface expression of CD4 allows distinction of CD4(+)CD25(+) antigen-specific effector T cells from CD4(+)CD25(+) regulatory T cells in murine experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;192:57–67. doi:10.1016/j.jneuroim.2007.09.004.
- Hegele A, Varga Z, von Knobloch R, Heidenreich A, Kropf J, Hofmann R. TGF-beta1 in patients with renal cell carcinoma. Urol Res. 2002;30:126–9. doi:10.1007/s00240-002-0245-6.
- Junker U, Haufe CC, Nuske K, Rebstock K, Steiner T, Wunderlich H, Junker K, Reinhold D. Elevated plasma TGF-β1 in renal diseases: cause or consequence? Cytokine. 2000;12:1084–91. doi:10.1006/cyto.1999.0645.
- Dedobbeleer O, Stockis J, van der Woning B, Coulie PG, Lucas S. Cutting edge: active TGF-beta1 released from GARP/TGF-beta1 complexes on the surface of stimulated human B lymphocytes increases class-switch recombination and production of IgA. J Immunol. 2017;199:391–6. doi:10.4049/jimmunol.1601882.
- Song J, Xiao L, Du G, Gao Y, Chen W, Yang S, Fan W, Shi B. The role of regulatory B cells (Bregs) in the Tregs-amplifying effect of Sirolimus. Int Immunopharmacol. 2016;38:90–6. doi:10.1016/j.intimp.2016.05.014.
- Taylor AW. Review of the activation of TGF-beta in immunity. J Leukoc Biol. 2009;85:29–33. doi:10.1189/jlb.0708415.
- Nouël A, Pochard P, Simon Q, Ségalen I, Le Meur Y, Pers JO, Hillion S. B-Cells induce regulatory T cells through TGF-β/IDO production in a CTLA-4 dependent manner. J Autoimmun. 2015;59:53–60. doi:10.1016/j.jaut.2015.02.004.
- Jin N, Malcherek G, Mani J, Zurleit R, Schmitt A, Chen B, Freund M, Ho AD, Schmitt M. Suppression of cytomegalovirus-specific CD8(+)T cells by everolimus. Leuk Lymphoma. 2014;55:1144–50. doi:10.3109/10428194.2013.822496.
- Zeng H, Chi H. mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol. 2017;46:103–11. doi:10.1016/j.coi.2017.04.005.
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–12. doi:10.1038/nature08155.
- Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–8. doi:10.1038/nm0196-52.
- Gilboa E, Vieweg J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol Rev. 2004;199:251–63. doi:10.1111/j.0105-2896.2004.00139.x.
- Cibula D, Rob L, Mallmann P, Knapp P, Klat J, Chovanec J, Minar L, Melichar B, Hein A, Kieszko D, et al. Dendritic cell-based immunotherapy (DCVAC/OvCa) combined with second-line chemotherapy in platinum-sensitive ovarian cancer (SOV02): a randomized, open-label, phase 2 trial. Gynecol Oncol. 2021;162:652–60. doi:10.1016/j.ygyno.2021.07.003.
- Zhong R, Ling X, Cao S, Xu J, Zhang B, Zhang X, Wang H, Han B, Zhong H. Safety and efficacy of dendritic cell-based immunotherapy (DCVAC/LuCa) combined with carboplatin/pemetrexed for patients with advanced non-squamous non-small-cell lung cancer without oncogenic drivers. ESMO Open. 2022;7:100334. doi:10.1016/j.esmoop.2021.100334.
- Rob L, Cibula D, Knapp P, Mallmann P, Klat J, Minar L, Bartos P, Chovanec J, Valha P, Pluta M, et al. Safety and efficacy of dendritic cell-based immunotherapy DCVAC/OvCa added to first-line chemotherapy (carboplatin plus paclitaxel) for epithelial ovarian cancer: a phase 2, open-label, multicenter, randomized trial. J ImmunoTher Cancer. 2022;10. doi:10.1136/jitc-2021-003190.
- Willemen Y, Versteven M, Peeters M, Berneman ZN, Smits ELJ. Ribonucleic acid engineering of dendritic cells for therapeutic vaccination: ready ‘N able to improve clinical outcome? Cancers. 2020;12:12. doi:10.3390/cancers12020299.
- Anguille S, Van de Velde AL, Smits EL, Van Tendeloo VF, Juliusson G, Cools N, Nijs G, Stein B, Lion E, Van Driessche A, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. 2017;130:1713–21. doi:10.1182/blood-2017-04-780155.
- Boudewijns S, Bloemendal M, de Haas N, Westdorp H, Bol KF, Schreibelt G, Aarntzen EHJG, Lesterhuis WJ, Gorris MAJ, Croockewit A, et al. Autologous monocyte-derived DC vaccination combined with cisplatin in stage III and IV melanoma patients: a prospective, randomized phase 2 trial. Cancer Immunol Immunother. 2020;69:477–88. doi:10.1007/s00262-019-02466-x.
- Tryggestad AMA, Axcrona K, Axcrona U, Bigalke I, Brennhovd B, Inderberg EM, Hønnåshagen TK, Skoge LJ, Solum G, Saebøe-Larssen S, et al. Long-term first-in-man phase I/II study of an adjuvant dendritic cell vaccine in patients with high-risk prostate cancer after radical prostatectomy. Prostate. 2022;82:245–53. doi:10.1002/pros.24267.
- Lau SP, van ‘t Land FR, van der Burg SH, Homs MYV, Lolkema MP, Aerts J, van Eijck CHJ. Safety and tumour-specific immunological responses of combined dendritic cell vaccination and anti-CD40 agonistic antibody treatment for patients with metastatic pancreatic cancer: protocol for a phase I, open-label, single-arm, dose-escalation study (REACtiVe-2 trial). BMJ Open. 2022;12:e060431. doi:10.1136/bmjopen-2021-060431.
- Storkus WJ, Maurer D, Lin Y, Ding F, Bose A, Lowe D, Rose A, DeMark M, Karapetyan L, Taylor JL, et al. Dendritic cell vaccines targeting tumor blood vessel antigens in combination with dasatinib induce therapeutic immune responses in patients with checkpoint-refractory advanced melanoma. J ImmunoTher Cancer. 2021;9:e003675. doi:10.1136/jitc-2021-003675.
- Adams AM, Carpenter EL, Clifton GT, Vreeland TJ, Chick RC, O’Shea AE, McCarthy PM, Kemp Bohan PM, Hickerson AT, Valdera FA, et al. Divergent clinical outcomes in a phase 2B trial of the TLPLDC vaccine in preventing melanoma recurrence and the impact of dendritic cell collection methodology: a randomized clinical trial. Cancer Immunol Immunother. 2022;72:1–9. doi:10.1007/s00262-022-03272-8.
- Lau SP, Klaase L, Vink M, Dumas J, Bezemer K, van Krimpen A, van der Breggen R, Wismans LV, Doukas M, de Koning W, et al. Autologous dendritic cells pulsed with allogeneic tumour cell lysate induce tumour-reactive T-cell responses in patients with pancreatic cancer: a phase I study. Eur J Cancer. 2022;169:20–31. doi:10.1016/j.ejca.2022.03.015.
- Caro AA, Deschoemaeker S, Allonsius L, Coosemans A, Laoui D. Dendritic cell vaccines: a promising approach in the fight against ovarian cancer. Cancers. 2022;14:14. doi:10.3390/cancers14164037.
- Fucikova J, Hensler M, Kasikova L, Lanickova T, Pasulka J, Rakova J, Drozenova J, Fredriksen T, Hraska M, Hrnciarova T, et al. An autologous dendritic cell vaccine promotes anticancer immunity in patients with ovarian cancer with low mutational burden and cold tumors. Clin Cancer Res. 2022;28:3053–65. doi:10.1158/1078-0432.CCR-21-4413.
- Guo Z, Yuan Y, Chen C, Lin J, Ma Q, Liu G, Gao Y, Huang Y, Chen L, Chen L-Z, et al. Durable complete response to neoantigen-loaded dendritic-cell vaccine following anti-PD-1 therapy in metastatic gastric cancer. NPJ Precis Oncol. 2022;6:34. doi:10.1038/s41698-022-00279-3.
- Popovic M, Matovina-Brko G, Jovic M, Popovic LS. Immunotherapy: a new standard in the treatment of metastatic clear cell renal cell carcinoma. World J Clin Oncol. 2022;13:28–38. doi:10.5306/wjco.v13.i1.28.
- George DJ, Lee CH, Heng D. New approaches to first-line treatment of advanced renal cell carcinoma. Ther Adv Med Oncol. 2021;13:17588359211034708. doi:10.1177/17588359211034708.
- Heng DY, Xie W, Regan MM, Harshman LC, Bjarnason GA, Vaishampayan UN, Mackenzie M, Wood L, Donskov F, Tan M-H, et al. External validation and comparison with other models of the international metastatic renal-cell carcinoma database consortium prognostic model: a population-based study. Lancet Oncol. 2013;14:141–8. doi:10.1016/S1470-2045(12)70559-4.
- Mekhail TM, Abou-Jawde RM, Boumerhi G, Malhi S, Wood L, Elson P, Bukowski R. Validation and extension of the memorial sloan-kettering prognostic factors model for survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol. 2005;23:832–41. doi:10.1200/JCO.2005.05.179.
- George DJ, Spigel DR, Gordan LN, Kochuparambil ST, Molina AM, Yorio J, Rezazadeh Kalebasty A, McKean H, Tchekmedyian N, Tykodi SS, et al. Safety and efficacy of first-line nivolumab plus ipilimumab alternating with nivolumab monotherapy in patients with advanced renal cell carcinoma: the non-randomised, open-label, phase IIIb/IV CheckMate 920 trial. BMJ Open. 2022;12:e058396. doi:10.1136/bmjopen-2021-058396.
- Buti S, Leonetti A, Dallatomasina A, Bersanelli M. Everolimus in the management of metastatic renal cell carcinoma: an evidence-based review of its place in therapy. Core Evid. 2016;11:23–36. doi:10.2147/CE.S98687.
- U.S. Food and Drug Administration. FDA approves nivolumab plus ipilimumab combination for intermediate or poor-risk advanced renal cell carcinoma. 2016, Apr 16
- Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, Plimack ER, Barthélémy P, Porta C, George S, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. 2018;378:1277–90. doi:10.1056/NEJMoa1712126.
- Shah AY, Kotecha RR, Lemke EA, Chandramohan A, Chaim JL, Msaouel P, Xiao L, Gao J, Campbell MT, Zurita AJ, et al. Outcomes of patients with metastatic clear-cell renal cell carcinoma treated with second-line VEGFR-TKI after first-line immune checkpoint inhibitors. Eur J Cancer. 2019;114:67–75. doi:10.1016/j.ejca.2019.04.003.
- Auvray M, Auclin E, Barthelemy P, Bono P, Kellokumpu-Lehtinen P, Gross-Goupil M, De Velasco G, Powles T, Mouillet G, Vano Y-A, et al. Second-line targeted therapies after nivolumab-ipilimumab failure in metastatic renal cell carcinoma. Eur J Cancer. 2019;108:33–40. doi:10.1016/j.ejca.2018.11.031.
- Saran U, Foti M, Dufour JF. Cellular and molecular effects of the mTOR inhibitor everolimus. Clin Sci (Lond). 2015;129:895–914. doi:10.1042/CS20150149.
- Pantuck AJ, Seligson DB, Klatte T, Yu H, Leppert JT, Moore L, O’Toole T, Gibbons J, Belldegrun AS, Figlin RA. Prognostic relevance of the mTOR pathway in renal cell carcinoma: implications for molecular patient selection for targeted therapy. Cancer. 2007;109:2257–67. doi:10.1002/cncr.22677.
- Porta C, Tortora G, Larkin JM, Hutson TE. Management of poor-risk metastatic renal cell carcinoma: current approaches, the role of temsirolimus and future directions. Future Oncol. 2016;12:533–49. doi:10.2217/fon.15.313.
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116:4256–65. doi:10.1002/cncr.25219.
- Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, Mellado B, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16:1473–82. doi:10.1016/S1470-2045(15)00290-9.
- Inman s. FDA grants priority review to lenvatinib/everolimus combo for RCC. 2016, Jan 18.
- Viana SD, Reis F, Alves R. Therapeutic use of mTOR inhibitors in renal diseases: advances, drawbacks, and challenges. Oxid Med Cell Longev. 2018;2018:3693625. doi:10.1155/2018/3693625.
- Ma MKM, Yung S, Chan TM. mTOR inhibition and kidney diseases. Transplantation. 2018;102:S32–s40. doi:10.1097/TP.0000000000001729.
- Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, Fitzgerald L, Shi S, Akrawi E, Lei J, et al. TGF-β-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol. 2014;44:1728–36. doi:10.1002/eji.201344062.
- Kim CS, Kim Y, Kwon T, Yoon JH, Kim KH, You D, Hong JH, Ahn H, Jeong IG. Regulatory T cells and TGF-β1 in clinically localized renal cell carcinoma: comparison with age-matched healthy controls. Urol Oncol. 2015;33:113 e19–25. doi:10.1016/j.urolonc.2014.11.004.
- Nguyen TL, Makhlouf NT, Anthony BA, Teague RM, Di Paolo RJ. In vitro induced regulatory T cells are unique from endogenous regulatory T cells and effective at suppressing late stages of ongoing autoimmunity. PLoS One. 2014;9:e104698. doi:10.1371/journal.pone.0104698.
- Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C. Cd19(+)cd24(hi)cd38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;32:129–40. doi:10.1016/j.immuni.2009.11.009.
- Matz M, Lehnert M, Lorkowski C, Fabritius K, Unterwalder N, Doueiri S, Weber UA, Mashreghi M-F, Neumayer H-H, Budde K. Effects of sotrastaurin, mycophenolic acid and everolimus on human B-lymphocyte function and activation. Transpl Int. 2012;25:1106–16. doi:10.1111/j.1432-2277.2012.01537.x.
- Ye L, Lee J, Xu L, Mohammed AU, Li W, Hale JS, Tan WG, Wu T, Davis CW, Ahmed R, et al. mTOR promotes antiviral humoral immunity by differentially regulating CD4 helper T cell and B cell responses. J Virol. 2017;91:91. doi:10.1128/JVI.01653-16.
- Gammon JM, Gosselin EA, Tostanoski LH, Chiu YC, Zeng X, Zeng Q, Jewell CM. Low-dose controlled release of mTOR inhibitors maintains T cell plasticity and promotes central memory T cells. J Control Release. 2017;263:151–61. doi:10.1016/j.jconrel.2017.02.034.