
ABSTRACT
Cryptococcus spp. has a polysaccharide capsule composed of glucuronoxylomannan-GXM, a major virulence factor that can prevent the recognition of fungi by immune cells. Chimeric Antigen Receptor (CAR) redirects T cells to target Cryptococcus spp. as previously demonstrated by a CAR specific to GXM, GXMR-CAR. The current study evaluated the strength of the signal transduction triggered by GXMR-CAR, composed of a distinct antigen-binding domain sourced from a single-chain variable fragment (scFv). GXM-specific scFv derived from mAbs 2H1 and 18B7, 2H1-GXMR-CAR and 18B7-GXMR-CAR, respectively, were designed to express CD8 molecule as hinge/transmembrane, and the costimulatory molecule CD137 (4-1BB) coupled to CD3ζ. The 2H1-GXMR-CAR or 18B7-GXMR-CAR Jurkat cells recognized soluble GXM from C. gattii and C. neoformans, and the levels of IL-2 released by the modified cells did not differ between the GXMR-CAR constructs after exposure to Cryptococcus spp. 18B7-GXMR-CAR triggered tonic signaling was more pronounced in modified Jurkat cells, and a protein kinase inhibitor of the Src family (dasatinib) significantly reduced GXMR-CAR tonic signaling and inhibited cell activation against ligands. 18B7 scFv showed a structural modification of the variable heavy (VH) chain that clarified the difference in the strength of tonic signaling and the level of cell activation between 2H1-GXMR-CAR and 18B7-GXMR-CAR. GXMR-CAR constructs induced T-cell activation against clinical isolates of Cryptococcus spp. and serum from patients with cryptococcosis induced high levels of IL-2, mainly in cells modified with 18B7-GXMR-CAR. Thus, 18B7-GXMR-CAR and 2H1-GXMR-CAR mediated T cell activation against Cryptococcus spp. and 18B7 and 2H1 scFv influenced the strength of tonic signaling.
HIGHLIGHTS
2H1-GXMR-CAR and 18B7-GXMR-CAR are efficiently expressed on the cell surface;
2H1-GXMR-CAR and 18B7-GXMR-CAR redirected T cells toward the ligands;
18B7-GXMR-CAR provided highest levels of tonic signaling;
Binding pocket of 18B7 scFv favored the tonic signaling triggered by GXMR-CAR;
Binding pocket of 18B7 scFv favored the tonic signaling triggered by GXMR-CAR;
Graphical Abstract
Introduction
The infection caused by Cryptococcus spp. is responsible for cryptococcosis that initiates after inhaling propagules, spores, or dehydrated yeasts that reach the pulmonary alveolus, establishing Cryptococcus spp. infection [Citation1,Citation2] Cryptococcus gattii and Cryptococcus neoformans are clinically relevant because of their ability to cause disease in humans [Citation3,Citation4]C. gattii can infect healthy individuals, 3whereas C. neoformans is the primary causative agent of fungal meningitis in individuals affected by the human immunodeficiency virus (HIV) [Citation5]. The mortality rate attributed to cryptococcosis is approximately 625,000 deaths annually [Citation5]. Cryptococcus spp. modulate different signaling pathways to adapt to the host immune response. Regulation of the expression of virulence factors is the primary mechanism of Cryptococcus spp. uses to impair the cytotoxic effect triggered by innate and adaptive immune cells [Citation6]. Among the virulence factors of Cryptococcus spp. is highly relevant the role of polysaccharide capsule localized around the fungal cell wall [Citation7], and this virulence factor has a structure that is highly hydrophilic and is composed mainly of glucuronoxylomannan (GXM), which represents 90% of the total capsular mass [Citation8]. GXM has immunomodulatory properties, such as inhibition of T lymphocyte activation, direct interference with macrophage proliferation, and induction of T cell and macrophage apoptosis [Citation7,Citation9–11]. CD4+ and CD8+ T cells play a crucial role in controlling cryptococcosis when T helper (Th) profiles of type 1, Th17, T cytotoxic (Tc) type 1, and Tc17 are differentiated [Citation12,Citation13]. However, pathogen-associated molecular patterns (PAMPs) such as GXM, chitin, and β-glucans sourced from Cryptococcus spp. compromise the antigen processing step involved in developing a specific adaptive immune response. Moreover, the impairment of the maturation and activation of dendritic cells caused by the virulence factors of Cryptococcus spp. contributes to the differentiation of the Th2-cell subset that favors the polarization of M2 macrophages, and this microenvironment cannot control Cryptococcus infection [Citation1].
Current treatments for cryptococcosis are limited and focus on three main classes of drugs, azoles, polyenes, and pyrimidine analogs, which promote strong adverse side effects and induce antifungal resistance [Citation14]. Cryptococcus spp. exhibit genomic plasticity and physiological adaptability, which are responsible for generating resistance to antifungal drugs available for the treatment, resulting in low therapeutic efficiency [Citation14]. Immunotherapeutic approaches have been used to treat cryptococcosis, and the application of chimeric antigen receptor (CAR) technology to redirect immune cells to target Cryptococcus spp. has advanced consistently. A previous study generated a CAR specific to Cryptococcus spp. that presented an antigen-recognition domain composed of a single-chain variable fragment (scFv) from an anti-GXM monoclonal antibody (mAb), clone 18B7, enabling the generation of a GXMR-CAR receptor targeting Cryptococcus spp [Citation15]. Genetically modified T-cells expressing GXMR-CAR were used to treat NSG mice infected with C. neoformans, reducing the percentage of Titan cells from C. neoformans in the lung tissue [Citation15]. The first construct of GXMR-CAR was composed of an IgG4 portion as a hinge domain, followed by CD28 as a transmembrane domain and a costimulatory portion coupled with CD3z as the activation domain [Citation15]. The optimization of GXMR-CAR to improve cell activation was initiated by modifying its hinge/transmembrane and signal transduction domains, as reported by Dos Santos et al. [Citation16], which resulted in a combination of CD8 (hinge/transmembrane region) and 4-1BB (costimulatory domain) molecules. These modifications in GXMR-CAR favored a higher expression of CAR on the T cell surface and triggered more robust signal transduction in response to the ligands [Citation16].
In addition, the antigen recognition domain can affect the strength of cell activation induced by the CAR, which contains the same target [Citation17]. Previous studies that evaluated the improvement in signal transduction triggered by GXMR-CAR considered an antigen recognition domain sourced from an anti-GXM monoclonal antibody from hybridoma 18B7 [Citation18]. Clone 18B7 has high affinity and specificity for GXM, and off-target effects on human cells or tissues obtained from healthy donors were not detected [Citation19]. Another anti-GXM monoclonal antibody from hybridoma 2H1, which presents a high similarity to the amino acid sequences of the variable chain of clone 18B7 [Citation20] and the anti-GXM clone 2H1 was the first monoclonal antibody specific for GXM developed and also favored a significant improvement in the opsonization of Cryptococcus spp [Citation21]. Previous studies have revealed that anti-GXM monoclonal antibodies from clones 18B7 and 2H1 have a high identity in the variable region, considering that only one amino acid differed in the complementarity-determining regions (CDR) of the light chain and nine amino acids were distinct in the CDR of the heavy chain. These variations between clones 18B7 and 2H1 resulted in somatic mutations and genetic rearrangements in their variable regions, which explains the higher affinity of clone 18B7 for GXM than for mAb 2H1 [Citation20].
Here, we report a comparison between two GXMR-CAR constructs, 2H1-GXMR-CAR and 18B7-GXMR-CAR, which differ in the antigen recognition domain derived from the variable portion of the GXM-specific monoclonal antibody clones 18B7 and 2H1. Then, the main goals of the current study were focused in the evaluation of the impact of different scFv in the expression rate, functionality, and strength of transduction signaling between GXMR-CAR constructs in the presence or absence of the ligands [Citation18]. We hypothesized that the biological activity triggered by GXMR-CAR in the presence or absence of ligands could be affected by antigen-binding domain. Our results demonstrated that T cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR showed differences in the recognition of soluble GXM obtained from C. gattii, and that the GXMR-CAR constructs were differentially expressed over time during cellular expansion. Moreover, the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs exhibited distinct tonic signaling strengths. Interestingly, tonic signaling and antigen recognition promoted by 2H1-GXMR-CAR and 18B7-GXMR-CAR were abolished when GXMR-CAR expressed only the scFv-specific heavy chain of GXM. In addition, the comparison between 2H1-GXMR-CAR and 18B7-GXMR-CAR-CAR regarding cellular activation against Cryptococcus spp. did not show any significant differences.
Materials and methods
Cell lines and cultivation of Cryptococcus spp
HEK (human embryonic kidney-293T) and Jurkat (clone E6–1) cell lines were purchased from the Rio de Janeiro Cell Bank (BCRJ). HEK-293T cells were cultured in DMEM High Glucose (Dulbecco’s Modified Eagle Medium) (Cat. No. 10–017-CM; Corning, USA) supplemented with 10% fetal bovine serum (Cat. No. 16000044; Gibco, Waltham, MA, USA), 1% ampicillin/streptomycin (Cat. No. P4333; Sigma- Aldrich, MO, USA), and 100 mM sodium pyruvate (Cat. No. 11360039; Gibco, Waltham, MA, USA). Jurkat cells were cultured in RPMI medium (Roswell Park Memorial Institute) (Cat. No. 11875093; Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum, 1% ampicillin/streptomycin, and 100 mM sodium pyruvate. HEK-293T and Jurkat cell lines were maintained within a humidified 5% CO2 atmosphere at 37°C.
Cryptococcus neoformans H99 (serotype A) and Cryptococcus gattii R265 (molecular genotype VGII) strains stocked at −80°C were thawed and grown in Sabouraud dextrose medium (Cat. No. K25–1205; KASVI) at 30°C with vigorous shaking (150 rpm) for 20 h. The cell suspension was washed twice with phosphate-buffered saline (PBS; Cat. No. 10010023; Gibco, Waltham, MA, USA)) by centrifugation (3500 × g, 10 min, RT) and resuspended in PBS. C. neoformans and C. gattii were heat-killed (70°C for 60 min), washed with PBS (3500 × g, 10 min, RT), and resuspended in PBS to determine cell concentration using a Neubauer chamber. Clinical isolates of C. gattii (455, 478, and 474) and C. neoformans (523,525,521) were kindly provided by Prof. Dr. Roberto Martinez, Ribeirão Preto Medical School, University of São Paulo, Brazil. Clinical isolates of C. neoformans (455 and 478) and C. gattii (521, 523, and 525) were obtained from the cerebrospinal fluid (CSF), and clinical isolates of C. neoformans (474) were obtained from bronchoalveolar lavage (BAL). These clinical isolates were obtained from samples used for diagnostic that were availabled to make a library of clinical isolates of Cryptococcus spp., and the Committee of Ethics of the School Hospital of the Ribeirão Preto Medical School at the University of São Paulo (protocol no. 12247/2010 approved) allowed to work with clinical isolates for research without an informed consent.
Design of GXMR-CAR constructs to target Cryptococcus spp
The GXMR-CAR constructs specific to the GXM of Cryptococcus spp. were synthesized using GenScript (GenScript, NJ, USA), and the sequence encoding GXMR-CAR was subcloned into a second-generation lentiviral plasmid backbone (pLentiCas9-EGFP, GenScript, NJ, USA) containing the GFP sequence as a reporter. GXMR-CAR constructs contain a variable region sequence sourced from the monoclonal antibody anti-GXM, 18B7 [Citation19] and 2H1 20clones responsible for the recognition of GXM. The single chain variable fragment (scFv) specific to GXM was linked to the hinge and transmembrane domains composed of the CD8a molecule (UniProt P01732, 136–206 aa position). The signal transduction domain contains the cytoplasmic portion of the human 4-1BB costimulatory molecule (UniProt Q07011, amino acids 214–255), followed by the intracellular portion of the human CD3ζ molecule (UniProt P20963, amino acids 52–164). The GXMR-CAR constructs were designated 18B7-GXMR-CAR and 2H1-GXMR-CAR. The plasmid backbone containing the GFP sequence alone was a control lentiviral (lenti-mock) vector.
The two GXMR-CAR mutants contained a variable region sequence encoding only the variable heavy (VH) chain of the monoclonal antibody anti-GXM, 18B7, and 2H1 clones, followed by hinge/transmembrane and intracellular domains. The GXMR-CAR mutants were designated 18B7-VH-GXMR-CAR and 2H1-VH-GXMR-CAR.
Production of lentiviral particles containing a GXMR-CAR-encoding sequence
The helper plasmid, pMD2.G (Addgene, Cat. 12259) and psPAX2 (Addgene; cat. 12260) encoding VSV-G envelope proteins and structural and regulatory HIV-1 proteins, respectively, were used to produce lentiviral particles. These helper plasmids were combined with lentiviral vectors encoding 2H1-GXMR-CAR or 18B7-GXMR-CAR, which were co-transfected into HEK-293T cells using Lipofectamine 3000 according to the manufacturer’s instructions (Thermo Fischer Scientific; Cat. No. L3000008). HEK-293T cells were transfected with 1 μg of pMD2.G, 1.5 μg of psPAX2, and 2.5 μg of GXMR-CAR plasmid. HEK-293T cells were cultured in DMEM supplemented with 10% FBS and cultivated in 25-T flasks at 37°C, and once 90–95% confluence was reached, the transfection protocol was performed. The cell culture supernatant containing the lentiviral particles was collected 24, 48, and 72 h post-transfection, and the fresh medium was replaced each time. The cell culture supernatant was centrifuged (300× g, 10 minutes, 4°C), and the supernatant was incubated overnight with Lenti X concentratorTM reagent (Takarabio; Cat. No. 631232), according to the manufacturer’s instructions, to precipitate the lentiviral particles that were resuspended in 1 mL of PBS and aliquots were stored at −80°C.
Determination of titer of the lentiviral particles and transduction of jurkat cell line
Jurkat cells were seeded at a concentration of 4 × 10 [Citation5] cells/mL (final volume of 250 μL) in a 48-well plate, and serial dilutions of lentiviral particles were performed before incubation with Jurkat cells. RPMI supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and one mM sodium pyruvate was used for the cell culture. The cells incubated with lentiviral particles were submitted to the spinoculation method (850 × g, 22°C, 65 min) for transduction, and subsequently, the cells were maintained within a humidified 5% CO2 atmosphere at 37°C. After 72 h of incubation, the levels of the GFP expression were measured by flow cytometry (Guava EasyCyte, Guava Technologies, Millipore, Burlington, MA, USA), and the lentiviral titer was determined using the formula: Transduction unit (TU)/mL = (C × %GFP × D)/V; ‘C’ represents the number of cells seeded for each well, ‘%GFP’ expressed in decimal, ‘D’ represents the dilution of lentiviral particles, and ‘V’ represents the final volume mL of cell culture. The formula should consider the percentage of GFP between 10% and 40%.
The spinoculation protocol (850× g, 65 min, 22°C) cited above was used for the transduction of Jurkat cells, and multiplicity of infection (MOI) of 1 or 3 lentiviral particles of 18B7-GXMR-CAR, 2H1-GXMR-CAR, 18B7-VH-GXMR-CAR, 2H1-VH-GXMR-CAR, and lenti-mock were considered. Cells (1 × 10 [Citation5] cells/well) were seeded in 48-well plates and incubated with lentiviral particles before spinoculation. The transduction efficiency based on GFP expression was measured using flow cytometry at 72 h post-transduction. The cells were either used for the experiments or maintained in culture.
Recognition of soluble GXM by 2H1-GXMR-CAR and 18B7-GXMR-CAR localized on the cell surface
The interaction between the GXMR-CAR constructs expressed on the Jurkat cell surface and soluble GXM was performed as previously reported [da Silva et al.; dos Santos et al.]. Cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR were incubated with soluble GXM (10 µg/mL) obtained from C. gattii and C. neoformans as described by Wozniak and Levitz [Citation22]. Jurkat cells expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR (1 × 10 [Citation6] cells) were incubated with soluble GXM and maintained on ice for 45 min, and the cells were washed with 1 mL of PBS (300 × g, 10 min, 4°C) and added 100 μL of anti-GXM murine monoclonal antibody (clone 18B7 Sigma; Cat. No. #MABF2069) at a dilution of 1:100 in PBS. After 45 min, the cells were washed with 1 mL of PBS and added 100 μL of biotinylated goat anti-murine IgG monoclonal antibody diluted 1:100 in PBS, followed by 45 min of incubation on ice. The cells were washed with PBS (1 mL), incubated with streptavidin-conjugated phycoerythrin (PE; eBioscienceTM; Cat. No. 12-4317-87) diluted in PBS (1:100), and after 30 min, the cells were washed with PBS. The cells were evaluated by flow cytometry, and the recognition of soluble GXM by Jurkat cells expressing GXMR-CAR was demonstrated by dot plots represented by double-positive cells for GFP and anti-GXM antibody detected by PE-streptavidin. Jurkat cells modified with pLenti-Mock were used as negative controls.
Activation of jurkat cells modified with GXMR-CAR constructs in the presence of ligands
Jurkat cells expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR after transduction using an MOI 3 were seeded at a concentration of 2 × 10 [Citation5] cells/mL (final volume of 200 μL) in 96-well plates. The cells were incubated with C. gattii or C. neoformans (effector (E) to target (T) ratio of 1:1) that were previously heat-killed. Cryptococcus spp. clinical isolates and ATCC references were were detailed in M &M section in the topic ‘Cell lines and cultivation of Cryptococcus spp..’ The co-culture was maintained in a humidified 5% CO2 atmosphere at 37°C for 24 and 48 h, and the cell concentration and viability of GXMR-CAR Jurkat cells and the expression rate of GXMR-CAR were quantified using flow cytometry. Furthermore, the cell culture supernatant was collected in the periods of 24 and 48 h of co-culture and stored at −20°C, and subsequently, the levels of IL-2 were measured by ELISA (BD OptEIA ™; BD Biosciences). Jurkat cells expressing 2H1-VH-GXMR-CAR or 18B7-VH-GXMR-CAR were also co-cultured with C. gattii (R265 strain) or C. neoformans (H99 strain) under the same conditions described above. After 24 and 48 h of incubation, the cell culture supernatant was collected to measure the levels of IL-2 by ELISA. Jurkat cells modified with pLenti-Mock were used as negative controls. The activation of modified Jurkat cells was also assessed by the expression of CD69 or CD25 on the cell surface after co-culture with heat-killed yeast from C. gattii and C. neoformans. Moreover, to evaluate the exhaustion markers the expression of TIM-3 and PD-1 were quantified. After 24 h of incubation, the cells were washed with PBS and incubated with PE-conjugated anti-CD69 antibody (BD Biosciences; FN50 clone) CD25 (BD Biosciences; M-A251 clone), TIM-3 (BD Biosciences; 7D3 clone), or PD-1 (BD Biosciences; EH12.1 clone) for 40 minutes. Subsequently, the cells were washed with PBS, and the expression of activation and exhaustion markers was measured by flow cytometry. Values are expressed as median fluorescence intensity (MFI). Jurkat cells were transduced with GXMR-CAR at an MOI of 3.
Activation of cells modified with 2H1-GXMR or 18B7-GXMR in the presence of tyrosine kinase inhibitor (dasatinib) or serum from cryptococcosis patients
GXMR-CAR Jurkat cells and non-modified cells were seeded at a concentration of 2 × 10[Citation5] cells/mL in 200 μL in 96-well plates, and previously the co-culture with heat-killed yeast form of C. gattii (R265) and C. neoformans (H99) was added 50 nM of Dasatinib (pharmacological inhibitor for Src family kinase) (Cat. No. SML2589; Sigma- Aldrich, MO, USA)and GXMR-CAR Jurkat cells were incubated for three hours at 37°C. The co-culture of cells expressing GXMR-CAR and Cryptococcus spp. used an E:T ratio of 1:1, and after 24 and 48 h of incubation, the cell culture supernatant was collected to measure the levels of IL-2 by ELISA. Jurkat cells modified with GXMR-CAR or pLenti-Mock were cultured with the same concentration of vehicle-DMSO in the presence or absence of Cryptococcus spp. as a control to evaluate the effect of dasatinib. Jurkat cells expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR at a concentration of 2 × 10[Citation5] cells/mL in 200 μL in 96-well plates were incubated with serum obtained from a patient infected with C. neoformans using a dilution of 1:10. Sera obtained from healthy donors and Jurkat cells modified with pLenti-Mock were used as negative controls. The cells incubated with serum were maintained in a humidified 5% CO2 atmosphere at 37°C for 24 and 48 h, and the levels of IL-2 in the cell culture supernatant were measured using ELISA. The procedures to obtain serum from healthy donor and cryptococcosis patients were previously approved by the Committee of Ethics of the School Hospital of the Ribeirão Preto Medical School at the University of São Paulo with protocol no. 4.277.966.
Fluorescence microscopy of modified jurkat cells 2H1-GXMR-CAR and 18B7-GXMR-CAR
Jurkat cells that were modified with 2H1-GXMR-CAR or 18B7-GCMR-CAR expressing mCherry fluorescent protein were plated on slides that were previously treated with 50 μg/ml of Poly-lysine (Cat. No. 6282; Sigma- Aldrich, MO, USA) and incubated for one hour at 37°C for adherence of Jurkat cells. The slides were mounted using ProLong Gold (Cat. No. P36930; Thermo Fisher Scientific, Waltham, MA, USA), and the images were analyzed using fluorescence microscopy at a magnification of 63× and 6× zoom. Images were acquired using a Leica TCS SP5 confocal microscope, and a laser 594 nm was used for excitation.
Molecular dynamics and docking of GXMR-CAR scFvs
Molecular Dynamics (MD) simulations of both 2H1-scFv and 18B7-scFv were performed using the GROMACS 2020.4 package [Citation23–27] and the CHARMM36 [Citation28] force field to describe the atomic interactions. The initial scFv structures of both simulated systems were obtained using variable domains (VL and VH) from the 2H1 Fab fragment structure (PDB 2H1P) [Citation29] as a template for homology modeling and modeling of the linker using RoseTTAFold [Citation30]. The atomic coordinates of both scFv models were submitted to the H ++ server [Citation31] and then placed in a solvated dodecahedral box filled with TIP3P [Citation32] water molecules. The distance between the protein and box edges was set to 2 nm for all axes. Protein-binding lengths were controlled using the LINCS [Citation33,Citation34] algorithm, and water was controlled using the SETTLE [Citation35] algorithm. The van der Waals and Coulomb interactions between unbound atoms were evaluated with a cutoff radius of 1.3 nm, and corrections for electrostatic interactions were calculated using the particle mesh Ewald method (PME) [Citation36].
The first step of energy minimization was performed using the steepest descent integrator [Citation37] at 10,000 steps and an integration step of 2 fs, with the atomic coordinates of the protein restrained to allow better adjustment of the solvent around the protein. Energy system minimization was continued through short NVT and NPT ensemble simulations at 310 K. The minimization began in NVT with a 17.5 ps MD (time step of 0.5, 1 fs), keeping the atomic coordinates of the scFvs restrained in each system. Then, NPT was continued with an integration time of 1 fs for a total time of 20 ps with the backbone still restrained, followed by 50 ps (time step of 2 fs) with the protein-free. The temperature was controlled using a V-rescale thermostat [Citation38] with the protein, solvent, and ions separately coupled with a coupling constant of 0.2 ps. Likewise, the pressure of the systems was controlled by Berendsen barostat [Citation39] with a constant of 0.2 ps in 1 bar. Trajectory acquisition was performed using the NPT ensemble for 700 ns with a time step of 2 fs. The MD coordinates were recorded every 100 ps.
Molecular docking was performed to compare the interfaces between the simulated 2H1-scFv and 18B7-scFv structures and mimotope (PA1) co-crystallized with the 2H1-Fab fragment (Young et al., 1997). The HADDOCK 2.4 [Citation40,Citation41] webserver was used. Docking was performed by selecting the residues of the 2H1-Fab antibody binding site [Citation29] as active residues to obtain the most likely docking site. The best model for each structure was selected by evaluating the HADDOCK, RMSD, and Z-scores.
Molecular dynamics analysis
The resulting trajectories of both simulations were processed using the gmx trjconv program. Root-mean-square deviations (RMSD) and solvent-accessible surface areas (SASA) were calculated using the GROMACS 2020.4 programs [Citation23,Citation24,Citation42–44]. The intermolecular interaction potential (IIP) energy profiles were determined using the gmx energy program, including the long-range (electrostatic) and short-range (Lennard-Jones) potentials [Citation45] between the scFv variable domains (VH and VL) within a 0.5 nm cutting radius. The structural conformation during the simulation was clustered with the g cluster program using the Gromos algorithm [Citation46]. A threshold value of 0.2 nm was used to obtain the most representative structure for VLVH during the simulation from 17,500 frames. Structural representations of the analyzed systems were generated using PyMOL 2.2.0 (Schrödinger). All graphs were plotted using the Origin 8.0® program.
Statistical analysis
Data analysis was conducted using Prism 9.0 software from GraphPad. The normality assumption of statistical assessments was evaluated utilizing the Shapiro-Wilk test, while the equality of variances was assessed through the F-test when comparing two groups or the Bartlett test when comparing three or more groups. In cases where data displayed Gaussian distributions, Student’s t-test was employed for experiments involving two groups, and ANOVA was utilized for experiments involving three or more groups. For datasets demonstrating non-normal distributions, the Mann-Whitney test was employed for two-group experiments, and the Kruskal-Wallis test was utilized for experiments involving three or more groups. Differences in the means of groups were examined using a one-way ANOVA, followed by subsequent implementation of Tukey’s multiple comparison tests. Alternatively, for non-normally distributed data, the Kruskal-Wallis test was followed by Dunn’s multiple comparison test. Statistically significant differences were identified when the p-value fell below 0.05. The presentation of results follows the format of mean ± standard deviation (SD).
Results
2H1-GXMR-CAR and 18B7-GXMR-CAR mediate the recognition of soluble GXM from C. gattii and C. neoformans
The capsule of Cryptococcus spp. contains the GXM polysaccharide, which is usually targeted in several immunotherapeutic strategies. A pioneering cell therapy approach developed a chimeric antigen receptor (CAR) containing a binding domain specific to GXM, known as GXMR-CAR. Human T cells expressing the GXMR-CAR construct were redirected to interact with Cryptococcus spp. in the lungs, favoring a reduction in the frequency of titan cells form of C. neoformans [Citation15]. Our group recently demonstrated that the modifications in the hinge, transmembrane, and signal transduction domains of GXMR-CAR improved CAR expression and its strength signaling transduction [Citation16]. The current study worked with T cells modified with GXMR-CAR containing distinct antigen-binding domains; clones 2H1 and 18B7 of monoclonal antibodies specific to GXM were used to generate two single-chain variable fragments (scFv) that comprised the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs (). Then, the main goal of this work was the investigation of the functionality of GXMR-CAR constructs containing different antigen-binding domain specific to GXM and sharing the same hinge/transmembrane and signal transduction domains (), and the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs were evaluated the presence or absence of the ligands. This proposal was based on the hypothesis that distinct scFv influences the expression rate, functionality, and strength of transduction signaling between GXMR-CAR constructs. In general, T cells were modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR and the level of cell activation against the ligands and the strength of tonic signaling were measured. Moreover, the molecular mechanisms involved in the recognition of target by 2H1-GXMR-CAR or 18B7-GXMR-CAR were associated with the T cell activation induced by GXMR-CAR constructs.
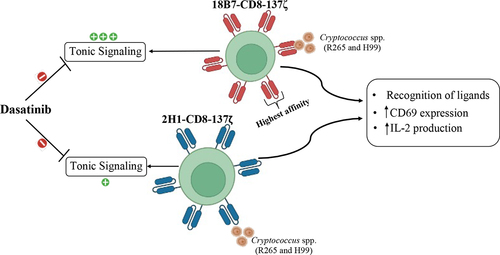
Figure 1. 2H1-GXMR-CAR and 18B7-GXMR-CAR recognize soluble GXM from C. gattii and C. neoformans. (a) scheme of the GXMR-CAR constructs containing antigen binding domains sourced from clones 18B7 (blue) or 2H1 (red) of anti-GXM monoclonal antibody followed by CD8 molecule as hinge/transmembrane domain (black), CD137 as a costimulatory portion (purple), and CD3ζ (yellow) as signaling transduction domain. The enhanced green fluorescence protein (EGFP) is a marker separated by T2A peptide from GXMR-CAR expression. The constructs were called 18B7-GXMR-CAR and 2H1-GXMRCAR. (b) steps of the protocol of co-transfection of GXMR-CAR plasmids and helper plasmids in HEK-293T cells to produce lentiviral particles carrying the DNA sequence of 2H1-GXMR-CAR or 18B7-GXMR-CAR and the lentiviral particles were obtained from the cell supernatant after 24, 48, and 72h post-transduction. The lentiviral particles were concentrated, the titer was calculated as described in the M&M, and the Jurkat cell line was modified by transduction to express GXMR-CAR constructs. (c) Jurkat cells (1×10 [Citation5] cells/well) distributed in a 48-well plate were incubated with lentiviral particles and performed the spinoculation method for transduction, and after 72h the percentage of GFP+ cells were determined by flow cytometry to calculate the lentiviral particles titer for each GXMR-CAR construct. (d) binding assay between Jurkat cells expressing 18B7-GXMR-CAR or 2H1-GXMR-CAR and soluble GXM (10µg/mL) obtained from C. gattii or C. neoformans. After incubation between soluble GXM and GXMR-CAR Jurkat cells was added an anti-GXM murine monoclonal antibody (clone 18B7) followed by the addition of goat anti-mouse IgG secondary antibody biotin conjugated. The presence of GXM on the cell surface was revelead by incubation with streptavidin-PE, and the mean fluorescence intensity (MFI) was measured by flow cytometry. As a negative control, Jurkat cells modified with lentiviral particles carrying GPF alone (pLenti-MOCK) were considered in all steps. Jurkat cells expressing GXMR-CAR or GFP alone (pLenti-Mock) were gated, and the MFI of the interaction of GXM was represented in histograms.
![Figure 1. 2H1-GXMR-CAR and 18B7-GXMR-CAR recognize soluble GXM from C. gattii and C. neoformans. (a) scheme of the GXMR-CAR constructs containing antigen binding domains sourced from clones 18B7 (blue) or 2H1 (red) of anti-GXM monoclonal antibody followed by CD8 molecule as hinge/transmembrane domain (black), CD137 as a costimulatory portion (purple), and CD3ζ (yellow) as signaling transduction domain. The enhanced green fluorescence protein (EGFP) is a marker separated by T2A peptide from GXMR-CAR expression. The constructs were called 18B7-GXMR-CAR and 2H1-GXMRCAR. (b) steps of the protocol of co-transfection of GXMR-CAR plasmids and helper plasmids in HEK-293T cells to produce lentiviral particles carrying the DNA sequence of 2H1-GXMR-CAR or 18B7-GXMR-CAR and the lentiviral particles were obtained from the cell supernatant after 24, 48, and 72h post-transduction. The lentiviral particles were concentrated, the titer was calculated as described in the M&M, and the Jurkat cell line was modified by transduction to express GXMR-CAR constructs. (c) Jurkat cells (1×10 [Citation5] cells/well) distributed in a 48-well plate were incubated with lentiviral particles and performed the spinoculation method for transduction, and after 72h the percentage of GFP+ cells were determined by flow cytometry to calculate the lentiviral particles titer for each GXMR-CAR construct. (d) binding assay between Jurkat cells expressing 18B7-GXMR-CAR or 2H1-GXMR-CAR and soluble GXM (10µg/mL) obtained from C. gattii or C. neoformans. After incubation between soluble GXM and GXMR-CAR Jurkat cells was added an anti-GXM murine monoclonal antibody (clone 18B7) followed by the addition of goat anti-mouse IgG secondary antibody biotin conjugated. The presence of GXM on the cell surface was revelead by incubation with streptavidin-PE, and the mean fluorescence intensity (MFI) was measured by flow cytometry. As a negative control, Jurkat cells modified with lentiviral particles carrying GPF alone (pLenti-MOCK) were considered in all steps. Jurkat cells expressing GXMR-CAR or GFP alone (pLenti-Mock) were gated, and the MFI of the interaction of GXM was represented in histograms.](/cms/asset/81ebbb5a-4999-4493-97cb-f2301e528076/kbie_a_2281059_f0001_oc.jpg)
Lentiviral particles containing the coding sequences for 2H1-GXMR-CAR or 18B7-GXMR-CAR were produced in HEK-293T cells after co-transfection with GXMR-CAR constructs and helper plasmids, and the lentiviral particle titer was determined by transduction of Jurkat cells (). Lentiviral particles with the 18B7-GXMR-CAR coding sequence showed higher titers than those with the 2H1-GXMR-CAR construct (). Initially, Jurkat cells were modified to express 2H1-GXMR-CAR or 18B7-GXMR-CAR at an MOI 3 of lentiviral particles, and the ability of the GXMR-CAR constructs to recognize soluble GXM obtained from C. gattii and C. neoformans showed the expression of 2H1-GXMR-CAR or 18B7-GXMR-CAR on the cell surface (). The interaction between soluble GXM and GXMR-CAR constructs was detected using a monoclonal anti-GXM antibody after flow cytometry analysis using a previously reported protocol [Citation16]. Both 2H1-GXMR-CAR (blue)- and 18B7-GXMR-CAR (red)-modified cells recognized soluble GXM of C. gattii and C. neoformans as represented in the histograms, and 18B7-GXMR-CAR had a higher recognition of GXM obtained from C. gattii than 2H1-GXMR-CAR (). In contrast, GXM polysaccharides of C. neoformans were similarly recognized between GXMR-CAR constructs (). Jurkat cells modified with mock-GFP lentiviral particles (Lenti-Mock; black line) were used as negative controls and were incubated with GXM obtained from both Cryptococcus species.
18B7-GXMR-CAR shows highest transduction efficiency, and 2H1-GXMR-CAR expression favored a cellular expansion
Jurkat cells were modified with GXMR-CAR constructs considering a MOI of 1 and 3 to determine the transduction efficiency based on the percentage of GFP-positive cells, and the mean fluorescence intensity (MFI) was used to evaluate the GXMR-CAR expression rate. The transduction efficiency of 18B7-GXMR-CAR was significantly higher compared to 2H1-GXMR-CAR at both MOI of 1 and 3, as verified three days post-transduction (), and the 18B7-GXMR-CAR expression rate was higher than that of the 2H1-GXMR-CAR construct on day three (). Cells modified with GXMR-CAR constructs were evaluated over nine days, and the percentage of 18B7-GXMR-CAR Jurkat cells was significantly higher than that of 2H1-GXMR-CAR Jurkat cells throughout the analysis period (). In contrast, the MFI of cells modified with 2H1-GXMR-CAR and 18B7-GXMR-CAR did not differ significantly at most of the time points considered (), and cell modification at an MOI of 3 was adopted to evaluate the capacity of GXMR-CAR constructs to induce cell activation against the target, as shown in . In addition, the cellular expansion of 2H1-GXMR-CAR and 18B7-GXMR-CAR Jurkat cells demonstrated that the 2H1-GXMR-CAR construct favored cellular expansion, as evidenced by the population doubling level (PDL), compared to Jurkat cells modified with 18B7-GXMR-CAR at MOI of 1 and 4 (). Together, these results demonstrated that cells modified with 2H1-GXMR-CAR showed high levels of CAR expression, as observed with 18B7-GXMR-CAR, and Jurkat cells expressing 2H1-GXMR-CAR had satisfactory cellular expansion.
Figure 2. Expression levels of 2H1-GXMR-CAR and 18B7-GXMR-CAR in Jurkat cells were modified over nine days of cell culture. Jurkat cells were modified with 2H1-GXMR-CAR (blue line), 18B7 GXMR-CAR (red line), or Mock (negative control; black line) using an MOI of 1 (a, c, e) and 3 (b, d, f). The percentage of GFP positive cells (A and B) and their expression rate represented by mean fluorescence intensity (MFI) (C and D) were measured by cytometry every three days post-transduction. (e, f) the cellular expansion was monitored over nine days of cell culture, and the population doubling level (PDL) was determined using the formula: [Log2 (number of CAR+ cells at the analyzed time) – Log2 (number of CAR+ cells at the initial time). ‘#’ denotes statistical significance between 18B7-GXMR-CAR and Mock, ‘*’ denotes statistical significance between 2H1-GXMR-CAR and 18B7-GXMR-CAR, ‘+’ denotes statistical significance between 2H1-GXMR-CAR and Mock. Values are expressed as mean ± SD. Each experiment was performed in triplicate. +,#, *p < 0.05; ##; ++,##, ** p < 0.01; +++,###, *** p < 0.001; ++++,####, **** p < 0.0001; according to one-way ANOVA.
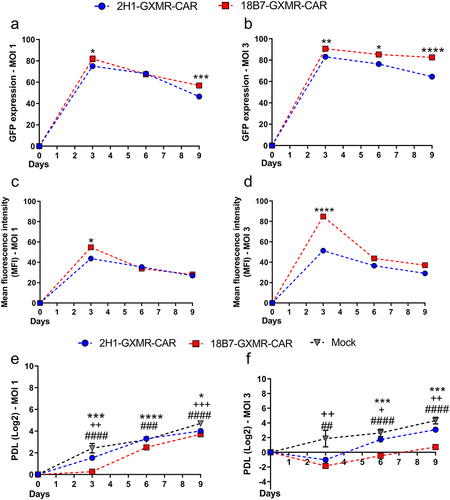
Figure 3. Recognition of Cryptococcus spp. By 2H1-GXMR and 18B7-GXM induced a similar cell activation. Jurkat cells (2×10 [Citation5] cells/mL) modified with 2H1-GXMR-CAR, 18B7-GXMR-CAR, or pLenti-Mock (negative control) at an MOI of 3 were distributed in 96-well plates and incubated with 1 μg/ml of soluble GXM of C. gattii or C. neoformans (a-b) and also co-cultured with inactivated C. gattii or C. neoformans yeasts using a ratio of 1:1 between effector: target cells (c-g). After 24 (a and c) and 48 h (b and d) of incubation, the levels of IL-2 in the cell culture supernatant were measured by ELISA and expressed in pg/mL. After 24 hours of incubation with C. gattii or C. neoformans yeasts (1:1 cell: yeast ratio), the CD69 expression on the cell surface was measured by flow cytometry (e). Moreover, the concentration of viable cells over time during 48 h of co-culture between modified cells and Cryptococcus spp. was determined by flow cytometry using propidium iodide to exclude dead cells (f-g). Values are expressed as mean ± SD; each experiment was performed in triplicate. #, *p < 0.05; ##; ##, ** p < 0.01; ###, *** p < 0.001; ####, **** p < 0.0001; according to one-way ANOVA. (A-D) ’*’ comparison between 18B7-GXMR-CAR or 2H1-GXMR-CAR modified cells versus Mock (negative control), and “#” statistical significance between 2H1-GXMR-CAR and 18B7-GXMR-CAR groups. (f and g) ’*’ and ‘#’ represents statistical significance between GXMR-CAR Jurkat cells incubated with medium (blue lines) in comparison to modified cells co-cultured in the presence of C. gatti (green line) or C. neoformans (red line), respectively.
![Figure 3. Recognition of Cryptococcus spp. By 2H1-GXMR and 18B7-GXM induced a similar cell activation. Jurkat cells (2×10 [Citation5] cells/mL) modified with 2H1-GXMR-CAR, 18B7-GXMR-CAR, or pLenti-Mock (negative control) at an MOI of 3 were distributed in 96-well plates and incubated with 1 μg/ml of soluble GXM of C. gattii or C. neoformans (a-b) and also co-cultured with inactivated C. gattii or C. neoformans yeasts using a ratio of 1:1 between effector: target cells (c-g). After 24 (a and c) and 48 h (b and d) of incubation, the levels of IL-2 in the cell culture supernatant were measured by ELISA and expressed in pg/mL. After 24 hours of incubation with C. gattii or C. neoformans yeasts (1:1 cell: yeast ratio), the CD69 expression on the cell surface was measured by flow cytometry (e). Moreover, the concentration of viable cells over time during 48 h of co-culture between modified cells and Cryptococcus spp. was determined by flow cytometry using propidium iodide to exclude dead cells (f-g). Values are expressed as mean ± SD; each experiment was performed in triplicate. #, *p < 0.05; ##; ##, ** p < 0.01; ###, *** p < 0.001; ####, **** p < 0.0001; according to one-way ANOVA. (A-D) ’*’ comparison between 18B7-GXMR-CAR or 2H1-GXMR-CAR modified cells versus Mock (negative control), and “#” statistical significance between 2H1-GXMR-CAR and 18B7-GXMR-CAR groups. (f and g) ’*’ and ‘#’ represents statistical significance between GXMR-CAR Jurkat cells incubated with medium (blue lines) in comparison to modified cells co-cultured in the presence of C. gatti (green line) or C. neoformans (red line), respectively.](/cms/asset/276a5a85-1dba-461c-976a-ead9e5034d7f/kbie_a_2281059_f0003_oc.jpg)
18B7-GXMR-CAR triggers strong tonic signaling in modified jurkat cells
The ability of 2H1-GXMR-CAR and 18B7-GXMR-CAR to induce cell activation was evaluated in Jurkat cells modified with GXMR-CAR constructs followed by incubation with soluble GXM or Cryptococcus spp. yeast. Jurkat cells expressing GXMR-CAR were initially incubated with soluble GXM (1 ìg/mL) obtained from C. gattii or C. neoformans. The levels of IL-2 were measured in the cell supernatant collected after 24 h of culture, and 18B7-GXMR-CAR and 2H1-GXMR-CAR constructs mediated a significant increase in the production of IL-2 in response to soluble GXM compared to modified cells cultured in the absence of ligand (medium) (). The 18B7-GXMR-CAR construct induced the production of IL-2, which was significantly higher than the levels produced by cells modified with 2H1-GXMR-CAR and incubated with soluble GXM for 48 h (). In addition, the levels of IL-2 induced by 2H1-GXMR-CAR and 18B7-GXMR-CAR were evaluated in a co-culture of Jurkat cells expressing GXMR-CAR and heat-killed C. neoformans and C. gattii. After 24 and 48 h of incubation, high levels of IL-2 were induced by 2H1-GXMR-CAR and 18B7-GXMR-CAR in the presence of C. neoformans and C. gattii yeasts (), and the 18B7-GXMR-CAR constructs induced IL-2 levels that were significantly higher than those mediated by 2H1-GXMR-CAR when incubated with C. gattii for 24 h (). Moreover, Jurkat cells expressing the GXMR-CAR constructs showed no significant difference in the levels of IL-2 released between C. neoformans and C. gattii (). In addition, the activation of Jurkat cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR was evaluated through the expression of CD69 on the cell surface after 24 h of co-culture with C. gattii or C. neoformans yeasts. An increase in CD69 expression was observed in cells expressing GXMR-CAR compared to unmodified Jurkat cells in response to Cryptococcus spp. (). Interestingly, 2H1-GXMR-CAR and 18B7-GXMR-CAR Jurkat cells showed higher levels of CD69 expression than unmodified Jurkat cells, even without the ligand ().
The strength of cell activation mediated by 18B7-GXMR-CAR was more pronounced than that observed in cells modified with 2H1-GXMR-CAR, as evidenced by the reduction in the cellular concentration of Jurkat cells expressing 18B7-GXMR-CAR, even in the absence of the ligand (). This event was corroborated by the high levels of IL-2 induced by 18B7-GXMR-CAR in the absence of its target, as highlighted after 24 and 48 h of incubation with medium alone (), and at the time point 48-hour, the production of IL-2 by Jurkat cells expressing 18B7-GXMR-CAR was significantly higher than that of cells modified with 2H1-GXMR-CAR and incubated in the absence of the ligand (medium) (). These results indicate that 18B7-GXMR-CAR expressed by Jurkat cells triggers pronounced tonic signaling and that the GXMR-CAR constructs mediate strong cell activation in response to the targets.
Expression of 18B7-GXMR-CAR markedly favored the PD-1 expression by modified jurkat cells incubated with Cryptococcus spp
The evident involvement of GXMR-CAR constructs in the activation of T cells against ligands led to the investigation of the expression of activation and exhaustion markers in Jurkat cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR and incubated with Cryptococcus spp yeasts. CD25 (alpha chain of the IL-2 receptor) is not constitutively expressed in Jurkat cells and does not change after cell modification with 2H1-GXMR-CAR or 18B7-GXMR-CAR (). However, the expression of 2H1-GXMR-CAR and 18B7-GXMR-CAR by Jurkat cells contributed to an increase in the percentage of cells positive for the PD-1 exhaustion molecule compared to non-modified Jurkat cells (), whereas the expression of TIM-3, another exhaustion molecule, did not change in cells expressing GXMR-CAR constructs (). In addition, the expression level of the PD-1 molecule was also evaluated in Jurkat cells expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR and incubated with inactivated yeast of C. neoformans (H99) or C. gattii (R265). After 24 h of co-culture, the percentage of 18B7-GXMR-CAR Jurkat cells expressing PD-1 was significantly increased compared to 2H1-GMXR-CAR Jurkat cells (). These data indicated that the strength of tonic signaling induced by 18B7-GXMR-CAR triggered the prominent expression of PD-1 in modified cells, even without a ligand.
Figure 4. Expression of the PD-1 exhaustion marker was enhanced in 18B7-GXMR-CAR Jurkat cells incubated with ligands. (a-c) Jurkat cells (5×10 [Citation5] cells) expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR were incubated with monoclonal anti-CD25 (a), anti-TIM-3 (b), or anti-PD-1 (c) antibodies conjugated with PE, and the detection of these molecules on the cell surface was determined by flow cytometry. The dot plots represent the percentage of positive cells for GXMR-CAR and CD25, TIM-3, or PD-1 expression. Non-modified cells were considered as a negative control. (d) 2H1-GXMR-CAR and 18B7-GXMR-CAR Jurkat cells (2×10 [Citation5] cells/mL) were incubated with C. gattii or C. neoformans (ratio of 1:1 of the effector to target cells), and after 24 h of incubation, the cells were obtained to perform the staining of PD-1. The percentage of positive cells for PD-1 expression was measured by flow cytometry, and non-modified Jurkat cells were used as a negative control. The data are expressed as mean ± SD, and each experiment was performed in triplicate.
![Figure 4. Expression of the PD-1 exhaustion marker was enhanced in 18B7-GXMR-CAR Jurkat cells incubated with ligands. (a-c) Jurkat cells (5×10 [Citation5] cells) expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR were incubated with monoclonal anti-CD25 (a), anti-TIM-3 (b), or anti-PD-1 (c) antibodies conjugated with PE, and the detection of these molecules on the cell surface was determined by flow cytometry. The dot plots represent the percentage of positive cells for GXMR-CAR and CD25, TIM-3, or PD-1 expression. Non-modified cells were considered as a negative control. (d) 2H1-GXMR-CAR and 18B7-GXMR-CAR Jurkat cells (2×10 [Citation5] cells/mL) were incubated with C. gattii or C. neoformans (ratio of 1:1 of the effector to target cells), and after 24 h of incubation, the cells were obtained to perform the staining of PD-1. The percentage of positive cells for PD-1 expression was measured by flow cytometry, and non-modified Jurkat cells were used as a negative control. The data are expressed as mean ± SD, and each experiment was performed in triplicate.](/cms/asset/8a532d00-cf87-4350-ada1-ca77b55341a4/kbie_a_2281059_f0004_oc.jpg)
Tonic signaling induced by 18B7-GXMR-CAR and the cell activation mediated by GXMR-CAR against Cryptococcus spp. were attenuated by the dasatinib
Previous studies evidenced that the protein tyrosine kinase of the Src family is involved with the tonic signaling triggered by CAR, and Dasatinib is a pharmacological inhibitor of the kinases of the Src family that attenuated the signaling pathway induced by costimulatory molecule 4-1BB and CD3ζ localized in the intracellular domain of CAR [Citation47]. Jurkat cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR were incubated with dasatinib before adding inactivated yeasts of C. neoformans or C. gattii. After 24 and 48 h of co-culture, the concentration of GXMR-CAR Jurkat cells was determined, and the levels of IL-2 in the cell supernatant were measured. Jurkat cells expressing 18B7-GXMR-CAR and incubated with dasatinib showed a resumption of cellular expansion in the presence or absence of Cryptococcus spp. compared to 18B7-GXMR-CAR cells not treated with dasatinib (). In addition, dasatinib reduced the production of IL-2 in cells modified with 18B7-GXMR-CAR in the absence of the ligand, indicating a direct effect on tonic signaling event (). In contrast, cells expressing 2H1-GXMR-CAR incubated with dasatinib did not significantly change the levels of IL-2 in the absence of the ligand ().
Figure 5. The activation of cells expressing 2H1-GXMR or 18B7-GXMR against Cryptococcus spp. was attenuated by dasatinib. GXMR-CAR Jurkat cells (2X10 [Citation5] cells/mL) were distributed in 96-well plates and were incubated or not with pharmacological inhibitor dasatinib (50 nM) for three hours before the addition of inactivated yeasts of C. neoformans or C. gattii using a ratio of 1:1 of effector to target cells. After 24 and 48 h of incubation, the viable cells (a-Ff) concentration was determined by flow cytometry after the exclusion of dead cells using propidium iodide, and the cell culture supernatant was used to measure the IL-2 levels by ELISA (g-l). The cells were incubated with only the culture medium (medium) as a negative control. Values are represented as mean ± SD, with * p < 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001 according to one-way ANOVA.
![Figure 5. The activation of cells expressing 2H1-GXMR or 18B7-GXMR against Cryptococcus spp. was attenuated by dasatinib. GXMR-CAR Jurkat cells (2X10 [Citation5] cells/mL) were distributed in 96-well plates and were incubated or not with pharmacological inhibitor dasatinib (50 nM) for three hours before the addition of inactivated yeasts of C. neoformans or C. gattii using a ratio of 1:1 of effector to target cells. After 24 and 48 h of incubation, the viable cells (a-Ff) concentration was determined by flow cytometry after the exclusion of dead cells using propidium iodide, and the cell culture supernatant was used to measure the IL-2 levels by ELISA (g-l). The cells were incubated with only the culture medium (medium) as a negative control. Values are represented as mean ± SD, with * p < 0.05; ** p ≤ 0.01; *** p ≤ 0.001; **** p ≤ 0.0001 according to one-way ANOVA.](/cms/asset/ab3e94cf-831d-4b92-9364-fe7d50102000/kbie_a_2281059_f0005_oc.jpg)
18B7-GXMR-CAR and 2H1-GXMR-CAR Jurkat cells previously treated with dasatinib showed a significant reduction in the production of IL-2 in response to co-culture with C. gattii or C. neoformans for 24 h () and 48 h () of incubation. Therefore, dasatinib can mitigate tonic signaling induced by 18B7-GXMR-CAR expression, and the activation of cells modified with GXMR-CAR was strongly attenuated against Cryptococcus spp.
Stability of scFv of 18B7 and 2H1 did not promote oligomerization of GXMR-CAR, and the VH domain of 18B7 had a role in the strength of GXMR-CAR tonic signaling
The 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs differed only in the antigen recognition domain characterized by the scFv of clones 2H1 and 18B7, respectively, which could explain the difference in the strength of tonic signaling between the GXMR-CAR constructs. Initially, GXMR-CAR was modified to express the mCherry fluorescence marker to localize the distribution of GXMR-CAR on the cell surface, which was evaluated using fluorescence microscopy. Jurkat cells modified with 18B7-GXMR-CAR and 2H1-GXMR-CAR showed intense spots on the cell surface, and both GXMR-CAR constructs showed similar distribution on the cell surface (). The aggregation of GXMR-CAR constructs caused by scFv was discarded after Molecular Dynamics (MD) simulation data were obtained for scFv of 18B7 and 2H1. MD analysis revealed that both scFv of 18B7 and 2H1 exhibited structural stability, and RMSD profiles demonstrated that the relative movement between the VL/VH domains was more pronounced in the scFv of 18B7 than in the scFv of 2H1 (Figures S1C-S1D). Specifically, we observed more expressive oscillations in the RMSD of VH after overlapping with VL in 18B7 (0.55 ± 0.13 nm) compared to 2H1 (0.32 ± 0.07 nm) (Table s1). These results indicate that cell surface puncta formation is not associated with scFv oligomerization of clones 18B7 and 2H1 that could be responsible for aggregation of GXMR-CAR constructs.
Figure 6. scFv from 2H1-GXMR-CAR and 18B7-GXMR-CAR are not responsible for the intensity of aggregation of GXMR-CAR, and the absence of the VL chain from scFv from GXMR-CAR constructs abolished the intense levels of activation. Jurkat cells modified with 2H1-GXMR-CAR (a) or 18B7-GXMR-CAR (b) containing in frame mCherry as fluorescence marker were seeded on polylysine slide coated (50 μg/mL) and incubated for one hour at 37°C to obtain a cell attachment. Slides were mounted using ProLong gold, and the images were acquired by fluorescence microscopy using a 63× objective lens for high-resolution imaging, and the green arrows indicate puncta of aggregates of GXMR-CAR on the cell surface. (c-e) Jurkat cells modified with 2H1-VH-GXMR-CAR, 18B7-VH-GXMR-CAR, or Mock (negative control) at a concentration of 2 × 105cells/mL were cultured in the presence of culture medium (c) or inactivated yeasts of C. neoformans (d) or C. gattii (e), using 1:1 ratio of effector to target cells. The co-culture was performed in 96-well plates, and after 48 h of incubation, the cell culture supernatant was used to measure the levels of IL-2 by ELISA. (f) histograms represent the CD69 expression on the surface of Jurkat cells modified or not with 2H1-VH-GXMR-CAR or 18B7-VH-GXMR-CAR after 24 hours of co-culture with C. gattii or C. neoformans heat-killed (1:1 cell: yeast ratio) or in the absence of the ligand (medium). CD69 expression on the cell surface was evaluated by cytometry. According to one-way ANOVA, values are represented as mean ± SD, with * p < 0.05.
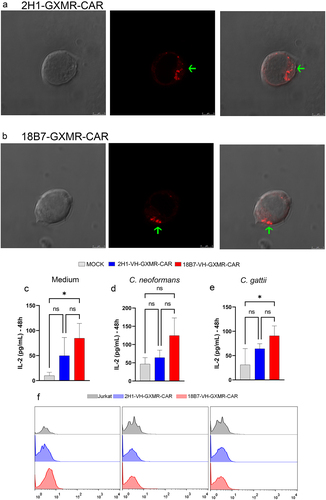
In addition, the RMSD profiles demonstrated a similar oscillation pattern between VLVH and VH for the scFv of both 18B7 and 2H1 (Figures S1A and 1B), and a small change in the oscillation pattern of the scFv structure was more closely related to the VH domain. The RMSDs of the VL domain of 18B7 (0.15 ± 0.02 nm) and 2H1 (0.11 ± 0.02 nm) are smaller than those amounts detected for the VH domain in both 18B7 and 2H1 (18B7 = 0.20 ± 0.01 nm and 2H1 = 0.22 ± 0.03 nm) (Table S1). To correlate the impact of the VH domain of scFv of 18B7 and 2H1 on the strength of tonic signaling induced by 2H1-GXMR-CAR and 18B7-GXMR-CAR, the antigen recognition portion of the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs was modified to express only the variable region of the heavy (VH) chain. These novel GXMR-CAR variants were named 2H1-VH-GXMR-CAR and 18B7-VH-GXMR-CAR. Jurkat cells modified with the GXMR-CAR variants were generated and co-cultured with C. gattii or C. neoformans yeasts. Interestingly, the levels of IL-2 and CD69 expression induced by 18B7-VH-GXMR-CAR in the absence of the ligand were significantly higher than those levels found in cells modified with pLenti-Mock (negative control), whereas IL-2 levels and CD69 expression in cells expressing 2H1-VH-GXMR-CAR did not significantly differ from the negative control (). However, 2H1-VH-GXMR-CAR and 18B7-VH-GXMR-CAR lost their ability to induce cell activation against the Cryptococcus spp. (). Thus, the oscillation pattern of the scFv VH domain in 18B7 and 2H1 analyzed by RMSD was consistent with the strength of tonic signaling induced by 18B7-GXMR-CAR.
2H1-GXMR-CAR mediated an intense cell activation against clinical isolates of Cryptococcus spp., and the responsiveness of cells modified with 2H1-GXMR-CAR was more reduced against serum from cryptococcosis patients
To further evaluate the capacity of the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs to induce cell activation in response to the ligand, we used clinical isolates of C. gattii and C. neoformans incubated with Jurkat cells modified with GXMR-CAR. This co-culture was performed between cells expressing 2H1-GXMR-CAR or 18B7-GXMR-CAR and inactivated yeasts. After 24 and 48 h of incubation, the cell culture supernatant was used to quantify the levels of IL-2. Both the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs mediated cell activation against all evaluated clinical isolates, as demonstrated by the high levels of IL-2 produced by GXMR-CAR Jurkat cells compared to unmodified cells (). Co-culture of 18B7-GXMR-CAR Jurkat cells and clinical isolates of C. gattii (478 or 474) or C. neoformans (525) resulted in higher levels of IL-2 than in cells modified with 2H1-GXMR-CAR (). Therefore, T cell activation mediated by the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs against clinical isolates of Cryptococcus spp. was satisfactory.
Figure 7. 2H1-GXMR-CAR and 18B7-GXMR-CAR expressed in Jurkat cells mediated the activation against clinical isolates of C. gattii and C. neoformans and in the presence of serum of cryptococcosis patients. Jurkat cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR (2 × 10 5 cells/ml) were co-cultured with clinical isolates of C. gattii (478, 474, and 455) or C. neoformans (525, 521, and 523) at a 1:1 ratio of effector to target cells. Jurkat cells not modified with GXMR-CAR were used as negative controls (CTRL). After 24 h (a) and 48 h (B) of incubation, the cell culture supernatant was collected and used to measure the levels of IL-2 by ELISA. (c, d) cells with or without 2H1-GXMR-CAR or 18B7-GXMR-CAR were seeded in a 96-well plate and incubated with serum obtained (diluted 1:10) from a healthy donor or a patient diagnosed with cryptococcosis. After 24 (c) and 48 h (d), IL-2 levels in the cell culture supernatant were measured using ELISA. (a, b) ’*’ denotes statistical significance between 18B7-GXMR-CAR and 2H1-GXMR-CAR groups versus non-modified cells (control) and ‘#’ denotes statistical significance between 2H1-GXMR-CAR and 18B7-GXMR-CAR Jurkat cells. Values are expressed as the mean ± DE. Each experiment was performed in triplicates. #, *p < 0.05; ##; ##, ** p < 0.01; ###, *** p < 0.001; ####, **** p < 0.0001; according to one-way ANOVA.
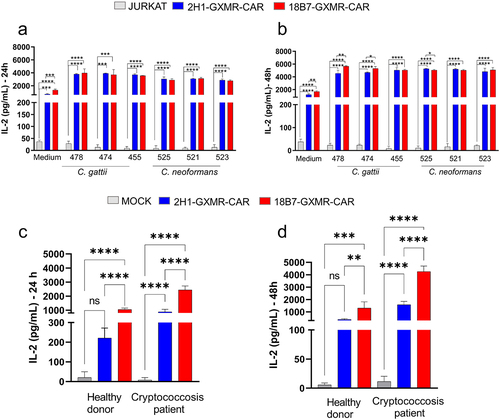
It is well established that during the progression of Cryptococcus spp. infection, the levels of GXM, which could be a ligand of GXMR-CAR, are higher in the serum of patients. This hypothesis was evaluated by the addition of serum obtained from the peripheral blood of patients with cryptococcosis to the culture of cells modified with 2H1-GXMR-CAR or 18B7-GXMR-CAR, and the measurement of the levels of IL-2 was considered to investigate cell activation. The incubation of Jurkat cells modified with GXMR-CAR and serum from a cryptococcosis patient led to high levels of IL-2 after 24 and 48 h of culture compared to the cells incubated with serum alone (). Interestingly, the 2H1-GXMR-CAR construct induced levels of IL-2 significantly lower than those produced by cells expressing 18B7-GXMR-CAR (). Therefore, these data demonstrate that the expression of 2H1-GXMR-CAR triggered a lower cell activation against serum from cryptococcosis patients, whereas 18B7-GXMR-CAR induced strong cell activation.
Binding pocket of 18B7 scFv has an enlarged surface to interact with GXM
A comparative analysis between the sequences of 2H1 and 18B7 scFv was performed to elucidate the structural differences between 2H1 and 18B7 scFv that could contribute to the strength of tonic signaling and cell activation against the ligands. The Molecular Dynamics (MD) simulations of the isolated 2H1 and 18B7 scFv were considered, and the RMSD profiles of each variable region of 2H1 and 18B7 were simulated, as shown in Figures S1A-B. The RMSD analysis did not consider the link between VL and VH because its flexibility impaired the simulation system. The isolated VL and VH domains and the VLVH pair were used to measure RMSD profiles. The RMSD data demonstrated that the solution equilibrium time was teq = 330 ns for the 2H1 and 18B7 scFv structures in both systems; this condition was used to evaluate the structural and energetic properties. The average RMSDs (Table S1) were below 0.25 nm, indicating high structural stability for the variable domains of 2H1 and 18B7. The RMSD profile of 18B7 exhibited significant fluctuations at the beginning of the simulation because of stabilization movements of the modified structure of the homology model using 2H1 as a model (Figure S1B). These data demonstrated that mutations in 2H1 to propose the 18B7 template resulted in the accommodation of the structure up to 330 ns. In contrast, the RMSD profile of 2H1 showed significantly lower fluctuations, which were related to greater structural stability, mainly in the VL domain (Figure S1A). RMSD profiles showed similar oscillation patterns between the VLVH pair and VH for 18B7 and 2H1 scFv (Figures S1A-B). These data revealed that small changes in the scFv structure were due to the structural modifications of VH.
The RMSDs of VL in 18B7 (0.15 ± 0.02 nm) and 2H1 (0.11 ± 0.02 nm) are smaller than VH (18B7 = 0.20 ± 0.01 nm and 2H1 = 0.22 ± 0.03 nm) (Table S1). Considering that VH favors greater structural changes, the relative movement between VL and VH domains was investigated, and RMSD profiles of VH after VL overlay showed more prominent oscillations in 18B7 (0.55 ± 0.13 nm) versus 2H1 (0.32 ± 0.07 nm) (Figure S1C-D). Moreover, when fitting the structures of VH to calculate the RMSD of VL (Figure S1C), a smaller amplitude was observed in the oscillations for VL than in those for VH (Figure S1D). The relative movement between the VL and VH domains of 18B7 was more evident and was related to the greater oscillation of 18B7 (Figure S1C-D), which impacted the interface between VL and VH, increasing structural instability. Furthermore, CDR H3 of 18B7 had more mutations than 2H1 and was located at the VL/VH interface. In addition, the profiles demonstrated an attractive interaction between VL and VH more pronounced in 2H1 (−143 ± 23 kcal.mol −1) compared to 18B7 (−112 ± 21 kcal.mol −1) (Table S1). The 2H1 profile features an IIP of ~ −205 kcal.mol −1 that was not observed for the 18B7 profile, reinforcing the drastic reduction in the stability of the VL/VH interface 18B7 compared to 2H1. Therefore, the position of CDR H3 of 18B7 was changed (Figure S3), and the CDR H3 of 18B7 was slightly displaced compared to the CDR H3 of 2H1, confirming that the structural modification of 18B7 was associated with the instability of the VL/VH interface.
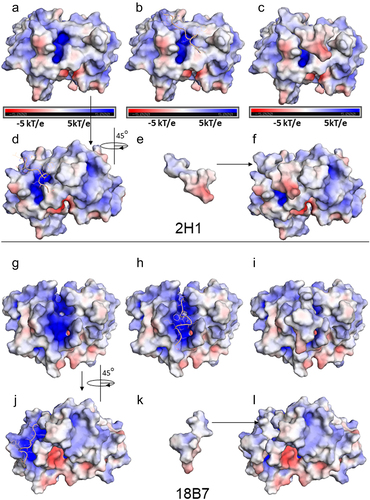
The investigation of clustered structures was based on a 0.2 nm slice (Figures S4A-B), cluster 1 was the most representative, and the MD configuration was used to couple a PA17 peptide to the binding site of 18B7 and 2H1 scFv. Figure S5A shows the positively charged region, determined to be the binding site of the PA1 peptide (GLQYTPSWMLVG), which was previously defined as a mimotope of the cryptococcal capsular polysaccharide [Citation29]. We used the last MD structure of 2H1 for docking PA1 and evaluated the differences in interaction with the target when the crystallographic structure was compared with the MD. The best redocking results for Fab and PA1 to validate the docking methodology using HadDock, in which the RMSD of the ligand was null, are shown in Figure S5B. The PA1 binding region in the MD of 2H1 showed a subtle reduction in the binding pocket compared to the crystallographic structure (Figure S5A, C). This result led to the modification of the orientation of PA1 (Figure S5C), and the structural motif that interacted with L3 Val99 and Trp101 and H2 Asn182 and Tyr189 was maintained. Otherwise, the binding pocket of 18B7 had an enlarged surface compared to the binding pocket of 2H1, the mimotope was better accommodated by 18B7 scFv, and different contact residues with the target were observed between 18B7 and 2H1 scFv. The docked PA1 was accommodated in the positively charged binding pocket between the CDRs H1, H2, and H3 of 18B7, and the CDRs residues from the VH domain, such as H1 Phe162, H2 Asn181, H2 Lys186, and H3 Arg228, had a greater impact (Figure S5D). The interaction potentials of PA1 with 2H1 and 18B7 scFv were −86.7 and −101.5 kcal mol-1, respectively, indicating that the mimotope has an enhanced attractive interaction with 18B7. The surface electrostatic maps obtained by APBS-PyMOL compared the pockets of structures 2H1 and 18B7 MD (). A larger positively charged pocket was observed in 18B7 (), demonstrating that 18B7 improved the structural conditions for interaction with carbohydrates. Likewise, the mimotope also fits better in the binding pocket of 18B7, highlighting the possibility of a higher affinity for 18B7 than for 2H1 ().
Figure 8. Electrostatic potential surfaces are calculated by the Adaptative Poisson Boltzmann Solver (APBS), using the APBS module in PyMol (−5kT/e a 5kT/e). (a-l) potential representation surfaces of 2H1 (a-f) and 18B7 (g-l). The mimotope PA1 is represented in the cartoon, light yellow, or electrostatic potential surface. a) top view of the scFv CDRs, with identification of the pocket in the middle of the structure with a positively charged region (blue); b) docked PA1 to the scFv binding pocket; c) docked PA1 surface to the scFv binding pocket, d) 45º rotation of the scFv from a new view of the PA1 in the binding pocket; e) PA1 represented in surface and f) docked PA1 surface to the scFv binding pocket. For 18B7, g) top view of the scFv CDRs, with identification of a prominent pocket in the middle of the structure and with a positively charged region (blue); h) docked PA1 to the scFv binding pocket, i) docked PA1 surface to the scFv binding pocket; j) 45º rotation of the scFv from g with the PA1 in the binding pocket; k) PA1 represented in surface and i) docked PA1 surface to the scFv binding pocket.
Discussion
The genus Cryptococcus spp. is one of the major pathogens responsible for invasive fungal infections [Citation48]. Treatment with antifungal drugs against Cryptococcus spp. has low efficacy and causes adverse side effects in patients [Citation14]. Several studies have investigated distinct approaches to control cryptococcosis and antifungal drug resistance, and the development of vaccines and immunotherapeutic strategies has been highlighted as promising for the control of cryptococcosis. The application of CAR to fight against invasive fungal infections has emerged as a relevant approach [Citation49–51], and GXMR-CAR has demonstrated high potential to redirect T cells to target Cryptococcus spp. The optimization of GXMR-CAR to improve T cell activation against Cryptococcus spp. was initiated by modification of hinge/transmembrane and signaling transduction domains, as reported by dos Santos et al., who demonstrated that CD8 molecules as hinge/transmembrane and 4-1BB as costimulatory domains favored T cell activation in response to Cryptococcus spp. yeasts [Citation16]. The current study evaluated the effect of distinct antigen recognition domains belonging to GXMR-CAR on the strength of T cell activation using the Jurkat cell line as an important model for comparing these GXMR-CAR constructs against the target. To this end, we evaluated two scFv specifically for GXM, which comprised 18B7-GXMR-CAR and 2H1-GXMR-CAR constructs. This study demonstrated that 2H1-GXMR-CAR could redirect Jurkat cells to target Cryptococcus spp., as observed in cells modified with 18B7-GXMR-CAR. The cellular activation induced by 2H1-GXMR-CAR showed similar activation marker levels observed in 18B7-GXMR-CAR Jurkat cells co-cultured with reference strains and clinical isolates of Cryptococcus spp. In contrast, tonic signaling triggered by 18B7-GXMR-CAR was significantly more pronounced, and the aggregation level of GXMR-CAR was strongly associated with the strength of tonic signaling. The full activation triggered by 2H1-GXMR-CAR and 18B7-GXMR-CAR in response to Cryptococcus spp. depends on the Src family tyrosine kinase; the GXMR-CAR constructs expressing scFv without a light chain impaired the redirection to Cryptococcus spp., and tonic signaling was abolished. Thus, 2H1-GXMR-CAR showed an advantage in the strength of tonic signaling, maintaining a high capacity to redirect T cells to target Cryptococcus spp.
Evaluation of the modifications in the hinge/transmembrane and intracellular domains of GXMR-CAR, considering the impact on cell activation, was extensively characterized by dos Santos et al. [Citation16] and initially demonstrated an improvement in GXMR-CAR expression was observed oncCD8 replaced IgG4/CD28 as a hinge/transmembrane region. In this study, GXMR-CAR containing the costimulatory molecule CD137 (4-1BB) favored GXMR-CAR expression, cellular expansion, and strong cell activation in response to the target compared to GXMR-CAR containing CD28 as a costimulatory domain [Citation16]. These results led us to work with GXMR-CAR expressing 4-1BB and CD8 as the costimulatory and hinge/transmembrane domains to generate GXMR-CAR variants related to the antigen-binding domain. The GXM-specific 18B7 and 2H1 monoclonal antibodies showed strong reactivity against the antigen [Citation20]. The sequences of the antigen-binding variable region between the 18B7 and 2H1 clones showed high similarity, and the portions that differed in the 18B7 and 2H1 sequences suggested that the 18B7 clone had the highest affinity for GXM [Citation20]. Interestingly, 18B7-GXMR-CAR showed better recognition of soluble GXM obtained from C. gattii, whereas GXM purified from C. neoformans was equally recognized by 18B7-GXMR-CAR and 2H1-GXMR-CAR. However, the ability of 18B7-GXMR-CAR and 2H1-GXMR-CAR to mediate the production of high levels of IL-2 by modified Jurkat cells after incubation with C. gattii or C. neoformans did not correlate with the strength of the interaction with soluble GXM observed for 18B7-GXMR-CAR and 2H1-GXMR-CAR. GXMR-CAR containing 18B7 or 2H1 clones as antigen recognition domains maintained a similar capacity to induce cell activation against Cryptococcus spp. yeast, as validated by CD69 expression and IL-2 production.
A critical phenomenon triggered mainly by the antigen recognition domain of the CAR is associated with tonic signaling, which is characterized by the activation of T cells modified with the CAR, even in the absence of a target. After modifying Jurkat cells, tonic signaling was detected for scFv 2H1 and 18B7 present in the GXMR-CAR constructs. Notably, cells modified with 18B7-GXMR-CAR showed higher levels of tonic signaling than Jurkat cells expressing 2H1-GXMR-CAR. Tonic signaling has been reported in T cells expressing distinct CAR, and tonic signaling promotes the benefits or losses of cell functionality depending on the CAR evaluated and the cell type [Citation52]. A previous study demonstrated that the modification of human CD19-specific scFv with a similar interaction to CD19 abolished tonic signaling and CAR clustering on the cell surface [Citation53], and the modification of the framework regions of scFv affected the CAR tonic signaling that was associated with scFv destabilization and its clustering [Citation54]. In addition, a comparative study between different CD19-CAR constructs containing distinct signal transduction domains demonstrated that tonic signaling changed significantly between the constructs, which affected the in vitro and in vivo proliferation of the modified cells [Citation55]. This study demonstrated that CD19-CAR tonic signaling enhanced the antitumor activity of modified cells; however, CD19-CAR tonic signaling induced negative effects, such as the propensity of modified T cells to enter an exhaustion process that limited the cytotoxic activity on the target [Citation56].
Our results indicate that tonic signaling levels differ significantly between the 2H1-GXMR-CAR and 18B7-GXMR-CAR constructs, reflected in their cellular capacity. The expression of 18B7-GXMR-CAR promoted the highest tonic signaling, which impaired cellular expansion, whereas cells modified with 2H1-GXMR-CAR showed moderate tonic signaling and the expected cellular expansion rate. These findings suggested a correlation between the level of GXMR-CAR tonic signaling induced in modified Jurkat cells and their capacity to proliferate, which was corroborated by a previous study [Citation52]. The expression level of the PD-1 exhaustion marker was significantly increased in cells modified with 18B7-GXMR-CAR compared to that in cells expressing 2H1-GXMR-CAR, confirming that the impairment in cell survival was mainly caused by 18B7-GXMR-CAR expression in Jurkat cells. Elevated PD-1 expression is a well-known parameter related to the reduction of cellular expansion of activated T cells [Citation57].
To investigate the involvement of the signal transduction domains of 18B7-GXMR-CAR and 2H1-GXMR-CAR in the induction of tonic signaling found in T cells, the pharmacological inhibitor dasatinib is usually considered because of its capacity to inhibit the phosphorylation of protein kinases of the Src family [Citation47]. 18B7-GXMR-CAR and 2H1-GXMR-CAR tonic signaling were drastically reduced in response to dasatinib treatment, indicating that the signal transduction domain of GXMR-CAR was responsible for the induction of high levels of IL-2 by modified cells in the absence of ligands. The inhibitor, dasatinib, also mitigated the activation of cells modified with GXMR-CAR in the presence of ligands, and the cellular expansion of cells modified with GXMR-CAR in the absence or presence of Cryptococcus spp. also changed in response to dasatinib treatment. Thus, the strength of tonic signaling and cell activation induced by 18B7-GXMR-CAR and 2H1-GXMR-CAR was reflected in the cellular expansion of modified Jurkat cells. In addition, pioneering studies have evaluated the role of the inhibitor dasatinib in CD19 CAR-T cell therapy, and there was a promising improvement in therapeutic efficacy based on the reported T cell differentiation and exhaustion markers [Citation47]. The application of dasatinib treatment associated with CAR T-cell therapy is made possible by the ability to modulate the effect of dasatinib using different doses, which can regulate the CAR activation rate, reflecting the control of adverse effects arising from therapy with CAR [Citation58].
Considering the distinct CAR with similar specificity, the modification of scFv directly affects the rate of CAR expression on the cell surface and contributes to the aggregation of CAR in the cell membrane, which interferes with antigen recognition. Furthermore, the avidity of CAR for the antigen and the efficiency and stability of CAR expression on the cell surface are influenced by scFv [Citation17]. In addition, additional factors related to post-translational modification can affect protein folding, causing an alteration in the stability of CAR that leads to CAR aggregation on the cell surface, even for constructs containing scFv with high affinity and specificity to the ligands [Citation17].
The absence of a constant region in the antibody that links the light (VL) and heavy (VH) variable chains could be responsible for the alteration in the stability of scFv, favoring the oligomerization of the VL and VH chains, which is a significant mechanism in the induction of CAR tonic signaling [Citation52]. The current study demonstrated that puncta formation on the surface of Jurkat cells modified with 18B7-GXMR-CAR or 2H1-GXMR-CAR suggested a non-uniform distribution of GXMR-CAR (). Initially, we hypothesized that GXMR-CAR tonic signaling was affected by scFv instability, leading to the formation of puncta on the cell surface. Previous studies have reported that CAR tonic signaling is strongly associated with scFv oligomerization on the cell surface, whereas homogeneous CAR distribution does not correlate with tonic signaling events [Citation54]. However, our data demonstrated a similar puncta distribution on the surface of cells expressing 2H1-GXMR-CAR and 18B7-GXMR-CAR, which did not clarify the difference in the strength of tonic signaling between the GXMR-CAR constructs. To further investigate the impact of scFv on puncta formation after GXMR-CAR expression, the Molecular Dynamics (MD) simulations of the scFv of 2H1 and 18B7 were performed. Neither 18B7 nor 2H1 scFv presented an instability that should justify GXMR-CAR oligomerization, even though 18B7 scFv exhibited greater relative movement than 2H1 scFv (Figure S1C and S1D).
The RMSD profiles between the VLVH and VH domains of the antibodies 18B7 and 2H1 (Figure S1A-B) indicated that small changes in the 2H1 and 18B7 scFv structures were mainly caused by modifications in the VH structure. Modification of the GXMR-CAR constructs to express only the VH domain showed that 18B7-GXMR-CAR tonic signaling was significantly reduced but was not abolished, whereas 2H1-GXMR-CAR eliminated tonic signaling. Furthermore, the CDR H3 region located at the VL/VH interface of 18B7 scFv showed more variation than 2H1, and our data demonstrated that the H3 region of 18B7 favors a structural change that affects the stability of 18B7 scFv. In addition, 18B7 scFv contains mutations in the CDR H3 region that strongly alter the binding surface, allowing the expansion of the binding pocket to PA1, which showed a pronounced affinity for GXM compared to 2H1 scFv. Therefore, 18B7-GXMR-CAR and 2H1-GXMR-CAR depend on the VH domain to redirect T cells to target Cryptococcus spp., and the VH region of 18B7 scFv alters its stability, providing more robust tonic signaling.
The current study proposed a comparison between GXMR-CAR constructs containing distinct scFv based on the functional and structural evaluation of the 18B7-GXMR-CAR and Jurkat cells, and this human T cell lineage is often used to the CAR expression aiming the comprehension of essential biological activities of T cells that could be altered after modification with CAR. Moreover, Jurkat cells optimize the study of CAR expression owing to the easy handling of the human T-cell lineage for the transduction and expansion of CAR-positive cells over a short time compared to primary T-cells. As expected in primary CAR T cells, Jurkat cells reproduce the main functional and structural properties induced by CAR expression [Citation59]. The present work opens new perspectives for evaluating PBMC modified with GXMR-CAR constructs. The main findings differed between 18B7-GXMR-CAR and 2H1-GXMR-CAR when expressed in Jurkat cells. Our hypothesis is that the 2H1-GXMR-CAR has some advantage in mediating a better T-cell effector function over time during in vivo Cryptococcus spp. infection, based on the following two points: (i) the 2H1-GXMR-CAR construct triggered a lower strength of tonic signaling and induced high levels of cell activation in the presence of ligands; (ii) 2H1-GXMR-CAR induced lower levels of cell activation after exposure to serum from patients with cryptococcosis that presented soluble GXM, which avoids an early expression of exhaustion markers. Thus, the current study demonstrates that 18B7-GXMR-CAR and 2H1-GXMR-CAR have potential applications in the therapeutic approach against cryptococcosis, which reinforces the need for further studies comparing GXMR-CAR constructs as mediators of the fungicidal activity of T cells.
Conclusion
18B7-GXMR-CAR and 2H1-GXMR-CAR constructs induced high activation of modified Jurkat cells against soluble GXM and Cryptococcus spp. yeasts, and the binding pocket of 18B7 scFv is the major responsible for tonic signaling strength of 18B7-GXMR-CAR and its highest affinity to GMX. The lower levels of cell activation induced by 2H1-GXMR-CAR after incubation with serum from cryptococcosis patients indicated that 2H1-GXMR-CAR has some advantage in the cell therapy using GXMR-CAR.
Author contributions
Conceptualization, M.P.M and T.A.S; methodology, M.P.M, M.H.S, J.G.G, G.Y.C, P.K.M.O.B, C.M.G.F, C.P.R, N.F.F, S.G.S, P.R.K, M.R.L and T.A.S; software, M.P.M, M.H.S, J.G.G, G.Y.C, P.K.M.O.B, C.M.G.F, C.P.R, N.F.F, S.G.S, P.R.K, M.R.L and T.A.S.; writing – original draft preparation, M.P.M, N.F.F, M.R.L and T.A.S; writing – review and editing, M.P.M and T.A.S; supervision, T.A.S.; funding acquisition, P.R.K and T.A.S. All authors have read and agreed to the published version of the manuscript.
Supplemental Material
Download Zip (3.1 MB)Acknowledgments
We thank Prof. Dr. Roberto Martinez (Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil) for providing clinical isolates of C. gattii and C. neoformans. We also thank Profa. Dra. Maria Cristina Roque-Barreira and Prof. Dr. Paulo Coelho (Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil) for providing infra-structure support. We thank Technology Platform of Bioinformatic at the Fiocruz, Ceará, Brazil. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are available from the corresponding author, da Silva TA, upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21655979.2023.2281059
Additional information
Funding
References
- May RC, Stone NRH, Wiesner DL, et al. Cryptococcus: from environmental saprophyte to global pathogen. Nat Rev Microbiol. 2016;14(2):106. doi: 10.1038/nrmicro.2015.6
- Skolnik K, Huston S, Mody CH. Cryptococcal Lung Infections. Clin Chest Med. 2017;38(3):451–26. doi: 10.1016/j.ccm.2017.04.007
- Maziarz EK, Perfect JRIDCNA. Cryptococcosis. Infect Dis Clin North Am. 2016;30(1):179. doi: 10.1016/j.idc.2015.10.006
- Harris J, Lockhart S, Chiller T. Cryptococcus gattii: where do we go from here? Med Mycol. 2012;50(2):113–129. doi: 10.3109/13693786.2011.607854
- O’Halloran JA, Powderly WG, Spec A. Cryptococcosis today: it is not all about HIV infection. Curr Clin Microbiol Rep. 2017;4(2):88. doi: 10.1007/s40588-017-0064-8
- Kozubowski L, Lee SC, Heitman J. Signalling pathways in the pathogenesis of Cryptococcus. Cell Microbiol. 2009;11(3):370–380. doi: 10.1111/j.1462-5822.2008.01273.x
- Shourian M, Qureshi ST. Resistance and tolerance to cryptococcal infection: an intricate balance that controls the development of disease. Front Immunol. 2019;10:66. doi:10.3389/fimmu.2019.00066
- Cherniak R, Valafar H, Morris LC, et al. Cryptococcus neoformans chemotyping by quantitative analysis of 1H nuclear magnetic resonance spectra of glucuronoxylomannans with a computer-simulated artificial neural network. Clin Diagn Lab Immunol. 1998;5(2):146–159. doi: 10.1128/CDLI.5.2.146-159.1998
- Cherniak R, Sundstrom JB. Polysaccharide antigens of the capsule of Cryptococcus neoformans. Infect Immun. 1994;62(5):1507. doi: 10.1128/iai.62.5.1507-1512.1994
- Urai M, Kaneko Y, Ueno K, et al. Evasion of innate immune responses by the highly virulent Cryptococcus gattii by altering capsule glucuronoxylomannan structure. Front Cell Infect Microbiol. 2015;5:101. doi: 10.3389/fcimb.2015.00101
- Ben-Abdallah M, Sturny-Leclère A, Avé P, et al. Fungal-induced cell cycle impairment, chromosome instability and apoptosis via differential activation of NF-κB. PLOS Pathog. 2012;8(3):e1002555. doi: 10.1371/journal.ppat.1002555
- Chai LYA, Van De Veerdonk F, Marijnissen RJ, et al. Anti-Aspergillus human host defence relies on type 1 T helper (Th1), rather than type 17 T helper (Th17), cellular immunity. Immunology. 2010;130(1):46. doi: 10.1111/j.1365-2567.2009.03211.x
- Elsegeiny W, Marr KA, Williamson PR. Immunology of cryptococcal infections: developing a rational approach to patient therapy. Front Immunol. 2018;9:651. doi:10.3389/fimmu.2018.00651
- Iyer KR, Revie NM, Fu C, et al. Treatment strategies for cryptococcal infection: challenges, advances and future outlook. Nat Rev Microbiol. 2021;19(7):454. doi: 10.1038/s41579-021-00511-0
- da Silva TA, Hauser PJ, Bandey I, et al. Glucuronoxylomannan in the Cryptococcus species capsule as a target for Chimeric Antigen Receptor T-cell therapy. Cytotherapy. 2021;23(2):119–130. doi: 10.1016/j.jcyt.2020.11.002
- dos Santos MH, Machado MP, Kumaresan PR, et al. Modification of hinge/transmembrane and signal transduction domains improves the expression and signaling threshold of GXMR-CAR specific to Cryptococcus spp. Cells. 2022;11(21):3386. doi: 10.3390/cells11213386
- Fujiwara K, Masutani M, Tachibana M, et al. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem Biophys Res Commun. 2020;527(2):350–357. doi: 10.1016/j.bbrc.2020.03.071
- Martinez LR, Moussai D, Casadevall A. Antibody to Cryptococcus neoformans glucuronoxylomannan inhibits the release of capsular antigen. Infect Immun. 2004;72(6):3674. doi: 10.1128/IAI.72.6.3674-3679.2004
- Larsen RA, Pappas PG, Perfect J, et al. Phase I evaluation of the safety and pharmacokinetics of murine-derived anticryptococcal antibody 18B7 in subjects with treated cryptococcal meningitis. Antimicrob Agents Chemother. 2005;49(3):952–958. doi: 10.1128/AAC.49.3.952-958.2005
- Casadevall A, Cleare W, Feldmesser M, et al. Characterization of a murine monoclonal antibody to Cryptococcus neoformans polysaccharide that is a candidate for human therapeutic studies. Antimicrob Agents Chemother. 1998;42(6):1437. doi: 10.1128/AAC.42.6.1437
- Casadevall A, Mukherjee J, Devi SJN, et al. Antibodies elicited by a Cryptococcus neoformans-tetanus toxoid conjugate vaccine have the same specificity as those elicited in infection. J Infect Dis. 1992;165(6):1086–1093. doi: 10.1093/infdis/165.6.1086
- Wozniak KL, Levitz SM. Isolation and purification of antigenic components of Cryptococcus. Methods Mol Biol. 2009;470:71.
- Abraham MJ, Murtola T, Schulz R, et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001
- Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput Phys Commun. 1995;91(1–3):43–56. doi: 10.1016/0010-4655(95)00042-E
- Hess B, Kutzner C, Van Der Spoel D, et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable Molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q
- Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055
- Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291
- Huang J, Rauscher S, Nawrocki G, et al. charmm36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods. 2016;14(1):71–73. doi: 10.1038/nmeth.4067
- Young ACM, Valadon P, Casadevall A, et al. The three-dimensional structures of a polysaccharide binding antibody to Cryptococcus neoformans and its complex with a peptide from a phage display library: implications for the identification of peptide mimotopes. J Mol Biol. 1997;274(4):622–634. doi: 10.1006/jmbi.1997.1407
- Baek M, DiMaio, F, Anishchenko, I, Dauparas, J, Ovchinnikov, S, Lee, G.R, Wang, J, Cong, Q., Kinch, L.N, Schaeffer, R.D Millán, C et al. Accurate prediction of protein structures and interactions using a three-track neural network. Sci (1979). 2021;373:871–876.
- Anandakrishnan R, Aguilar B, Onufriev AV. H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012;40(W1):W537–W541. doi: 10.1093/nar/gks375
- Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79(2):926–935. doi: 10.1063/1.445869
- Hess B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J Chem Theory Comput. 2007;4(1):116–122. doi: 10.1021/ct700200b
- Hess B, Bekker H, Berendsen HJC, et al. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1997;18(12):1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463:AID-JCC4>3.0.CO;2-H
- Miyamoto S, Kollman PAS. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem. 1992;13(8):952–962. doi: 10.1002/jcc.540130805
- Essmann U, Perera L, Berkowitz ML, et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103(19):8577–8593. doi: 10.1063/1.470117
- Haug EJ, Arora JS, Matsui K. A steepest-descent method for optimization of mechanical systems. J Optim Theory Appl. 1976;19(3):401–424. doi: 10.1007/BF00941484
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126(1). doi: 10.1063/1.2408420
- Berendsen HJC, Postma JPM, Van Gunsteren WF, et al. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. doi: 10.1063/1.448118
- Daura X, Gademann K, Jaun B, et al. Peptide folding: when simulation meets experiment. Angew Chem Int Ed Engl. 1999;38(1–2):236–240. doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<236:AID-ANIE236>3.0.CO;2-M
- Honorato RV, Koukos PI, Jiménez-García B, et al. Structural Biology in the clouds: the WeNMR-EOSC ecosystem. Front Mol Biosci. 2021;8:729513. doi: 10.3389/fmolb.2021.729513
- Hess B, Kutzner C, Van Der Spoel D, et al. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable Molecular simulation. 2008. doi: 10.1021/ct700301q
- Pronk S, Páll S, Schulz R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845. doi: 10.1093/bioinformatics/btt055
- Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291
- Berendsen HJC, Postma JPM, van Gunsteren WF, et al. Interaction models for water in relation to protein hydration. 1981;pp. 331–342. doi: 10.1007/978-94-015-7658-1_21
- Van Zundert GCP, Rodrigues JPGLM, Trellet M, et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428(4):720–725. doi: 10.1016/j.jmb.2015.09.014
- Zhang H, Hu Y, Shao M, et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J Hematol Oncol. 2021;14(1). doi: 10.1186/s13045-021-01117-y
- Bongomin F, Gago S, Oladele RO, et al. Global and Multi-National Prevalence of Fungal Diseases—Estimate Precision. Journal Of Fungi. 2017;3(4):57. doi: 10.3390/jof3040057
- Dos Santos MH, Machado MP, Kumaresan PR, et al. Titan cells and yeast forms of Cryptococcus neoformans and Cryptococcus gattii are recognized by GXMR-CAR. Microorganisms. 2021;9(9):1886. doi: 10.3390/microorganisms9091886
- Seif M, Kakoschke TK, Ebel F, et al. CAR T cells targeting Aspergillus fumigatus are effective at treating invasive pulmonary aspergillosis in preclinical models. Sci Transl Med. 2022;14(664). doi: 10.1126/scitranslmed.abh1209
- Kumaresan PR, Manuri PR, Albert ND, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci U S A. 2014;111(29):10660–10665. doi: 10.1073/pnas.1312789111
- Ajina A, Maher J. Strategies to address chimeric antigen receptor tonic signalling. Mol Cancer Ther. 2018;17(9):1795. doi: 10.1158/1535-7163.MCT-17-1097
- Sommermeyer D, Hill T, Shamah SM, et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia. 2017;31(10):2191. doi: 10.1038/leu.2017.57
- Landoni E, Fucá G, Wang J, et al. Modifications to the framework regions eliminate chimeric antigen receptor tonic signaling. Cancer Immunol Res. 2021;9(4):441–453. doi: 10.1158/2326-6066.CIR-20-0451
- Milone MC, Fish JD, Carpenito C, et al. Chimeric Receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453. doi: 10.1038/mt.2009.83
- Gomes-Silva D, Mukherjee M, Srinivasan M, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector dependent. Cell Rep. 2017;21(1):17. doi: 10.1016/j.celrep.2017.09.015
- Ostrand-Rosenberg S, Horn LA, Haile ST. The programmed death-1 immune suppressive pathway: barrier to anti-tumor immunity. J Immunol. 2014;193(8):3835. doi: 10.4049/jimmunol.1401572
- Weber EW, Lynn RC, Sotillo E, et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. 2019;3(5):711. doi: 10.1182/bloodadvances.2018028720
- Bloemberg D, Nguyen T, MacLean S, et al. A high-throughput method for characterizing novel chimeric antigen receptors in Jurkat cells. Mol Ther Methods Clin Dev. 2020;16:238–254. doi: 10.1016/j.omtm.2020.01.012