
Abstract
Objective
To evaluate safety, dose response, and preliminary efficacy of reldesemtiv over 12 weeks in patients with amyotrophic lateral sclerosis (ALS). Methods: Patients (≤2 years since diagnosis) with slow upright vital capacity (SVC) of ≥60% were randomized 1:1:1:1 to reldesemtiv 150, 300, or 450 mg twice daily (bid) or placebo; active treatment was 12 weeks with 4-week follow-up. Primary endpoint was change in percent predicted SVC at 12 weeks; secondary measures included ALS Functional Rating Scale-Revised (ALSFRS-R) and muscle strength mega-score. Results: Patients (N = 458) were enrolled; 85% completed 12-week treatment. The primary analysis failed to reach statistical significance (p = 0.11); secondary endpoints showed no statistically significant effects (ALSFRS-R, p = 0.09; muscle strength mega-score, p = 0.31). Post hoc analyses pooling all active reldesemtiv-treated patients compared against placebo showed trends toward benefit in all endpoints (progression rate for SVC, ALSFRS-R, and muscle strength mega-score (nominal p values of 0.10, 0.01 and 0.20 respectively)). Reldesemtiv was well tolerated, with nausea and fatigue being the most common side effects. A dose-dependent decrease in estimated glomerular filtration rate was noted, and transaminase elevations were seen in approximately 5% of patients. Both hepatic and renal abnormalities trended toward resolution after study drug discontinuation. Conclusions: Although the primary efficacy analysis did not demonstrate statistical significance, there were trends favoring reldesemtiv for all three endpoints, with effect sizes generally regarded as clinically important. Tolerability was good; modest hepatic and renal abnormalities were reversible. The impact of reldesemtiv on patients with ALS should be assessed in a pivotal Phase 3 trial. (ClinicalTrials.gov Identifier: NCT03160898)
Introduction
Fast skeletal muscle troponin activators (FSTAs) sensitize the sarcomere to calcium and increase muscle force. This mechanism is of potential relevance in amyotrophic lateral sclerosis (ALS) and other neuromuscular disorders that cause weakness and muscle fatigue. A first-generation FSTA, tirasemtiv, showed promise in phase 2a studies in ALS (Citation1–3) and myasthenia gravis (Citation4). Additionally, a large phase 2b study of tirasemtiv in ALS suggested efficacy by slowing the rates of decline of slow vital capacity (SVC) and isometric muscle strength (Citation5). Dizziness was the most common adverse event (AE) of tirasemtiv, which often resulted in dropout from the study. A subsequent phase 3 trial was designed to reduce the incidence of early termination. The trial failed to show a statistically significant effect on any endpoint; however, large numbers of dose-dependent, early terminations due to poor tolerability still occurred and confounded the interpretation of the trial results (Citation6).
Reldesemtiv is a second generation FSTA derived from a different chemical scaffold than tirasemtiv, with limited penetration of the blood-brain barrier to minimize off-target effects. A single-dose study in healthy participants showed that reldesemtiv had a greater pharmacodynamic effect on muscle force generation with submaximal nerve stimulation frequencies than tirasemtiv, and central nervous system side effects were not noted (Citation7, Citation8). Based on these data, the phase 2b trial, FORTITUDE-ALS (Functional Outcomes in a Randomized Trial of Investigational Treatment with CK-2127107 to Understand Decline in Endpoints – in ALS), was designed to study the safety, tolerability, and preliminary efficacy of three doses of reldesemtiv versus placebo in patients with ALS.
Methods
Patients
This randomized, double-blind, multicentre, dose-ranging, placebo-controlled, phase 2b trial recruited patients from 65 clinical trial sites in the United States, Canada, Ireland, Spain, the Netherlands, and Australia. Patients were between 18 and 80 years of age, and diagnosed within 24 months with possible, laboratory-supported probable, probable, or definite ALS according to the revised El Escorial criteria (Citation9). An upright SVC ≥60% predicted for age, height, sex, and ethnic group at screening was required for inclusion. Patients on riluzole must have taken it for ≥30 days prior to screening. Following protocol amendment 2 (August 10, 2017), patients on edaravone were eligible to enroll in the trial and must have completed ≥2 cycles prior to screening. Exclusion criteria included prior use of reldesemtiv or tirasemtiv or receipt of stem cell or gene therapy for ALS.
Standard protocol approvals, registrations, and patient consents
All patients provided written informed consent, and all sites received institutional review board approvals prior to enrollment. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. FORTITUDE-ALS was registered with ClinicalTrials.gov (NCT03160898) and was conducted between August 2017 and March 2019.
Trial design and assessments
After a screening period of up to 14 days, eligible patients were randomized via an interactive web response system 1:1:1:1 to placebo or reldesemtiv (CK-2127107) oral tablets 150, 300, or 450 mg twice daily (bid) stratified by use and nonuse of riluzole and edaravone. All site clinical staff (investigators, pharmacists, support staff) involved with the study, patients, and the sponsor were blinded to treatment assignment. Study medication was to be taken twice daily, approximately 12 ± 2 hours apart and within 2 h following a meal. The active treatment period was 12 weeks with assessments at day 1 and weeks 2, 4, 8, and 12 and at a follow-up visit 4 weeks after the last dose.
The primary endpoint was the change in percent predicted SVC, from baseline to week 12. To qualify for the study, at screening in addition to having a minimum SVC of 60% of predicted based upon the global lung initiative values (Citation10), patients also had to demonstrate less than 10% variability of the two highest values in five or fewer attempts. All flow volume loops were reviewed by blinded pulmonologists. Secondary endpoints included changes in the ALS Functional Rating Scale-Revised (ALSFRS-R) Total Score and the slope of muscle strength mega-score from baseline to week 12 measured by hand-held dynamometer and by hand grip dynamometry. The following muscle groups were tested bilaterally: elbow flexion, wrist extension, first dorsal interosseous, hip flexion, knee extension, and ankle dorsiflexion. Plasma concentrations for each dose level of reldesemtiv were assessed at day 1 and weeks 2, 4, 8, and 12. Safety assessments included the incidence and severity of treatment-emergent AEs (TEAEs) as recorded using the Medical Dictionary for Regulatory Activities version 20.0 as well as vital signs, clinical laboratory tests, electrocardiograms, Beck Depression Inventory-Fast Screen, Ashworth Score, and physical and neurological examinations.
Statistical analysis
The full analysis set (FAS) consisted of all randomized patients who received any study drug and had a baseline and at least one post-baseline efficacy assessment during the double-blind period. The safety analysis set included all randomized patients who received any study drug. The pharmacokinetics analysis consisted of all randomized patients with at least one evaluable plasma concentration of reldesemtiv.
The primary efficacy analysis hypothesis was that reldesemtiv had a beneficial and dose-dependent effect on function as measured by the change from baseline to week 12 in the percent predicted SVC. To test this hypothesis, a mixed model for repeated measures (MMRM) with a contrast (−5, −1, 3, 3) for the placebo and reldesemtiv 150 mg bid, 300 mg bid, and 450 mg bid dose groups was used (SAS® version 9.4 or greater). The response variable in the model was the change in the percent predicted SVC from baseline to each post-baseline visit. The model also included the terms of treatment, baseline value, pooled site, visit, and randomization stratification factors of baseline riluzole and/or edaravone use/nonuse, as well as treatment-by-visit and baseline-by-visit interactions. An unstructured variance-covariance structure was used in the model. The model included all observed data points from baseline to week 12 for all patients in the FAS with missing values maintained as missing and imputed missing data under the missing at random paradigm and provided the estimates at week 12 from the observed data. For the secondary efficacy outcomes, change from baseline in the ALSFRS-R was analyzed using the same model as above.
The global null hypothesis for the primary and secondary efficacy endpoints were tested in a pre-specified order (as listed above) using a closed testing procedure, and maintained the family-wise error rate at two-sided significance level of 0.05 for all hypotheses tested. No adjustment for multiplicity was made for analyses of all reldesemtiv groups pooled versus placebo, and subgroups defined by patient characteristics that were post hoc exploratory; all p values of statistical significance are nominal. An estimated 445 patients had to be randomized to provide 90% power to detect a 2.75, 5.5, and 5.5 percentage point advantage over placebo for the 150 mg bid, 300 mg bid, and 450 mg bid reldesemtiv dose groups, respectively, in change from baseline of percent predicted SVC, at the end of the double-blind period (week 12). This calculation was based on a two-sided test with α set at 0.05 and an assumed common standard deviation of 14%.
Results
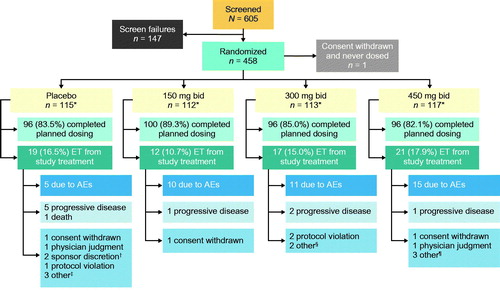
Of the 605 patients screened, 458 were randomized to placebo (n = 115) or reldesemtiv 150 mg bid (n = 112), 300 mg bid (n = 113), or 450 mg bid (n = 117) (one patient was randomized but withdrew before treatment; ); 84.7% completed planned dosing. While early termination from active treatment occurred at similar frequencies in all groups, there was a tendency for increased numbers of patients to withdraw from active treatment due to AEs as a function of increased dose. However, early terminations due to perceived disease progression were more common in the placebo group.
Figure 1 Patient disposition. *All patients randomized contributed to the primary and secondary efficacy analyses, except for 1 patient who withdrew consent right after randomization and did not receive any treatment in the placebo group; all dosed patients contributed to the safety analysis. †2 patients were off study drug too long due to prolonged hospitalization. ‡1 patient no longer wanted to participate in the study due to factors other than the study treatment or study procedures, 1 patient had difficulty traveling to clinic visits, 1 patient withdrew for personal reasons. §1 patient could not continue the study and required visits due to unforeseen work events, 1 patient withdrew due to family circumstances. ¶2 patients did not feel were benefiting from treatment and decided to discontinue, 1 patient had difficulty traveling to clinic visits. AE: adverse event; bid: twice daily; ET: early termination.
Patient characteristics in the reldesemtiv and placebo groups are detailed in . Patients were from the United States (n = 284), Canada (n = 101), Spain (n = 38), Australia (n = 20), the Netherlands (n = 11), and Ireland (n = 4). Baseline demographics were well balanced, with no meaningful differences among the 4 groups. The characteristics resemble those of most previous and current ALS trials, with the exception that time from first symptom to screening was 22.8 ± 19.1 months, which is longer than in many recent trials.(Citation6, Citation11–14) For some trials, symptom onset and not time since diagnosis was the basis of the inclusion criteria, which may contribute to at least some of the differences related to this patient characteristic.(Citation11–14) In addition, 113/457 (24.7%) of patients were on edaravone, either alone or in combination with riluzole. Edaravone was used only by patients in the United States and Canada.
Table 1 Baseline demographics and disease characteristics.
Efficacy
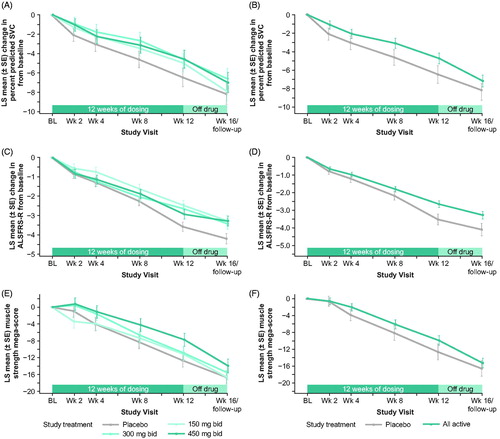
The primary efficacy analysis of change from baseline to week 12 in percent predicted SVC using MMRM did not show a statistically significant weighted dose-response relationship. Placebo-treated patients showed a decline in vital capacity of 6.46 percentage points at week 12, while the patients treated with 150 mg bid, 300 mg bid, and 450 mg bid reldesemtiv showed declines of 4.97 percentage points, 4.62 percentage points, and 4.58 percentage points, respectively (p = 0.11; ). Analyses of changes from baseline in the ALSFRS-R Total Score () and the muscle strength mega-score () using a similar mixed model also did not reach statistical significance (p = 0.09 for change from baseline to week 12 in ALSFRS-R Total Score; p = 0.31 for muscle strength mega-score slope from baseline through week 12), although both measures showed trends toward benefit at all doses. A post hoc comparison of all reldesemtiv-treated patients pooled together versus placebo showed reductions in decline by 27%, 25% and 21% for change from baseline to week 12 in SVC, change from baseline to week 12 in ALSFRS-R Total Score, and muscle strength mega-score from baseline through 12 weeks (nominal p = 0.10, 0.01, and 0.20, respectively; ). As seen in , at the week 16 visit, which was 4 weeks after the study drug was stopped, there was a tendency for beneficial effects for those assigned reldesemtiv to still be present for all three outcome measures, most notably for SVC and ALSFRS-R.
Figure 2 LS mean change from baseline for each group from a mixed model analysis of (A) percent predicted SVC (primary endpoint), (C) ALSFRS-R total score, and (E) muscle strength mega-score. Post hoc analysis of LS mean change from baseline from a mixed model analysis for all reldesemtiv-treated patients versus placebo for (B) percent predicted SVC, (D) ALSFRS-R Total Score, and (F) muscle strength mega-score. ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; bid, twice daily; BL: baseline; LS: least squares; SE: standard error; SVC: slow vital capacity; wk: week.
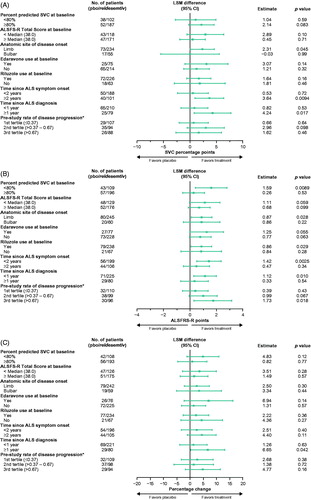
Post hoc comparisons of all reldesemtiv groups pooled versus placebo in subgroups defined by various patient characteristics at baseline showed that trends toward efficacy were seen with virtually all subgroups across all three outcome measures, and no subgroup appeared to be deleteriously affected by reldesemtiv, suggesting that a potentially beneficial effect of reldesemtiv was not driven by a single patient group (). Overall, patients appeared to benefit from reldesemtiv regardless of edaravone or riluzole treatment during the trial. There were no statistical differences between patients receiving reldesemtiv (all doses pooled) compared to the placebo group, irrespective of whether or not they were taking edaravone or riluzole (p = 0.055 to 0.57); patients taking riluzole and receiving reldesemtiv (all doses combined) seemed to have a nominal statistical significant reduction in the change in ALSFRS-R Total Score at 12 weeks compared to placebo (p = 0.029). A statistically significant reduction in the change in ALSFRS-R at 12 weeks on reldesemtiv (all dose levels pooled) versus placebo was observed among patients who had ALS symptom onset less than 2 years prior to baseline (least squares [LS] mean difference ± standard error [SE] 1.4 ± 0.5 for all patients treated with reldesemtiv [n = 199] versus placebo [n = 56]; p = 0.0025) ().
Figure 3 Forest plots from post hoc analyses of LS mean differences between treatment with reldesemtiv and placebo by subgroups for (A) percent predicted SVC, (B) ALSFRS-R, and (C) muscle strength mega-score. *Pretrial reduction of ALSFRS-R total score per month. ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; CI: confidence interval; LSM: least squares mean; pbo: placebo SVC: slow vital capacity.
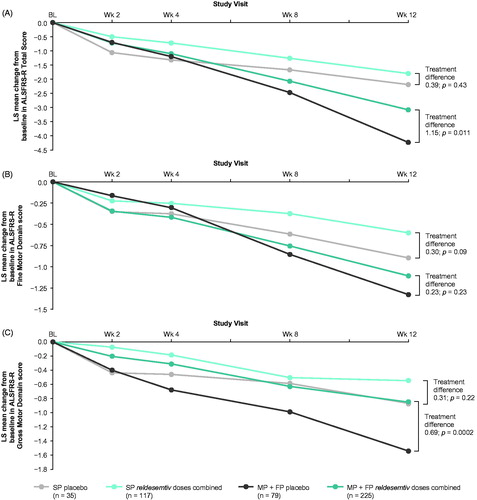
In post hoc analyses, outcomes were also evaluated by estimated rate of disease progression using the date of symptom onset and the ALSFRS-R Total Score at baseline. Patients were sorted into tertiles: slowest (pretrial reduction of ALSFRS-R Total Score ≤0.37 per month), middle (>0.37–0.67 per month), and fastest (>0.67 per month). In the fastest progressing tertile, there was a statistically significant difference between all reldesemtiv dose levels combined and placebo that favored reldesemtiv in the change from baseline in the ALSFRS-R (LS mean difference ± SE 1.7 ± 0.72 for all reldesemtiv-treated patients in that tertile [n = 96] versus the placebo-treated patients in that tertile [n = 30]; p = 0.018) (). In the combined middle and fastest tertiles, change from baseline in ALSFRS-R Total Score at week 12 was significantly smaller in patients who received any dose of reldesemtiv versus placebo (LS mean treatment difference 1.15, p = 0.011); no significant difference between reldesemtiv and placebo was observed in the slowest tertile (). Changes from baseline in the ALSFRS-R Fine and Gross Motor Domain scores showed a similar pattern ()); in the middle and fastest tertiles, a significantly smaller change from baseline in the ALSFRS-R Gross Motor Domain score at week 12 was observed in patients treated with reldesemtiv versus placebo (LS mean treatment difference 0.69, p = 0.0002).
Figure 4 Post hoc analysis of change from baseline in (A) ALSFRS-R Total Score, (B) ALSFRS-R Fine Motor Domain and (C) ALSFRS-R Gross Motor Domain score by progressor tertiles. ALSFRS-R: Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; FP: fastest progressors; LS: least squares; MP: middle progressors; SP: slowest progressors.
Safety
Overall, reldesemtiv was well tolerated (). Serious TEAEs were infrequent; a total of 34 serious TEAEs were equally distributed across all treatment groups and organ systems. The most common serious TEAEs (occurring in >1 patient) regardless of attribution of relationship to study drug are listed in . There was one death in the placebo group during the 12 weeks of active treatment, and two deaths in the 4-week follow-up period (one in the placebo group, one in the reldesemtiv 450 mg bid group). There were six serious TEAEs as defined by the principal investigator that were attributed to study drug; the event terms were hepatoxicity (two patients), alanine transaminase increased (one patient), urinary retention (one patient), transient ischemic attack (one1 patient) and one patient with increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), increased creatinine kinase, and decreased estimated glomerular filtration rate (eGFR, which was based on cystatin C). Clinical TEAEs were common, occurring in 403/457 (88%) patients. Except for fatigue and nausea, which showed a dose-dependent pattern, events were equally distributed across groups (). There was no difference between active treatment and the placebo arms regarding the Beck Depression Inventory results or the Ashworth spasticity scores (data not shown). No clinically relevant abnormalities were observed in ECG measurements during the 12-week active treatment and 4-week follow-up periods.
Table 2 Most common TEAEs (≥10 patients in any treatment group), serious TEAEs (>1 patient), and deaths during the trial.
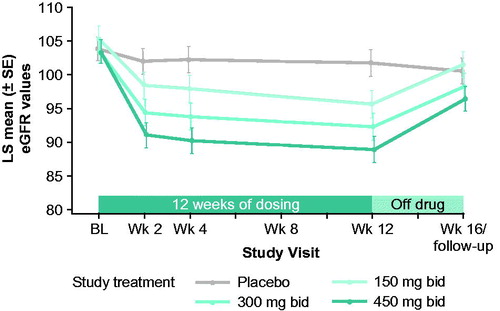
There was a significant dose-dependent relationship noted in both increases in cystatin C and decreases in eGFR determined by cystatin C, with patients showing average reductions in eGFR from baseline to week 12 of 9.2%, 11.2%, and 14.0% in the reldesemtiv 150, 300, and 450 mg bid dose groups, respectively, compared with a small reduction of 2.1% on placebo. Mean eGFR declined to an essentially stable level by 2 weeks of active treatment and tended to recover after 4 weeks off drug (). Manifestations of renal toxicity such as renal tubular casts or elevated urine protein were not seen. Decline in eGFR was the most common reason for early termination of study drug due to AEs, occurring in 7/457 (1.5%) patients. Elevated ALT and AST of at least five times the upper limit of normal were noted in a dose-dependent manner, occurring in six patients, four of whom were on 450 mg bid of reldesemtiv and one each on 150 mg bid and 300 mg bid. Four of the six patients were also on riluzole. For these six patients, when the drug was stopped, values returned to normal for five patients and were improving at the time of last follow-up for one patient. Elevations in ALT and AST were reported as TEAEs in 20/457 (4.4%) and 16/457 (3.5%) patients, respectively ().
Figure 5 eGFR (based on cystatin C) over time. bid: twice daily; BL: baseline; eGFR: estimated glomerular filtration rate; LS: least squares; wk: week.
Pharmacokinetics
Plasma concentrations of reldesemtiv increased with administration of higher dose levels. The highest levels were observed at the 3 h (± 30 min) post-dose time point at week 2 for all doses with geometric mean ± SE values of 1.05 ± 1.07 μg/mL, 2.4 ± 1.06 μg/mL, and 3.79 ± 1.07 μg/mL for reldesemtiv 150, 300, and 450 mg bid, respectively. Compared to a pharmacodynamic translational study in which the common fibular nerve was stimulated at varying frequencies in healthy participants and ankle dorsiflexion force was measured, all three dose levels reached concentrations active in that pharmacodynamic study (Citation7).
Discussion
This phase 2b trial of reldesemtiv in patients with ALS did not meet its pre-specified primary efficacy analysis, despite trends toward efficacy in all functional outcome measures at all dose levels. There are several possible reasons unrelated to the intrinsic effect of reldesemtiv that might explain why the primary endpoint was not reached. First, the primary outcome measure (SVC) in the placebo group in this trial declined more slowly than in most other ALS studies, due to inclusion criteria enriching for slow progressors. Most ALS clinical trials instead try to enrich rapid progressors and have observed a decline in percent predicted SVC of approximately 3 percentage points per month (Citation15–17). In this trial, the decline in SVC in the placebo arm over 12 weeks was 6.46 percentage points, or 2.15 percentage points per 4-week interval. This slower than expected rate of decline in percent predicted SVC reduced the ability to demonstrate a statistically significant effect of reldesemtiv on SVC. Second, the mixed model with multiple contrasts employed in the primary analysis assumed a negligible effect of the lowest dose (150 mg bid) compared with the two higher doses. As the lowest dose appeared to be active in all three key efficacy measures, this multiple contrast approach may not have been the best analysis to demonstrate a treatment effect of reldesemtiv versus placebo. Finally, this trial enrolled more slowly progressing patients than several recent trials (Citation6, Citation11–14), which may have somewhat diluted an efficacy signal by some patients not progressing appreciably during the relatively short 12-week duration of active treatment. Indeed, there appeared to be clear evidence of benefit of reldesemtiv among those patients progressing more rapidly prior to randomization and during the course of the trial.
Results of this trial are consistent across the three key outcome measures, all demonstrating reductions in the rates of decline of 20% or more. When a group of ALS experts was surveyed, slowing of the rate of decline of the ALSFRS-R Total Score by 20% was felt to be at least somewhat clinically meaningful by 93% of respondents, and slowing the decline by 25% or more was felt to be at least somewhat clinically meaningful by 100% of responding clinicians (Citation18). It is unknown whether the magnitude of effect of reldesemtiv will continue to grow with time, but the curves for both SVC and ALSFRS-R seem to be diverging at 12 weeks, so a maximum effect may not have been reached.
With respect to ALSFRS-R, patients manifesting more aggressive disease progression showed a stronger treatment effect than those with more slowly progressing disease. Nominally statistically significant effects of treatment with reldesemtiv were seen in the middle and fastest tertiles of progressors (based on the estimated pretrial rate of disease progression) in both the ALSFRS-R Total Score and ALSFRS-R Gross Motor Domain score at week 12; numerically smaller decreases from baseline were also observed following treatment with reldesemtiv in the middle and fastest progressors in ALSFRS-R Fine Motor Domain, percent predicted SVC, and muscle strength mega-score at week 12. Similarly, patients with symptoms of ALS for less than 2 years or diagnosed for less than 1 year showed a stronger treatment effect, likely because this group includes fewer patients with more indolent disease. In contrast, the change in percent predicted SVC in reldesemtiv-treated patients (all doses pooled) compared to placebo showed benefit favoring reldesemtiv in patients with symptoms for at least 2 years and with diagnosis of ALS for at least one year. This disparity may be related to how these measures change over time; ALSFRS-R decline generally follows a somewhat curvilinear pattern but with decline occurring throughout the disease (Citation19), while SVC may be relatively stable early in the disease (particularly given the mean baseline SVC was 84.7 ± 15.3%) and start to decline later in the disease course (Citation20). Altogether, it appears more likely to demonstrate an effect of reldesemtiv versus placebo in patients who are more rapidly progressing on any measure than in those who are not changing or are worsening slowly. In fact, this reasoning formed the basis for the stringent inclusion/exclusion criteria adopted for the phase 3 edaravone trial aimed at enriching for rapid progressors (Citation21).
Importantly, the estimated effect size of reldesemtiv was not different between patients taking or not taking either riluzole or edaravone, medications approved for ALS, as shown by the non-statistically significant p values for the treatment-by-existing therapy interaction. At week 12, the impact of reldesemtiv compared with placebo was similar regardless of the use/no-use of riluzole or edaravone on percent predicted change in SVC (treatment-by-riluzole use interaction p = 0.90 and treatment-by-edaravone use interaction p = 0.43), change in ALSFRS-R Total Score (treatment-by-riluzole use interaction p = 0.56 and treatment-by-edaravone use interaction p = 0.63), and muscle strength mega-score (treatment-by-riluzole use interaction p = 0.87 and treatment-by-edaravone use interaction p = 0.27, respectively). As ALS is heterogeneous from the pathogenesis standpoint, it is likely that effective ALS treatment will require multiple medications in combination, with any given medication exerting less benefit than the combination. Our data suggest that, should the efficacy of reldesemtiv be confirmed by a pivotal phase 3 trial, it can be effectively combined with existing approved therapies to provide ALS patients with additional benefit.
Also of interest is the observation that 4 weeks after active treatment was stopped, there was still a trend toward benefit in all outcome measures. A similar pattern was noted in the phase 2 BENEFIT-ALS trial of tirasemtiv in ALS, in which the reduction in rate of decline in SVC was also maintained 4 weeks after active treatment was discontinued. A similar persistence of effect 4 weeks after the last dose of reldesemtiv was observed on the 6 min walk test in older children and adults with spinal muscular atrophy (Citation22). This effect cannot be explained by the half-life of the drug or any of its metabolites, so it may represent a disease-modifying effect on either nerve or muscle. It is possible that increasing muscle force and endurance results in a muscle conditioning effect that persists after stopping the study drug. Alternatively, if peripheral nerves do not need to fire as rapidly to achieve a given muscle force, one can hypothesize that motor neuron function may be preserved longer.
The safety and tolerability of reldesemtiv also appear supportive of further development. While clinical AEs were frequent, they were primarily mild and, for the most part, balanced across treatment groups. Nausea and fatigue occurred slightly more frequently in active treatment arms but did not limit continued use of the drug. The reduction in eGFR (based on cystatin C) was clearly related to dose. However, it was not progressive over time, and tended toward resolution by 4 weeks after stopping the study drug. The lack of indicators of renal toxicity such as urinary casts or elevated protein suggests that the eGFR change may be a pharmacodynamic effect rather than true nephrotoxicity. Similarly, elevations of transaminases more than five times the upper limit of normal were rare and normalized after the drug was withdrawn. Overall, the safety findings do not appear limiting to further development of reldesemtiv.
In summary, reldesemtiv is well tolerated and appeared to have a consistent trend toward reducing rates of decline across multiple measures of ALS disease progression. Statistically significant differences were not observed for the pre-specified primary dose-response analyses of primary and secondary endpoints, thus additional studies are warranted to fully evaluate the effect and benefit of reldesemtiv in patients with ALS. Its distinct mechanism of action makes it possible to use in combination with existing and future ALS therapeutics, and further development in a phase 3 trial is planned.
Acknowledgments
We wish to thank the participants of FORTITUDE-ALS and their families for their contributions to this clinical trial, the investigators of FORTITUDE-ALS, and members of the Data Monitoring Committee and Steering Committee. We also wish to thank Dr. Elham Bayat (The George Washington University, Washington, DC), Dr. Richard Bedlack (Duke University School of Medicine, Durham, NC), Dr. Deborah Bradshaw (SUNY Syracuse, Syracuse), Dr. Robert Brown (University of Massachusetts Worcester, Worcester), Dr. Ted Burns (University of Virginia, Charlottesville), Dr. Peter Donofrio (Vanderbilt University Medical Center, Nashville, TN), Dr. Jonathan Glass (Emory University, Atlanta, GA), Dr. Kimberly Goslin (Providence ALS Center, Portland, OR), Dr. Jonathan Katz (California Pacific Medical Center, San Francisco), Dr. Dale Lange (Weill Medical College of Cornell University, New York, NY), Dr. Richard Lewis (Cedar Sinai Medical Center, Los Angeles, CA), Dr. Samuel Maiser (Twin Cities ALS Research Consortium, Minneapolis, MN), Dr. Nicholas Maragakis (Johns Hopkins Hospital, Baltimore, MD), Dr. Hiroshi Mitsumoto (Columbia University Medical Center, New York, NY), Dr. Daniel Newman (Henry Ford Hospital, Detroit, MI), Dr. Alan Pestronk (Washington University, St. Louis, MO), Dr. Ericka Simpson (Houston Methodist, Houston, TX), Dr. Yuen So (Stanford University Medical Center, Palo Alto, CA), Dr. Rup Tandan (University of Vermont, Burlington), Dr. John Turnbull (McMaster University Medical Center, Hamilton, ON), and Dr. Scott A. Vota (Virginia Commonwealth University, Richmond) for their contributions to the study.
Statistical analysis was performed by Lisa Meng and Jenny Wei (Cytokinetics, Inc., South San Francisco, CA, USA).
We thank Kakuri Omari, PhD, CMPP (Evidence Scientific Solutions, Philadelphia, PA) for assistance in collating/incorporating author comments, editing, and formatting, which was funded by Cytokinetics, Inc.
Declaration of interest
JMS received personal compensation from Avexis, Biogen, Brainstorm, Cytokinetics, Mitsubishi Tanabe Pharma America, Neurosense, Orphazyme, Otsuka, and Revalesio; and research support from Amylyx, Brainstorm, Cytokinetics, Medicinova, and Mitsubishi Tanabe Pharma America. JAA served as a consultant for ALS Pharma, Avexis, Biogen and Cytokinetics; is a former employee of Cytokinetics; and received research support from Biogen, Neuraltus, Orion, Roche, Novartis. AG served as a consultant for Alexion, AL-S Pharma, Biogen, Calico, and Cytokinetics. CJ has received grant support from Amylyx and Cytokinetics; served on the DSMB for Anelixis, Brainstorm, and Mallinckrodt; and served as a consultant for Cytokinetics, ITF Pharma, and Mitsubishi Tanabe Pharma America. NL is a consultant for Cytokinetics. TMM is a consultant for Cytokinetics and Disarm Therapeutics; has licensing agreements with C2N Diagnostics and Ionis Pharmaceuticals; and serves on the advisory board and receives research support from Biogen. CB served (uncompensated) on an advisory board for Argenx. BRB has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities from AB Science, Biogen, Biohaven, California ALS Research Summit, Cytokinetics, ITF Pharma, Mitsubishi Tanabe Pharma American, and for serving in an editorial capacity for American Journal of Managed Care; and has received research support from Acceleron, Alexion, Biogen, Biohaven, Boston Scientific, Center for Disease Control, Cytokinetics, ITF Pharma, Medicinova, Mitsubishi Tanabe Pharma America, Neuraltus, Orion, and Santhera. DF has served on an advisory board for Biogen Scientific. OH has received consulting fees from Novartis; research funding from Biogen, Cytokinetics, Ionis and Merck; and is Editor-in-Chief of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. GH served on speaker’s bureau and as a consultant for Alexion, CSL Behring, KabaFusion, and Mitsubishi Tanabe Pharma. TH-P has received consulting fees from Amylyx, Cytokinetics, ITF Pharma and Mitsubishi Tanabe Pharma. RDH served on SMA advisory board for Biogen and Pompe advisory board (Sanofi). WJ received honoraria from Mitsubishi Tanabe Pharma Canada advisory board; and served on clinical trials for Alexion Pharmaceuticals, ALS-Pharma SA, Biogen, Cytokinetics, Mallinckrodt, Mitsubishi Tanabe Pharma Development America, and Orion. CK served on advisory boards for Acceleron, Akcea, Alexion, Alnylam, Argenx, Biogen, CSL Behring, Cytokinetics, and Sanofi Genzyme. MCK receives funding from National Health & Medical Research Council of Australia. SJK served as a consultant for AveXis, Biogen, and Roche and receives grant support from AveXis. GM has research contracts with Cytokinetics, Mallinckrodt, Orion, and University of Calgary; and received fees for speaking, advisory board, and travel from Mitsubishi Tanabe Pharma Canada. MN received honoraria from Biogen, Novartis, and Sanofi for chairing and speaking at professional development meetings. BO serves as a consultant for Biogen, Biohaven, MediciNova, Mitsubishi, and Tsumura; and receives research funding from Biogen, Eisai, Genentech, Mitsubishi, and Orion. EPP received clinical trial and research funding from ALS Association, Iron Horse Diagnostics, and NIH/CDC, and served as consultant to Avanir Pharmaceuticals, Biohaven Pharmaceuticals, Cytokinetics, ITF Pharma, Mitsubishi Tanabe Pharma America, and Otsuka America. MP received honoraria from Argenx, Bioproducts Laboratory, CSL Behring, Catalyst, Grifols, and Stealth BioTherapeutics. DQ received research funding from Alnylam, Argenx, Cytokinetics, Flex Pharma, and Momenta. ZS is a consultant for Biogen and Cytokinetics, and received research support from Biogen. JS received grant funding from FSHD Society, Friends of FSH Research, MDA, and NINDS; is a consultant or on advisory boards for Acceleron, Dyne, FACIO, Fulcrum, Genzyme, Mitsubishi Tanabe Pharma, and Sarepta. AS received research support from Amylyx. LHV serves on scientific advisory boards for Biogen, Cytokinetics, and Orion; received an educational grant from Takeda; serves on the editorial boards of Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration and the Journal of Neurology, Neurosurgery, and Psychiatry; and receives research support from the Netherlands ALS Foundation, and The Netherlands Organization for Health Research and Development (Vici Scheme, JPND [SOPHIA, STRENGTH, ALSCare]). MW received funding from ALSA Association and ALS Finding a Cure; is a speaker for NuFactor; and is a consultant for Argenx and Ra Pharma. JW received research funding from Cytokinetics as part of the study. LZ received honoraria from Biogen, Cytokinetics, and Mitsubishi Tanabe Pharma. BMC owns stock in and was an employee of Cytokinetics during the conduct of the study. LM, JW, AAW, FIM, and SAR own stock and are employees of Cytokinetics. JC, AD, SAG, NAG, DH, LK, SL, JSM, GLP, KR, KLS, DS, CS, SS, TV, SV, and AW-R report no relevant disclosures.
Data availability statement
Data reported herein are part of a sponsor-led clinical development program that is ongoing, and thus complete datasets for the trial will not be made available with this report.
Additional information
Funding
References
- Shefner J, Cedarbaum JM, Cudkowicz ME, Maragakis N, Lee J, Jones D, et al. Safety, tolerability and pharmacodynamics of a skeletal muscle activator in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13:430–8.
- Shefner JM, Watson ML, Meng L, Wolff AA. A study to evaluate safety and tolerability of repeated doses of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:574–81.
- Shefner JM, Wolff AA, Meng L. The relationship between tirasemtiv serum concentration and functional outcomes in patients with ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14:582–5.
- Sanders DB, Rosenfeld J, Dimachkie MM, Meng L, Malik FI. A double-blinded, randomized, placebo-controlled trial to evaluate efficacy, safety, and tolerability of single doses of tirasemtiv in patients with acetylcholine receptor-binding antibody-positive myasthenia gravis. Neurotherapeutics. 2015;12:455–60.
- Shefner JM, Wolff AA, Meng L, Bian A, Lee J, Barragan D, et al. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:426–35.
- Shefner JM, Cudkowicz ME, Hardiman O, Cockcroft BM, Lee JH, Malik FI, et al. A phase III trial of tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;0:1–594.
- Andrews JA, Miller TM, Vijayakumar V, Stoltz R, James JK, Meng L, et al. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2018;57:729–34.
- Hansen R, Saikali KG, Chou W, Russell AJ, Chen MM, Vijayakumar V, et al. Tirasemtiv amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve. 2014;50:925–31.
- Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9.
- Quanjer PH, Stanojevic S, Cole TJ, Baur X, Hall GL, Culver BH, et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur Respir J. 2012;40:1324–43.
- Cudkowicz ME, van den Berg LH, Shefner JM, Mitsumoto H, Mora JS, Ludolph A, et al. Dexpramipexole versus placebo for patients with amyotrophic lateral sclerosis (EMPOWER): a randomised, double-blind, phase 3 trial. Lancet Neurol. 2013;12:1059–67.
- Cudkowicz ME, Titus S, Kearney M, Yu H, Sherman A, Schoenfeld D, et al. Safety and efficacy of ceftriaxone for amyotrophic lateral sclerosis: a multi-stage, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:1083–91.
- Aggarwal SP, Zinman L, Simpson E, McKinley J, Jackson KE, Pinto H, et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:481–8.
- Macchi Z, Wang Y, Moore D, Katz J, Saperstein D, Walk D, et al. A multi-center screening trial of rasagiline in patients with amyotrophic lateral sclerosis: possible mitochondrial biomarker target engagement. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:345–52.
- Andrews JA, Meng L, Kulke SF, Rudnicki SA, Wolff AA, Bozik ME, et al. Association between decline in slow vital capacity and respiratory insufficiency, use of assisted ventilation, tracheostomy, or death in patients with amyotrophic lateral sclerosis. JAMA Neurol. 2018;75:58–64.
- Miller RG, Moore DH, Forshew DA, Katz JS, Barohn RJ, Valan M, Bromberg MB, et al. Phase II screening trial of lithium carbonate in amyotrophic lateral sclerosis: examining a more efficient trial design. Neurology 2011;77:973–9.
- Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–53.
- Castrillo-Viguera C, Grasso DL, Simpson E, Shefner J, Cudkowicz ME. Clinical significance in the change of decline in ALSFRS-R. Amyotroph Lateral Scler. 2010;11:178–80.
- Thakore NJ, Lapin BR, Pioro EP. Trajectories of impairment in amyotrophic lateral sclerosis: Insights from the Pooled Resource Open-Access ALS Clinical Trials cohort. Muscle Nerve. 2018;57:937–45.
- Enache I, Pistea C, Fleury M, Schaeffer M, Oswald-Mammosser M, Echaniz-Laguna A, et al. Ability of pulmonary function decline to predict death in amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:511–8.
- Writing Group, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2017;16:505–12.
- Rudnicki SA, Andrews JA, Malik FI, Wolff AA, Day JW. Update of CY 5021: a phase 2 clinical trial of reldesemtiv, a fast skeletal muscle troponin activator (FSTA), for the potential treatment of spinal muscular atrophy. Presented at the 2018 SMA Researcher Meeting, June 14–16, 2018, Dallas, Texas.