
Abstract
Objective: To understand current genetic testing practices at Canadian ALS clinics. Methods: An online survey and phone interviews, with clinicians practicing in 27 ALS clinics in Canada, were employed to collect data. Quantitative and qualitative analyses were conducted. Results: Ninety-three percent (25/27) of ALS clinics in Canada are routinely ordering genetic testing for familial ALS, while 33% (9/27) of clinics are routinely ordering genetic testing for sporadic ALS. Barriers to genetic testing include a perceived lack of an impact on treatment plan, difficulty in obtaining approvals, primarily from provincial Ministries of Health, and limited access to genetic counseling. Predictive testing practices were found to be the most variable across the country. The average wait time for a symptomatic patient living with ALS to see a genetic counselor in Canada is 10 months (range 0–36 months). Conclusions: Access to genetic testing, and testing practices, vary greatly across Canadian ALS clinics. There may be patients with a monogenetic etiology to their ALS who are not being identified given that genetic testing for patients diagnosed with ALS is not routinely performed at all clinics. This study highlights potential inequities for patients with ALS that can arise from variability in health care delivery across jurisdictions, in a federally-funded, but provincially-regulated, health care system. Clinical trials for both symptomatic ALS patients and pre-symptomatic ALS gene carriers are ongoing, and ALS clinicians in Canada are motivated to improve access to genetic testing for ALS.
Introduction
Although most cases of ALS are believed to be sporadic (90–95%), a subset are familial (5–10%) (Citation1). There is no clear definition of familial ALS (fALS), and it is clinically indistinguishable from sporadic ALS (sALS) (Citation2,Citation3). Despite this, genetic testing practices for fALS and sALS vary; a global survey of ALS clinicians revealed that 90.2% order genetic testing for fALS, while only 49.4% offer testing for sALS (Citation4). There are numerous reasons why a family history of the disease could be missed (Citation3), and with similar clinical presentations between fALS and sALS, patients with a genetic mutation could be misclassified as an isolated, sporadic case of ALS. Depending on the genetic testing practices of the clinic, these patients could be excluded from access to genetic testing.
There is a marked increase in the number of genetically-targeted therapies entering the drug development pipeline (Citation5). Incidence of ALS-associated genetic mutations in seemingly sporadic ALS, where no family history is documented or apparent, is reported and not infrequent (Citation3,Citation6–8). There is an emerging appreciation for more widespread access to genetic testing in facilitating access to genetically-targeted therapies (Citation9,Citation10).
Addressing this issue requires a comprehensive understanding of local genetic testing practices and clinician perspectives. We hypothesize that genetic testing practices in Canadian ALS clinics are inconsistent because the Canadian health care system is federally funded, but provincially regulated. This pan-Canadian landscape assessment was performed as the initial step of a quality improvement initiative to improve access to genetic testing for Canadians living with ALS.
Methods
The protocol was reviewed by the McGill University Health Center Research Ethics Board (REB) – Neuroscience-Psychiatry Panel, and received a waiver given the quality improvement nature of the project. Given the waiver, consent from the participating clinicians was not required.
Sample
ALS clinics in Canada were primarily identified through the Canadian ALS Research Network (CALS), which is composed of 20 ALS clinics across the country. As per the network’s Terms of Reference, these are “multidisciplinary ALS clinics that participate in clinical research and adhere to the best practice clinical care guidelines.”
In addition, other Canadian ALS clinics that are not members of CALS, but provide routine care to patients with ALS, were identified through the provincial and national ALS societies. Ten such clinics were identified and will herein be referred to as non-CALS clinics.
Three community neurologists, from three different provinces, who routinely diagnose patients with ALS were interviewed, and revealed that genetic testing would not typically be ordered by a community or general neurologist who is referring the patient to a specialty clinic for their subsequent care. Therefore, data collection for this study were focused on ALS clinics alone, and not extended to the neurology community at large.
In some instances, multiple clinicians within a clinic participated in the survey and/or interview. Data presented are representative of the clinic as a whole.
Data collection
An online survey was developed covering the following four topics: (i) clinic demographics; (ii) genetic testing of symptomatic individuals; (iii) predictive testing for at-risk, asymptomatic family members; and (iv) pathways and barriers to genetic testing within the institution and/or province of practice. The survey consisted of 28 questions, including multiple choice using skip-logic, and free text responses. Some survey questions were conditional or optional.
Following completion of the survey, semi-structured phone interviews were conducted. Some questions were consistent for all clinician interviews, while others were developed based on individual survey responses, and tailored to the clinic and province of practice.
Data analysis
Quantitative data were analyzed using descriptive statistics. Qualitative data were analyzed using summative content analysis. Summative content analysis was achieved by counting and comparing keywords from survey free text and interview responses, followed by interpretation of the underlying context in which these keywords were used.
Results
All twenty (20/20) of the CALS clinics responded to the survey, and 7/10 non-CALS clinics responded. Of the 27 clinics that responded to the survey, 25 clinics accepted to participate in the phone interviews.
Clinic demographics
There are 2500–3000 Canadians living with ALS at any given time (Citation11). Patient volume from the responding non-CALS clinics represented less than 5% of total patient volume across all responding clinics. Therefore, assuming that the three non-CALS clinics that did not respond to the survey also follow a small volume of patients, and that all patients living with ALS in Canada are followed in an ALS clinic, it is estimated that the collected data are representative of care for more than 90% of Canadian ALS patients.
Two CALS clinics reported having a geneticist on their multidisciplinary care team, and one CALS clinic reported a genetic counselor on their team.
Genetic testing of symptomatic ALS cases
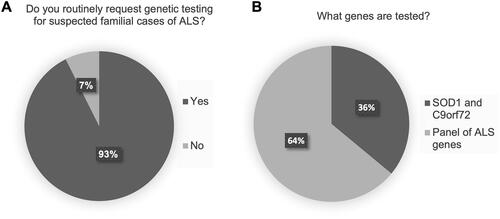
Ninety-three percent (25/27) of clinics routinely offer genetic testing for suspected cases of fALS (). Of those clinics that routinely offer genetic testing to fALS, more than half (64%, 16/25) order a comprehensive panel of ALS-associated genes. The remaining clinics (36%, 9/25) order a limited panel of SOD1 sequencing and C9orf72 repeat expansion analysis only (). The clinicians who are not routinely ordering genetic testing for fALS cited various reasons for the primary barrier to testing: “ALS genetics are poorly understood,” “pathway to request testing and have it approved is unclear,” “lack of implication for treatment plan,” “lack of access to genetic testing,” and “patients decline testing.”
Figure 1 Genetic testing practices for suspected familial cases of ALS (fALS). (A) Routine genetic testing for fALS. (B) For clinics routinely ordering genetic testing for fALS, proportion of clinics ordering SOD1 sequencing and C9orf72 repeat expansion testing only, versus those ordering a broad panel of ALS genes, including SOD1 and C9orf72 (note that composition of panels vary across the laboratories used).
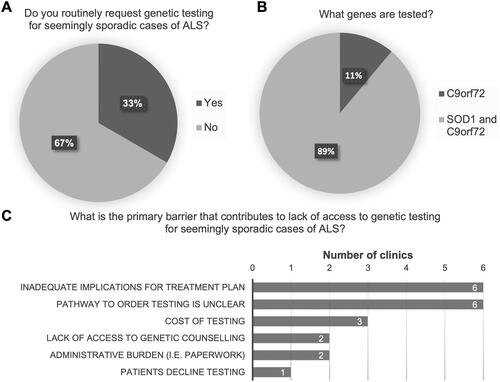
Thirty-three percent (9/27) of clinics are routinely ordering genetic testing for seemingly sporadic cases of ALS (); the majority of which (89%, 8/9) request both SOD1 sequencing and C9orf72 repeat expansion analysis (). The most frequently cited barriers were “inadequate implications for a treatment plan,” and the “pathway to request testing and have it approved is unclear” ().
Figure 2 Genetic testing practices for seemingly sporadic cases of ALS (sALS). (A) Routine genetic testing for sALS. (B) For clinics routinely ordering genetic testing for sALS, proportion testing C9orf72 only, versus those testing for both SOD1 and C9orf72. (C) Primary barriers cited to accessing genetic testing for sALS cases.
Approximately two-thirds of clinics (63%, 17/27) have access to a genetic counselor, either within their own clinic or through referral. For those clinics, the average reported wait time, from sending the referral, for a symptomatic ALS patient to see a genetic counselor was 10 months, with a range from 0 to 36 months.
Predictive genetic testing practices in ALS
Predictive testing practices vary greatly among the clinics surveyed. Thirty-seven percent (10/27) of clinics offer predictive testing to at-risk, asymptomatic family members of an ALS patient with a confirmed genetic mutation. Forty-one percent (11/27) of clinics indicated that predictive testing is performed “maybe/sometimes,” while the remaining 22% (6/27) selected “no” ().
Figure 3 Predictive testing practices for at-risk, asymptomatic family members. (A) Proportion of clinics offering predictive testing to at-risk, asymptomatic family members of a symptomatic ALS patient with a confirmed genetic etiology to their ALS. (B) Circumstances under which predictive testing is offered. (C) Specialties that handle predictive testing cases. (D) Primary barriers cited to accessing genetic testing for at-risk, asymptomatic family members.
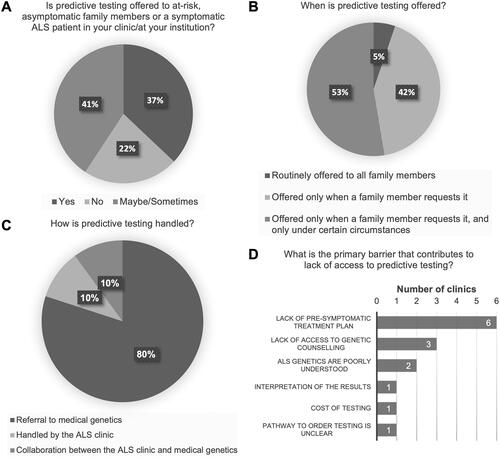
For those clinics that responded “maybe/sometimes,” the following questions were optional. Of those clinics that do offer predictive testing, the majority (95%, 18/19) offer predictive testing only when there is an explicit request from a family member (). Additionally, some of those clinics (10/18) will only offer testing following the request under limited circumstances (e.g., access to a medical genetics department to support counseling). Most clinics (80%, 16/20) refer at-risk, asymptomatic family members interested in predictive testing to a medical genetics department, or specialist, for a consult (). The remaining clinics either handle the predictive testing internally within the clinic (2/20), or work in collaboration with their medical genetics department (2/20). The most frequently cited barrier to offering predictive testing was that there is a “lack of pre-symptomatic treatment plan,” followed by the “lack of access to genetic counseling” (). If predictive testing confirms a gene carrier, only 30% (8/27) of clinics reported that they will provide prospective follow-up.
Other analyses
For provinces with more than one ALS clinic, data obtained from all the clinics within that province were compared. Genetic testing practices within a single province were found to be largely consistent. However, practices were more variable between the provinces. This data were not shown to maintain anonymity, as some provinces had only a single clinic.
Clinician perspectives on genetic testing and implications for treatment
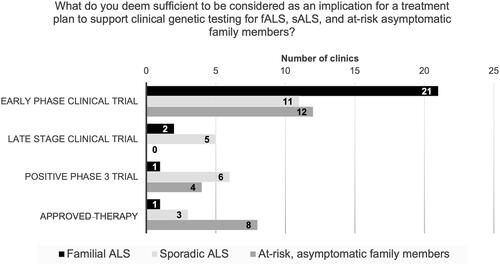
Clinicians were queried as to what they deemed sufficient to be considered an implication for a treatment plan, that would warrant clinical genetic testing: early-phase clinical trial, late-stage clinical trial, positive Phase 3 trial, or an approved therapy. Responses to this question were solicited with respect to fALS, sALS, and at-risk, asymptomatic family members.
Most clinicians (84%, 21/25) consider early phase clinical trials to be sufficient to warrant clinical genetic testing of fALS. Sixty-four percent (16/25) support testing of sALS for clinical trial purposes, for both early-phase (11/16) and late-stage trials (5/16). Fifty percent (12/24) want either a positive Phase 3 trial (4/24), or an approved therapy (8/24), before they would consider routinely offering genetic testing to at-risk, asymptomatic family members (). Of the remaining clinicians (12/24) who indicated that they would support routine clinical genetic testing for at-risk asymptomatic family members for clinical trials, 50% (6/12) stated that they would require the support of their medical genetics department.
Figure 4 Clinician perspectives on genetic testing and implication for a treatment plan. Clinicians were queried as to what they deem sufficient to be considered as an implication for a treatment plan to support clinical genetic testing in the familial ALS (fALS), sporadic (sALS), and at-risk, asymptomatic family member populations. Clinicians selected only one option out of: early phase clinical trial, late-stage clinical trial, positive Phase 3 trial, and approved therapy.
Summative content analysis
Survey free text and interview responses were analyzed using summative content analysis. The theme of “genetic counseling” was the most prevalent (23/27), with clinicians referencing “access to,” “lack of access,” its “importance in the process of ordering genetic testing,” and perceptions of “time sensitive” or “urgent” when evaluating “wait time.” “Lack of access” to (9/27) and “wait time” to see a genetic counselor (11/27) were often referred to when describing barriers to accessing genetic testing for their patients. When describing their process for ordering clinical genetic testing for a symptomatic ALS patient, 17/27 clinicians (63%) stated that they order the testing themselves, bypassing a genetics specialist entirely, if allowed by their province and institution, and perform the consent and pre-test counseling with the patient.
The two primary themes that emerged from data related to genetic testing of fALS were the “wait time [for genetic counseling and/or approvals]” (15/27) and references to “paperwork” (9/27). Selected quotes include: “Given the wait time for medical genetics (2+ years), the value is unclear.”; and “Not straight forward, lots of paperwork.”
Themes related to genetic testing of sALS were the “process [to have the testing performed]” (9/27) and “approvals” (14/27). Some selected quotes include: “Not currently testing, but considering changing practices. Uncertain if it would be approved”; “Each request requires Ministry of Health (MoH) approval. I fear that my testing practices will be flagged by the MoH if I increase my testing to include sporadic cases, and have my familial cases then denied.”; and “I’m not allowed to order testing myself, yet the wait time for a consult with medical genetics is 6 to 9 months.”
Across multiple aspects of genetic testing for ALS, the desire to “improve access to genetic testing” for ALS was raised by multiple clinicians (10/27), as well as the development of formal “consensus guidelines” (5/27).
Discussion
This study was designed to comprehensively assess genetic testing practices in Canadian ALS clinics, and it is estimated that it captured data and perspectives for more than 90% of ALS care in the country. A focused study of Canadian ALS clinics in this regard has not been previously conducted, nor have Canadian clinics been adequately represented in previously published studies. A 2017 global survey of genetic testing practices received 11 responses from Canadian clinicians (Citation4), whereas our study endeavored to identify all clinics in the country providing prospective care to patients living with ALS. Our findings on the proportion of clinics testing fALS and sALS are comparable to the data obtained from similar, global studies (Citation4,Citation12,Citation13).
In addition, this study evaluates genetic testing practices in ALS within a universal health care system. Canada has a federally-funded, but provincially regulated, health care system, and as such there are multiple different jurisdictions overseeing health care delivery. Our study highlights potential disadvantages that can arise from variability in health care delivery across jurisdictions (Citation14). Genetic testing practices among clinics within the same province were found to be, for the most part, consistent. However, differences in genetic testing practices between provinces are notable, as explained above. These differences can largely be attributed to provincial regulations surrounding ordering of genetic testing, leading to inequities in access for Canadian ALS patients. Access to genetic testing in Canadian provinces will be explored in a separate publication.
There are ongoing clinical trials with therapies targeting symptomatic individuals with ALS-associated genetic alterations, specifically SOD1 (NCT02623699) and FUS (NCT04768972) mutations, and C9orf72 (NCT03626012 & NCT04931862) and ATXN2 (NCT04494256) repeat expansions. Our data shows that not all cases of suspected fALS, where there is a reported family history, are being offered clinical genetic testing (). Given this, there are Canadian ALS patients, with clinically actionable mutations, who are not being identified. Of those clinics that are testing fALS, a substantial subset (36%) are ordering clinical genetic testing for SOD1 and C9orf72 only (), thereby overlooking other potentially important genetic alterations, such as ATXN2 and FUS. This may be due to test availability within a province, obtaining testing approval, or individual clinician practice.
Our data shows that more than a third of ALS clinics in Canada are routinely performing clinical genetic testing for seemingly sporadic cases of ALS, which accounts for 90–95% of ALS patients (). However, the most frequently cited barrier for those clinics not testing sALS—lack of implication for a treatment plan—is inconsistent with the current drug development pipeline and active clinical trials of genetically-targeted therapies. The fact that 64% of clinicians deemed early- and late-stage clinical trials to be sufficient to warrant clinical genetic testing of sALS points to possible deficiencies in access at some clinics. It should be noted that family history of ALS is not an inclusion criteria for any of the aforementioned clinical trials. For the four ALS-associated mutations with therapies currently in development, SOD1, C9orf72, FUS and ATXN2, markedly, the combined incidence in the sALS population ranges from 10% to 15% (Citation6,Citation15–17).
This study has highlighted the critical need to alleviate barriers to accessing genetic testing for all patients diagnosed with ALS in Canada. A recent study demonstrated that although the proportion of patients with SOD1 and C9orf72 is higher in fALS, the majority of cases may be found among sALS (Citation8). Feasibility of implementing more widespread testing in the sALS population needs to be prioritized.
Predictive testing practices were the most variable. Considerations for predictive testing in ALS have been previously published (Citation18–21); however, there is a need for global input and further development, given the recent advances in ALS genetics. In addition, there is a lack of research surrounding the psychological impact of predictive testing for ALS (Citation21). Seventy percent (19/27) of clinics indicated that they do not provide prospective follow-up to at-risk, asymptomatic family members who obtain positive predictive testing results. When these cases are referred to a medical genetics department for a consult (80%, 16/20 ), it is unclear to ALS clinicians what, if any, follow-up is provided. As such, it can be extrapolated that there are families with known genetic etiologies for ALS that could be lost to follow-up. A first-of-its-kind clinical trial for pre-symptomatic SOD1 gene mutation carriers is ongoing (NCT04856982), emphasizing the urgent need for better predictive testing practices in ALS.
The results of this survey also indicated that there are Canadians living with ALS who either do not have access to a genetic counselor, or for whom the wait time to be seen by a genetics specialist is too long for the rapid pace with which this disease progresses. With a prognosis of 2–3 years (Citation22), and an often lengthy time from symptom onset to diagnosis (Citation11), the average reported wait time of 10 months consumes a critical portion of early disease. Clinical trial criteria often emphasize reducing heterogeneity of participants, and treating early to reduce motor neuron loss, by selecting for patients who are earlier in their disease course (Citation23). Therefore, wait time to see a genetics specialist has the potential for a direct impact on clinical trial participation. Delays in access to genetic counseling are likely anticipated by clinicians, as 63% order genetic testing (where permitted) and perform pre-test counseling themselves.
One limitation of this study is recall bias due to it being survey based. Although comprehensive, the sample size precludes performing more complex or advanced statistical analyses. Another limitation is that these results represent perspectives of ALS clinicians only. Obtaining input from the genetic counseling and patient communities in Canada is an ongoing effort. Pre-natal testing was not within scope of this study, but represents a meaningful topic of future research.
Finally, a key point that was raised among Canadian ALS clinicians was a lack of recommendations for genetic testing in published guidelines for the clinical management ALS. The most recent Canadian and U.S. guidelines do not address genetic testing (Citation24,Citation25), and the European guidelines indicate that clinical genetic testing should be reserved for those patients with a family history of ALS (Citation18). Given the current therapeutic landscape, addressing genetic testing for ALS in the form of consensus guidelines is an area of great need.
Acknowledgements
The authors thank all clinicians that responded to the survey and took time to participate in the phone interviews, and Trisha Rao for copyediting during the drafting and revising of this manuscript.
Data availability statement
The data that support the findings of this study are available on request. The raw data are not publicly available to protect the anonymity of the clinics that participated.
Declaration of interest
K. Salmon, N. Anoja, A. Breiner, M. Chum, A. Dionne, N. Dupré, A. Fiander, D. Fok, A. Ghavanini, S. Gosselin, A. Izenberg, S. Kalra, G. Matte, M. Melanson, B. Ritsma, C. Shoesmith, S. Tremblay and H. Williams report no conflicts of interest. C. O’Connell has consulting agreements with Biogen and MT Pharma Canada. W. Johnston serves on advisory boards for Amylyx, Biogen, Cytokinetics, and MT Pharma Canada. K. Schellenberg has served on an advisory board for Biogen. A. Genge serves as the Chief Medical Officer of QurAlis, and has consulting agreements with AB Sciences, AL-S Pharma, Alexion, Biogen, Cytokinetics, MT Pharma America, MT Pharma Canada, Novartis, Orion, Revalesio, and Roche.
Additional information
Funding
References
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–55.
- Byrne S, Bede P, Elamin M, Kenna K, Lynch C, McLaughlin R, et al. Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:157–9.
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–15.
- Vajda A, McLaughlin RL, Heverin M, Thorpe O, Abrahams S, Al-Chalabi A, et al. Genetic testing in ALS: a survey of current practices. Neurology. 2017;88:991–9.
- Hardiman O, van den Berg LH. The beginning of genomic therapies for ALS. N Engl J Med. 2020;383:180–1.
- Perrone B, Conforti FL. Common mutations of interest in the diagnosis of amyotrophic lateral sclerosis: how common are common mutations in ALS genes? Expert Rev Mol Diagn. 2020;20:703–14.
- van Es MA, Dahlberg C, Birve A, Veldink JH, van den Berg LH, Andersen PM. Large-scale SOD1 mutation screening provides evidence for genetic heterogeneity in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2010;81:562–6.
- Brown CA, Lally C, Kupelian V, Flanders WD. Estimated prevalence and incidence of amyotrophic lateral sclerosis and SOD1 and C9orf72 genetic variants. Neuroepidemiology. 2021;55:1–12.
- Shepheard SR, Parker MD, Cooper-Knock J, Verber NS, Tuddenham L, Heath P, et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(5):510–518.
- Grassano M, Calvo A, Moglia C, Brunetti M, Barberis M, Sbaiz L, American Genomic Center, et al. Mutational analysis of known ALS genes in an Italian population-based cohort. Neurology. 2021;96:e600–e9.
- Hodgkinson VL, Lounsberry J, Mirian A, Genge A, Benstead T, Briemberg H, et al. Provincial differences in the diagnosis and care of amyotrophic lateral sclerosis. Can J Neurol Sci. 2018;45:652–9.
- Klepek H, Nagaraja H, Goutman SA, Quick A, Kolb SJ, Roggenbuck J. Lack of consensus in ALS genetic testing practices and divergent views between ALS clinicians and patients. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:216–21.
- Arthur KC, Doyle C, Chio A, Traynor BJ. Use of genetic testing in amyotrophic lateral sclerosis by neurologists. JAMA Neurol. 2017;74:125–6.
- Martin D, Miller AP, Quesnel-Vallée A, Caron NR, Vissandjée B, Marchildon GP. Canada's universal health-care system: achieving its potential. Lancet. 2018;391:1718–35.
- Srinivasan E, Rajasekaran R. A systematic and comprehensive review on disease-causing genes in amyotrophic lateral sclerosis. J Mol Neurosci. 2020;70:1742–1770.
- Wang MD, Gomes J, Cashman NR, Little J, Krewski D. Intermediate CAG repeat expansion in the ATXN2 gene is a unique genetic risk factor for ALS-a systematic review and meta-analysis of observational studies. PLoS One. 2014;9:e105534.
- Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88:540–9.
- , Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis A, P. M, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)-revised report of an EFNS task force. Eur J Neurol. 2012;19:360–75.
- Andersen PM, Borasio GD, Dengler R, Hardiman O, Kollewe K, Leigh PN, et al. Good practice in the management of amyotrophic lateral sclerosis: clinical guidelines. An evidence-based review with good practice points. EALSC Working Group. Amyotroph Lateral Scler. 2007;8:195–213.
- Benatar M, Stanislaw C, Reyes E, Hussain S, Cooley A, Fernandez MC, et al. Presymptomatic ALS genetic counseling and testing: experience and recommendations. Neurology. 2016;86:2295–302.
- Crook A, Williams K, Adams L, Blair I, Rowe DB. Predictive genetic testing for amyotrophic lateral sclerosis and frontotemporal dementia: genetic counselling considerations. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:475–85.
- Oskarsson B, Gendron TF, Staff NP. Amyotrophic lateral sclerosis: an update for 2018. Mayo Clin Proc. 2018;93:1617–28.
- van Eijk RPA, Westeneng HJ, Nikolakopoulos S, Verhagen IE, van Es MA, Eijkemans MJC, et al. Refining eligibility criteria for amyotrophic lateral sclerosis clinical trials. Neurology. 2019;92:e451–e460.
- Shoesmith C, Abrahao A, Benstead T, Chum M, Dupre N, Izenberg A, et al. Canadian best practice recommendations for the management of amyotrophic lateral sclerosis. CMAJ. 2020;192:E1453–E68.
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Quality Standards Subcommittee of the American Academy of Neurology, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73:1218–26.