
Abstract
Amyotrophic lateral sclerosis and Parkinson’s disease are neurodegenerative diseases of the motor system which are now recognized also to affect non-motor pathways. Non-motor symptoms have been acknowledged as important determinants of quality of life in Parkinson’s disease, and there is increasing interest in understanding the extent and role of non-motor symptoms in amyotrophic lateral sclerosis. We therefore reviewed what is known about non-motor symptoms in amyotrophic lateral sclerosis, using lessons from Parkinson’s disease.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration of motor neurons in the brain and spinal cord, leading to death in 3 to 5 years (Citation1). Genetic and environmental factors contribute to pathogenesis (Citation2), with a heritability of up to 60% (Citation3). There is considerable variation in motor symptoms, which may include limb weakness, slurred speech, swallowing difficulties, and muscle twitching (Citation4). Although cognitive involvement in the form of frontotemporal impairment is well recognized in ALS (Citation5,Citation6), other non-motor symptoms also cause significant distress and worsen prognosis and quality of life (Citation7). ALS can therefore be considered a multisystem disorder, although a clear characterization of its non-motor aspects and the mechanisms behind them are yet to be elucidated (Citation7).
Parkinson’s disease (PD) is an adult-onset neurodegenerative disorder with a largely unknown etiology. Its diagnosis is made by the presence of cardinal motor manifestations: bradykinesia, rigidity, and rest tremor (Citation8). The significant contribution of non-motor symptoms to the clinical picture is now well established, and research on the non-motor aspects of the disease has greatly increased during the last 20 years. Non-motor symptoms in PD can be present at any disease stage (Citation9) and have a major impact on health-related quality of life (Citation10). Moreover, some non-motor symptoms such as sleep disturbance, constipation and change in sense of smell have been identified as symptoms of the prodromal phase of PD (Citation11), a crucial finding in a condition where intervention in the disease mechanism is needed for a meaningful prognostic effect (Citation12).
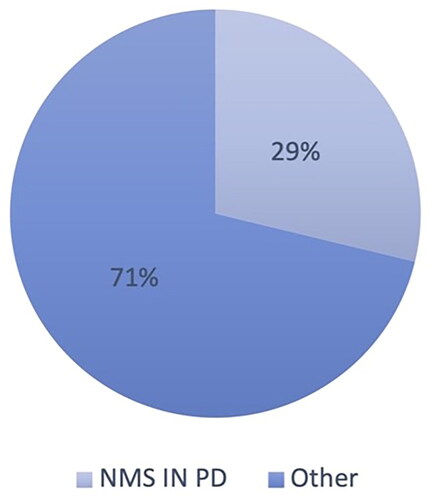
In contrast to PD, only 1% of ALS publications have focused on non-motor symptoms, the majority studying cognitive and behavioral changes ( and ).
Figure 1 Percentage of studies on non-motor symptoms in ALS, compared to studies focused on other aspects of ALS, based on publication counts. (a) The pie chart shows different areas of research in ALS. The search generated 2,210 papers dedicated to the study of non-motor symptoms in ALS, out of a total of 388,000 publications in ALS. (b) Research on different non-motor symptoms in ALS. The search generated 33551 papers for ALS, 3260 papers dedicated to cognition, psychiatric symptoms, and pain in ALS, out of a total of 5331 publications on non-motor symptoms of ALS. Search keywords: ‘ALS’, ‘non-motor symptoms’, ‘pain’, ‘cognition’, ‘sleep problems’, ‘psychiatric symptoms’, ‘gastrointestinal symptoms’, and ‘autonomic symptoms’. Search performed on 19/04/2023 (2010–2023, PubMed). ALS: amyotrophic lateral sclerosis.
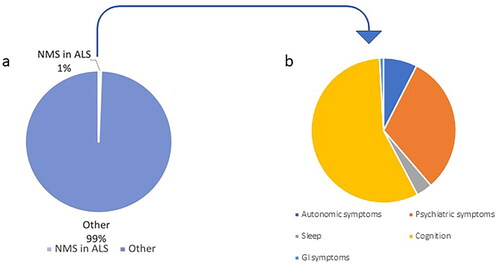
Figure 2 Percentage of studies on non-motor symptoms in PD, compared to studies focused on other aspects of PD, based on publication counts. Search keywords: ‘PD’, ‘non-motor symptoms’. Search performed on 16/05/2022 (2010–2022, Google scholar). PD: Parkinson’s disease.
Clinical trials in PD have been underpinned by non-motor symptoms-based research and have expanded the horizon of treatment, improving the lives of patients and careers. Furthermore, investigating non-motor symptoms in PD has helped in better understanding underlying disease mechanisms. These observations illustrate the importance of studying non-motor symptoms in ALS.
Non-motor symptoms in ALS: what we know so far
Estimates of the frequency of non-motor symptoms in ALS range from 5% to 80% (Citation7), largely dependent on the definitions used. Despite their importance, such symptoms have been poorly investigated and only a few population-based clinical studies on neuropsychiatric symptoms, weight loss and pain exist (Citation13). Some authors classify non-motor symptoms in ALS into five domains: neuropsychiatric, autonomic, gastrointestinal, vascular, and other system symptoms (Citation7).
Neuropsychiatric symptoms are probably the best characterized and include depression, anxiety, suicidal ideation, cognitive-behavioral impairment, fatigue, pseudobulbar affect, and sleep disturbances (Citation7). Cognitive and behavioral involvement may be in the form of frontotemporal dementia (FTD) or lesser frontotemporal involvement, which affects up to 80% of people with ALS, the frequency increasing with the disease stage (Citation14). Various factors contribute to the risk of cognitive impairment. For example, expansion in the C9orf72 gene is recognized as the most frequent genetic cause of ALS (10% of cases) and this variant also causes FTD. Variation in other ALS-related genes, such as VCP and ERBB4 among others, also increases the risk of ALS and cognitive impairment (Citation15). People with ALS-FTD may also experience psychosis and hallucinations, and there is a significant genetic overlap of ALS risk with schizophrenia (Citation16). Hypoventilation due to chronic type 2 respiratory failure can also contribute to cognitive impairment, and noninvasive ventilation may have a protective role in delaying its onset and progression (Citation17).
Autonomic symptoms include pain, urinary incontinence, and dyspnea (Citation7). Pain may be a result of immobility, postural changes and muscle responses and therefore related to the motor syndrome directly rather than representing an underlying neurodegenerative process distinct from the motor degeneration (Citation18,Citation19). Similarly, dyspnea is usually a result of neuromuscular diaphragmatic involvement, rather than having a central neurological component separate from the motor syndrome (Citation20). Urinary incontinence can represent urgency, which is either a result of spasticity affecting the bladder or motor weakness of the limbs slowing the ability to access toilet facilities (Citation21,Citation22). Only the first of these should be considered a non-motor symptom, as the bladder is not supplied by the corticospinal pathway but through the sacral autonomic outflow (Citation23). Other autonomic abnormalities, such as altered sympathetic skin response or vagus nerve atrophy, have been described in ALS (Citation24), and can contribute to heart rate variability, orthostatic hypotension and sudden falls, and have an impact on both survival and quality of life (Citation25,Citation26). Sexual dysfunction can have an autonomic cause too, together with decreased libido and reduced mobility (Citation27,Citation28).
In gastrointestinal symptoms, sialorrhea is one of the most frequent (prevalence 50%) (Citation29). Although it should legitimately be considered a motor symptom due to bulbar muscle weakness, there is also evidence of saliva flow rate changes (Citation30,Citation31) which should therefore be considered non-motor. Similarly, dysphagia is caused by motor dysfunction, but alongside muscle wasting, hypermetabolic state, and reduced appetite, contributes to weight loss (Citation32), while constipation is caused by several mechanisms, some motor (e.g., decreased nutrition or inactivity), together with non-motor ones (e.g., autonomic dysregulation) (Citation33).
Also pressure sores, which incidence seems to be higher in ALS patients compared to non-ALS-controls even in the younger ages, can be included in the list of non-motor symptoms in ALS, even if their primary cause is motor in nature (Citation34). Other non-motor symptoms exist, such as itching, for which the pathogenesis and progression are unknown (Citation7).
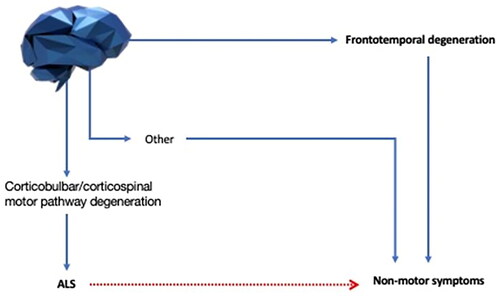
These examples illustrate the difficulty in defining what is meant by non-motor symptoms and the need for clarity ().
Figure 3 Defining non-motor symptoms in ALS. Non-motor symptoms in ALS can be caused by motor impairment (red line) or neurodegeneration outside the corticobulbar and corticospinal pathways and their lower motor neuron outflows (blue lines). Symptoms secondary to motor degeneration might have a trivial explanation, and our main interest should be focused on the non-motor symptoms arising from an underlying neurodegenerative process affecting areas different from the motor pathway. We therefore define non-motor symptoms as the ones that originate from these non-trivial processes. Among these, frontotemporal degeneration is the best studied, but it is now time to extend our knowledge to other non-motor symptoms.
Non-motor symptoms in PD: what we know so far
All patients with PD complain of one or more non-motor symptoms, independently of their disease stage (Citation35), and even in the early stages, such symptoms present a significant burden (Citation36). However, non-motor symptoms progress independently from motor symptoms (Citation37), and the load of non-motor symptoms increases with disease progression (Citation35).
It is only relatively recently that non-motor symptoms in PD have gained the deserved level of attention, considering the significant impact they have on patients’ quality of life, the increased risk of hospitalization and institutionalization, and socio-economic costs (Citation9). When looking at the impact of non-motor symptoms on patients’ health-related quality of life, they have, as a whole, a greater effect than motor symptoms (Citation10) and this finding has been demonstrated to be consistent even in the earliest stages before dopaminergic treatment is initiated (Citation38). Depression, apathy, fatigue, and sleep disturbances are the symptoms with the strongest negative association with health-related quality of life (Citation10,Citation38).
Several tools and scales, including part I of the Unified Parkinson’s Disease Rating Scale (UPDRS) (Citation39), are useful for the evaluation of PD non-motor symptoms in clinical practice. NMSQ (the non-motor symptoms questionnaire) is a patient-based screening tool that enables clinicians to complete a comprehensive assessment of a diverse range of non-motor symptoms (Citation40). Another widely known and used scale is the Non-Motor Symptoms Scale (NMSS) (Citation41), which has been recently updated in the Movement Disorder Society-sponsored Nonmotor Rating Scale (MDS-NMS) (Citation42). MDS-NMS groups PD non-motor symptoms into 13 domains: depression, anxiety, apathy, psychosis, impulse control and related disorders, cognition, orthostatic hypotension, urinary, sexual, gastrointestinal, sleep and wakefulness, pain and others. Additionally, a subscale for nonmotor fluctuations has been included in the MDS-NMS.
The development of similar tools, scales, and questionnaires for assessing non-motor symptoms in ALS is an important and currently unmet need. Each person with PD is affected by a combination of different non-motor symptoms, and each has an independent course (Citation37). Typically, some symptoms, such as fatigue or pain, characterize the early stages, while others, like dementia, apathy and dysautonomia, complicate and often dominate the clinical picture in advanced stages (Citation12,Citation43). Moreover, olfactory loss, rapid eye movement (REM) sleep behavior disorder (RBD) and constipation, are now established clinical markers of the premotor prodromal phase, due to their strong ability to predict conversion to PD, with the highest evidence for polysomnography-confirmed REM sleep behavior disorder (Citation11).
The spread of Lewy body pathology through the peripheral and central nervous systems is considered the possible cause of most non-motor symptoms in PD (Citation9,Citation12). Six different neuropathological stages of Lewy body pathology have been recognized, which are characterized by the initial involvement of the lower brainstem and olfactory bulb and a successive rostral diffusion to the cortex (Citation44). As a consequence of this spreading, several neurotransmitter systems are heterogeneously involved, with resulting deficits in multiple pathways, including dopaminergic, cholinergic, noradrenergic, and serotonergic, which contribute to the genesis of non-motor symptoms (Citation12,Citation45,Citation46). Moreover, there is evidence that the degeneration of non-dopaminergic pathways happens before the onset of dopaminergic motor symptoms, and this explains the presence of non-motor symptoms in the earliest stage of PD (Citation43,Citation47).
A similar spread of inclusion bodies is used for pathological staging in ALS and might also be found to correspond to non-motor symptoms in the future (Citation48). Observations derived from clinical practice, suggest different subtypes of PD, and this is supported by cluster analyses based on the co-occurrence of motor and non-motor symptoms (Citation49,Citation50). For instance, motor subtyping has identified non-tremor dominant subtype as associated with a higher load of autonomic involvement in the early stages and cognitive impairment in the later ones (Citation51). This specific motor subtype might reflect more diffuse and complex neurodegeneration, with a higher burden of cortical Lewy bodies and amyloid pathology (Citation52). However, non-motor subtyping has also been studied and reviewed extensively (Citation51,Citation53,Citation54). Using clustering analysis methods, several non-motor symptom-dominant phenotypes have been recognized, which reflect specific pathways of neurodegeneration, each related to a specific neurotransmitter dysfunction. For example, cholinergic loss, particularly in the nucleus basalis of Meynert and the pedunculopontine nucleus, characterizes a specific PD subtype, with prevalent gait disturbance and falls, levodopa-induced dyskinesias, cognitive impairment, REM sleep behavior disorder, psychosis, and olfactory loss (Citation55). Alternatively, when the serotonergic pathways are affected, fatigue, depression, anxiety, sleep dysfunction, and levodopa-induced dyskinesia predominate (Citation45,Citation56,Citation57).
The distinction in different subtypes not only has clinical significance for PD treatment (Citation57), but also prognostic value. For example, RBD and urinary dysfunction have been associated with a more malignant disease course, with faster progression to a more severe motor and non-motor phenotype (Citation51). Thus, Parkinson’s disease resembles a syndrome more than a single disease (Citation45).
Similarities and differences in non-motor symptoms in ALS and PD
As in PD, non-motor symptoms in ALS have a great impact on patient’s quality of life, and their burden increases with disease progression (Citation7,Citation10,Citation31,Citation35). However, non-motor symptoms in ALS have only recently gained the attention of the scientific community. As a result, their impact on ALS in terms of prognosis, disease progression and disease mechanisms, has yet to be well understood. Compared to PD, non-motor symptoms in ALS are poorly defined and the mechanisms are still unknown. Moreover, the distinction between motor and non-motor etiology is not always clear (Citation7) (). Immobility is clearly a contributor to the pathogenesis of some non-motor symptoms in ALS, such as pressure sores and constipation, and some authors refer to symptoms like dribbling, dysphagia, and insomnia, primarily as motor symptoms (Citation31). Similarly, even though it is well established that non-motor symptoms in PD are caused by the dysfunction of specific neurotransmitter pathways, a motor contribution to the etiology of some of them is also likely. Reduced mobility plays a role to a certain extent in the pathogenesis of symptoms such as constipation, nocturia or dysphagia (Citation58–60). Furthermore, as in ALS, the pathogenesis of some non-motor symptoms in PD, such as fatigue and apathy, is yet to be explained (Citation61).
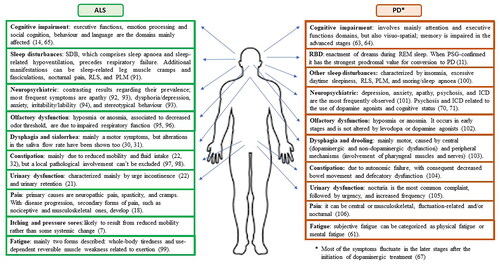
Some non-motor symptoms, such as depression and constipation, are common in both ALS and PD, while other symptoms, e.g. RBD for PD or itching and pressure sores for ALS, are found more in one of the two conditions (Citation7,Citation12). When considering a specific symptom present in both ALS and PD, the characteristics and the mechanisms behind it can differ in the two diseases. For example, cognitive impairment, despite being a common feature of both conditions, is characterized by two different profiles in ALS and PD. In PD, the cognitive decline initially affects executive functions and attention and is primarily subcortical in nature due to the depletion of dopamine in the fronto-striatal circuit (Citation62–64). In ALS is mainly characterized by impaired executive functions, emotion processing and social cognition, behavior and language, and is a consequence of the involvement of multiple cortical areas, in particular the frontal and temporal lobes (Citation65). Moreover, as previously mentioned, in ALS a contribution of reduced respiratory function and consequent hypoxia on impaired cognition has been demonstrated (Citation17), while in PD a similar relationship might be postulated only in the presence of obstructive sleep apnea (Citation66). The profile of non-motor symptoms in the two diseases is shown in .
Figure 4 The spectrum of non-motor symptoms in ALS and PD. ALS: amyotrophic lateral sclerosis; PD: Parkinson’s disease. SDB: sleep-disordered breathing. RLS: restless leg syndrome. PLM: periodic limb movements. REM: rapid eye movement. RBD: REM sleep behavior disorder. PSG: polysomnography. ICD: impulse control disorder.
In PD, non-motor symptoms fluctuate in relation to levodopa intake. In fact, chronic levodopa therapy is associated with the development of motor and non-motor complications, which include non-motor fluctuations. Non-motor fluctuations can occur together with motor fluctuations or can manifest in isolation, and have been classified as sensory/pain, cognitive/psychiatric, and autonomic, with psychiatric symptoms being the most likely to fluctuate in terms of frequency and severity (Citation67–69). The causes of such fluctuations are numerous and complex, and involve, among others, peripheral pharmacokinetic factors and altered neuronal plasticity in response to the discontinuous intake of exogenous dopamine (Citation67).
Additionally, in PD, the pathogenesis of some non-motor symptoms can be related to the use of medications, specifically dopamine agonists that can cause or precipitate symptoms such as impulse control disorder (ICD) or psychosis (Citation70,Citation71). On the other hand, none of the ALS non-motor symptoms fluctuates or has such a strong causal relationship with the use of specific medications, even in the more advanced disease stages. This may reflect the lack of an effective symptomatic therapy.
Finally, one of the main distinctions between the two diseases is the demonstration of defined non-motor symptoms characterizing the prodromal phase of PD. The acknowledgement that specific non-motor symptoms, such as RBD, olfactory impairment and constipation can precede the onset of motor symptoms by several years, has allowed the development of a specific score to estimate the probability of prodromal PD for research purposes, with potentially important therapeutic implications (Citation72). Even though the existence of a prodromal stage in ALS is recognized, the symptoms characterizing this phase have not yet been identified (Citation73), although the presence of pain and cramps in the two years before the onset of motor symptoms may be an ALS-specific prodromal feature (Citation74), as well as neuropsychiatric features, which seem to precede the onset of the motor phase of ALS (Citation75). There is also a problem of definition, since ALS following frontotemporal dementia could be regarded as demonstrating FTD as a prodromal feature of ALS.
What can research on non-motor symptoms in PD teach us about ALS?
The extensive investigation of non-motor symptoms in PD has enabled a better understanding of their underlying pathogenesis and has laid the foundations for the discovery of possible treatments, in order to improve quality of life (Citation12,Citation46,Citation57,Citation76). PD provides a valuable example of the way to approach the problem of neurodegenerative diseases. In fact, it has become increasingly clear that PD should be evaluated in a holistic manner, and consequently, a personalized medication regimen should be tailored for each patient according to their specific non-motor symptom profile and PD subtype (Citation77). A similar personalized approach would be particularly beneficial also in the clinical evaluation of people with ALS, since it is likely that each patient presents specific non-motor symptoms.
The identification of non-motor symptoms in ALS can be particularly challenging in everyday clinical practice for three reasons: possible lack of knowledge of the clinician, reduced patient awareness, and the absence of validated instruments to assess the presence of non-motor symptoms. Clinicians, especially in the past, have underestimated the burden of non-motor symptoms in PD because of a lack of awareness or time constraints in clinics, while patients with PD have tended to underreport their non-motor symptoms, either because of embarrassment or the misinterpretation of such symptoms being part of the PD clinical picture (Citation43,Citation78,Citation79). The same is likely to be true in ALS, in both the clinical and research contexts. This has been recently highlighted in a systematic review, which confirmed how non-motor symptoms have not been regularly investigated in ALS clinical trials and pointed out the use of inappropriate tools (Citation80). The validation of specific scales and questionnaires for non-motor symptoms in PD represents a fundamental milestone, allowing a comprehensive evaluation of the symptoms and a better estimation of the effect of different treatments on their severity (Citation81).
With the availability of ALS clinical staging systems such as the King’s and Milano–Torino (MiToS) staging systems (Citation82–85), researchers are now able to determine the timing of non-motor symptoms in ALS. However, one of the biggest challenges for the study of non-motor symptoms in ALS, is related to its natural history. In contrast to PD, which has a long disease progression with an overall longer life expectancy (Citation86,Citation87), ALS is characterized by an extremely severe clinical presentation and a short disease duration (Citation74). In this context, non-motor symptoms are likely to be underestimated, because the attention is mainly focused on the motor symptoms, particularly respiratory, which are the main determinants of survival (Citation88), and their pathogenesis is more likely to be due to a combination of overlapping mechanisms, including a contribution of motor dysfunction.
Following the example of PD, the study of non-motor symptoms in ALS should first involve the generation of specific questionnaires and scales. A self-assessed questionnaire, which the patient can complete in the waiting room, such as the NMSQ in PD (Citation40), can help in quickly identifying the non-motor symptoms. During the consultation, specific scales can help in better characterising the symptoms and their burden, such as the NMSS or the MDS-NMS for PD (Citation41,Citation42). Finally, symptom-specific scales, can investigate the symptom further, such as the Parkinson’s Disease Sleep Scale (PDSS) (Citation89) or the King’s Pain Scale in PD (Citation90).
Success in the study of non-motor symptoms in PD indicates that similar studies in ALS are likely to improve treatment outcomes, together with improved understanding of the disease.
Conclusions
Non-motor symptoms have been understudied in ALS and represent an unmet need in the care of patients. Following the example of PD, better definition, understanding frequency and characterization of their natural history will enable clinicians and researchers to provide a comprehensive overview of ALS, and potentially develop future therapies to address these important symptoms.
Acknowledgments
AAC is an NIHR Senior Investigator (NIHR202421). This work is in part an EU Joint Programme - Neurodegenerative Disease Research (JPND) project. The project is supported through the Motor Neurone Disease Association, My Name’5 Doddie Foundation, and Alan Davidson Foundation. This study represents an independent research part funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London.
Declaration of interest
AS, SR, and AAK do not have competing interests to disclose. KRC received honoraria for advisory boards from AbbVie, Britannia Pharmaceuticals, UCB, Pfizer, Jazz Pharma, GKC, Bial, Cynapsus, Novartis, Lobsor, Stada, Medtronic, Zambon, Profile Pharma, Sunovion, Roche, Theravance Biopharma, and Scion; honoraria for lectures from AbbVie, Britannia Pharmaceuticals, UCB, Mundipharma, Zambon, Novartis, Boeringer Ingelheim Neuroderm, and Sunovion; grants from Britannia Pharmaceuticals, AbbVie, UCB, GKC, Bial. AAC reports consultancies or advisory boards for Amylyx, Apellis, Biogen, Brainstorm, Cytokinetics, GenieUs, GSK, Lilly, Mitsubishi Tanabe Pharma, Novartis, OrionPharma, Quralis, Sano, Sanofi, and Wave Pharmaceuticals.
Additional information
Funding
References
- Brown RH, Jr., Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602.
- Martin S, Al Khleifat A, Al-Chalabi A. What causes amyotrophic lateral sclerosis? F1000Res. 2017;6:371.
- Al-Chalabi A, Kwak S, Mehler M, Rouleau G, Siddique T, Strong M, et al. Genetic and epigenetic studies of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14 Suppl 1:44–52.
- Tiryaki E, Horak HA. ALS and other motor neuron diseases. Continuum. 2014;20:1185–207.
- Rusina R, Vandenberghe R, Bruffaerts R. Cognitive and behavioral manifestations in ALS: beyond motor system involvement. Diagnostics. 2021;11:624.
- Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:9–14.
- Fang T, Jozsa F, Al-Chalabi A. Nonmotor symptoms in amyotrophic lateral sclerosis: A systematic review. Int Rev Neurobiol 2017;134:1409–41.
- Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–601.
- Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5:235–45.
- Martinez-Martin P, Rodriguez-Blazquez C, Kurtis MM, Chaudhuri KR, Group NV. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov Disord. 2011;26:399–406.
- Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol. 2016;12:622–34.
- Schapira AHV, Chaudhuri KR, Jenner P. Non-motor features of Parkinson disease. Nat Rev Neurosci. 2017;18:509.
- Urso D, Zoccolella S, Gnoni V, Logroscino G. Amyotrophic lateral sclerosis-the complex phenotype-from an epidemiological perspective: a focus on extrapyramidal and non-motor features. Biomedicines 2022;10:2537.
- Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018;91:e1370–e80.
- Al Khleifat A, Iacoangeli A, van Vugt J, Bowles H, Moisse M, Zwamborn RAJ, et al. Structural variation analysis of 6,500 whole genome sequences in amyotrophic lateral sclerosis. NPJ Genom Med. 2022;7:8.
- McLaughlin RL, Schijven D, van Rheenen W, van Eijk KR, O'Brien M, Kahn RS, et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun. 2017;8:14774.
- Huynh W, Sharplin LE, Caga J, Highton-Williamson E, Kiernan MC. Respiratory function and cognitive profile in amyotrophic lateral sclerosis. Eur J Neurol. 2020;27:685–91.
- Chio A, Mora G, Lauria G. Pain in amyotrophic lateral sclerosis. Lancet Neurol. 2017;16:144–57.
- Handy CR, Krudy C, Boulis N, Federici T. Pain in amyotrophic lateral sclerosis: a neglected aspect of disease. Neurol Res Int. 2011;2011:403808.
- Nichols NL, Van Dyke J, Nashold L, Satriotomo I, Suzuki M, Mitchell GS. Ventilatory control in ALS. Respir Physiol Neurobiol. 2013;189:429–37.
- Lopes de Carvalho ML, Motta R, Battaglia MA, Brichetto G. Urinary disorders in amyotrophic lateral sclerosis subjects. Amyotroph Lateral Scler. 2011;12:352–5.
- Nubling GS, Mie E, Bauer RM, Hensler M, Lorenzl S, Hapfelmeier A, et al. Increased prevalence of bladder and intestinal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:174–9.
- Kihira T, Yoshida S, Yoshimasu F, Wakayama I, Yase Y. Involvement of Onuf’s nucleus in amyotrophic lateral sclerosis. J Neurol Sci. 1997;147:81–8.
- Papadopoulou M, Bakola E, Papapostolou A, Stefanou MI, Moschovos C, Salakou S, et al. Autonomic dysfunction in amyotrophic lateral sclerosis: a neurophysiological and neurosonology study. J Neuroimaging. 2022;32:710–9.
- Brown AA, Ferguson BJ, Jones V, Green BE, Pearre JD, Anunoby IA, et al. Pilot study of real-world monitoring of the heart rate variability in amyotrophic lateral sclerosis. Front Artif Intell. 2022;5:910049.
- Jaafar N, Malek E, Ismail H, Salameh J. Nonmotor symptoms in amyotrophic lateral sclerosis and their correlation with disease progression. J Clin Neuromuscul Dis. 2021;23:1–6.
- Wasner M, Bold U, Vollmer TC, Borasio GD. Sexuality in patients with amyotrophic lateral sclerosis and their partners. J Neurol. 2004;251:445–8.
- Piccione EA, Sletten DM, Staff NP, Low PA. Autonomic system and amyotrophic lateral sclerosis. Muscle Nerve. 2015;51:676–9.
- Wang Y, Yang X, Han Q, Liu M, Zhou C. Prevalence of sialorrhea among amyotrophic lateral sclerosis patients: a systematic review and meta-analysis. J Pain Symptom Manage. 2022;63:e387–e96.
- Baltadzhieva R, Gurevich T, Korczyn AD. Autonomic impairment in amyotrophic lateral sclerosis. Curr Opin Neurol. 2005;18:487–93.
- Gunther R, Richter N, Sauerbier A, Chaudhuri KR, Martinez-Martin P, Storch A, et al. Non-motor symptoms in patients suffering from motor neuron diseases. Front Neurol. 2016;7:117.
- Jackson CE, McVey AL, Rudnicki S, Dimachkie MM, Barohn RJ. Symptom management and end-of-life care in amyotrophic lateral sclerosis. Neurol Clin. 2015;33:889–908.
- Liu G, Hrabe J, Sanchez R. Colostomy as a definitive treatment in an ALS patient with acute colonic pseudo-obstruction refractory to medical management, a case report. BMC Neurol. 2022;22:366.
- Chen JH, Wu SC, Chen HJ, Kao CH, Tseng CH, Tsai CH. Risk of developing pressure sore in amyotrophic lateral sclerosis patients – a nationwide cohort study. J Eur Acad Dermatol Venereol. 2018;32:1589–96.
- Martinez-Martin P, Schapira AH, Stocchi F, Sethi K, Odin P, MacPhee G, et al. Prevalence of nonmotor symptoms in Parkinson’s disease in an international setting; study using nonmotor symptoms questionnaire in 545 patients. Mov Disord. 2007;22:1623–9.
- Mollenhauer B, Trautmann E, Sixel-Doring F, Wicke T, Ebentheuer J, Schaumburg M, et al. Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology 2013;81:1226–34.
- Antonini A, Barone P, Marconi R, Morgante L, Zappulla S, Pontieri FE, et al. The progression of non-motor symptoms in Parkinson’s disease and their contribution to motor disability and quality of life. J Neurol. 2012;259:2621–31.
- Muller B, Assmus J, Herlofson K, Larsen JP, Tysnes OB. Importance of motor vs. non-motor symptoms for health-related quality of life in early Parkinson’s disease. Parkinsonism Relat Disord. 2013;19:1027–32.
- Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–70.
- Chaudhuri KR, Martinez-Martin P, Schapira AH, Stocchi F, Sethi K, Odin P, et al. International multicenter pilot study of the first comprehensive self-completed nonmotor symptoms questionnaire for Parkinson’s disease: the NMSQuest study. Mov Disord. 2006;21:916–23.
- Chaudhuri KR, Martinez-Martin P, Brown RG, Sethi K, Stocchi F, Odin P, et al. The metric properties of a novel non-motor symptoms scale for Parkinson’s disease: results from an international pilot study. Mov Disord. 2007;22:1901–11.
- Chaudhuri KR, Schrag A, Weintraub D, Rizos A, Rodriguez-Blazquez C, Mamikonyan E, et al. The movement disorder society nonmotor rating scale: initial validation study. Mov Disord. 2020;35:116–33.
- Todorova A, Jenner P, Ray Chaudhuri K. Non-motor Parkinson’s: integral to motor Parkinson’s, yet often neglected. Pract Neurol. 2014;14:310–22.
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
- Titova N, Padmakumar C, Lewis SJG, Chaudhuri KR. Parkinson’s: a syndrome rather than a disease? J Neural Transm. 2017;124:907–14.
- Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–74.
- Jellinger KA. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Mov Disord. 2012;27:8–30.
- Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38.
- Erro R, Vitale C, Amboni M, Picillo M, Moccia M, Longo K, et al. The heterogeneity of early Parkinson’s disease: a cluster analysis on newly diagnosed untreated patients. PLoS One. 2013;8:e70244.
- Mu J, Chaudhuri KR, Bielza C, de Pedro-Cuesta J, Larrañaga P, Martinez-Martin P. Parkinson’s disease subtypes identified from cluster analysis of motor and non-motor symptoms. Front Aging Neurosci. 2017;9:301.
- Marras C, Chaudhuri KR. Nonmotor features of Parkinson’s disease subtypes. Mov Disord. 2016;31:1095–102.
- Selikhova M, Williams DR, Kempster PA, Holton JL, Revesz T, Lees AJ. A clinico-pathological study of subtypes in Parkinson’s disease. Brain. 2009;132:2947–57.
- Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov Disord. 2015;30:229–37.
- Sauerbier A, Jenner P, Todorova A, Chaudhuri KR. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat Disord. 2016;22 Suppl 1: S41–S6.
- Perez-Lloret S, Barrantes FJ. Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. NPJ Parkinsons Dis 2016;2:16001.
- Huot P, Fox SH, Brotchie JM. The serotonergic system in Parkinson’s disease. Prog Neurobiol. 2011;95:163–212.
- Marras C, Chaudhuri KR, Titova N, Mestre TA. Therapy of Parkinson’s disease subtypes. Neurotherapeutics. 2020;17:1366–77.
- Batla A, Phe V, De Min L, Panicker JN. Nocturia in Parkinson’s disease: why does it occur and how to manage? Mov Disord Clin Pract. 2016;3:443–51.
- Frazzitta G, Ferrazzoli D, Folini A, Palamara G, Maestri R. Severe constipation in parkinson’s disease and in parkinsonisms: prevalence and affecting factors. Front Neurol. 2019;10:621.
- Kim JS, Youn J, Suh MK, Kim TE, Chin J, Park S, et al. Cognitive and motor aspects of parkinson’s disease associated with dysphagia. Can J Neurol Sci. 2015;42:395–400.
- Lazcano-Ocampo C, Wan YM, van Wamelen DJ, Batzu L, Boura I, Titova N, et al. Identifying and responding to fatigue and apathy in Parkinson’s disease: a review of current practice. Expert Rev Neurother. 2020;20:477–95.
- de la Fuente-Fernandez R. Frontostriatal cognitive staging in Parkinson’s disease. Parkinsons Dis. 2012;2012:1–8.
- Vasconcellos LF, Pereira JS. Parkinson’s disease dementia: diagnostic criteria and risk factor review. J Clin Exp Neuropsychol. 2015;37:988–93.
- Watson GS, Leverenz JB. Profile of cognitive impairment in Parkinson’s disease. Brain Pathol. 2010;20:640–5.
- Benbrika S, Desgranges B, Eustache F, Viader F. Cognitive, emotional and psychological manifestations in amyotrophic lateral sclerosis at baseline and overtime: a review. Front Neurosci. 2019;13:951.
- Harmell AL, Neikrug AB, Palmer BW, Avanzino JA, Liu L, Maglione JE, et al. Obstructive sleep apnea and cognition in Parkinson’s disease. Sleep Med. 2016;21:28–34.
- Ray Chaudhuri K, Poewe W, Brooks D. Motor and nonmotor complications of levodopa: phenomenology, risk factors, and imaging features. Mov Disord. 2018;33:909–19.
- Storch A, Schneider CB, Wolz M, Sturwald Y, Nebe A, Odin P, et al. Nonmotor fluctuations in Parkinson disease: severity and correlation with motor complications. Neurology 2013;80:800–9.
- Witjas T, Kaphan E, Azulay JP, Blin O, Ceccaldi M, Pouget J, et al. Nonmotor fluctuations in Parkinson’s disease: frequent and disabling. Neurology 2002;59:408–13.
- Ffytche DH, Aarsland D. Psychosis in Parkinson’s Disease. Int Rev Neurobiol 2017;133:585–622.
- Poletti M, Bonuccelli U. Impulse control disorders in Parkinson’s disease: the role of personality and cognitive status. J Neurol. 2012;259:2269–77.
- Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30:1600–11.
- Benatar M, Turner MR, Wuu J. Defining pre-symptomatic amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:303–9.
- Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: A critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Turner MR, Goldacre R, Talbot K, Goldacre MJ. Psychiatric disorders prior to amyotrophic lateral sclerosis. Ann Neurol. 2016;80:935–8.
- Seppi K, Ray Chaudhuri K, Coelho M, Fox SH, Katzenschlager R, Perez Lloret S, et al. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov Disord. 2019;34:180–98.
- Titova N, Chaudhuri KR. Personalized medicine in Parkinson’s disease: Time to be precise. Mov Disord. 2017;32:1147–54.
- Chaudhuri KR, Prieto-Jurcynska C, Naidu Y, Mitra T, Frades-Payo B, Tluk S, et al. The nondeclaration of nonmotor symptoms of Parkinson’s disease to health care professionals: an international study using the nonmotor symptoms questionnaire. Mov Disord. 2010;25:704–9.
- LeWitt PA, Chaudhuri KR. Unmet needs in Parkinson disease: motor and non-motor. Parkinsonism Relat Disord. 2020;80 Suppl 1: S7–S12.
- Beswick E, Forbes D, Hassan Z, Wong C, Newton J, Carson A, et al. A systematic review of non-motor symptom evaluation in clinical trials for amyotrophic lateral sclerosis. J Neurol. 2022;269:411–26.
- Martinez-Martin P, Rodriguez-Blazquez C, Forjaz MJ, Kurtis MM, Skorvanek M. Measurement of nonmotor symptoms in clinical practice. Int Rev Neurobiol 2017;133:291–345.
- Fang T, Al Khleifat A, Stahl DR, Lazo La Torre C, Murphy C, Young C, Uk-Mnd Lical S, et al. Comparison of the King’s and MiToS staging systems for ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:227–32.
- Balendra R, Jones A, Jivraj N, Knights C, Ellis CM, Burman R, et al. Estimating clinical stage of amyotrophic lateral sclerosis from the ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:279–84.
- Chio A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86:38–44.
- Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135:847–52.
- Hassan A, Wu SS, Schmidt P, Simuni T, Giladi N, Miyasaki JM, et al. The profile of long-term parkinson’s disease survivors with 20 years of disease duration and beyond. J Parkinsons Dis. 2015;5:313–9.
- Ishihara LS, Cheesbrough A, Brayne C, Schrag A. Estimated life expectancy of Parkinson’s patients compared with the UK population. J Neurol Neurosurg Psychiatry. 2007;78:1304–9.
- Corcia P, Pradat PF, Salachas F, Bruneteau G, Forestier N, Seilhean D, et al. Causes of death in a post-mortem series of ALS patients. Amyotroph Lateral Scler. 2008;9:59–62.
- Chaudhuri KR, Pal S, DiMarco A, Whately-Smith C, Bridgman K, Mathew R, et al. The Parkinson’s disease sleep scale: a new instrument for assessing sleep and nocturnal disability in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2002;73:629–35.
- Chaudhuri KR, Rizos A, Trenkwalder C, Rascol O, Pal S, Martino D, et al. King’s Parkinson’s disease pain scale, the first scale for pain in PD: an international validation. Mov Disord. 2015;30:1623–31.
- Boentert M. Sleep disturbances in patients with amyotrophic lateral sclerosis: current perspectives. Nat Sci Sleep. 2019;11:97–111.
- Grossman AB, Woolley-Levine S, Bradley WG, Miller RG. Detecting neurobehavioral changes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8:56–61.
- Lillo P, Mioshi E, Zoing MC, Kiernan MC, Hodges JR. How common are behavioural changes in amyotrophic lateral sclerosis? Amyotroph Lateral Scler. 2011;12:45–51.
- Wei Q, Chen X, Cao B, Ou R, Zhao B, Wu Y, et al. Associations between neuropsychiatric symptoms and cognition in Chinese patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:358–65.
- Viguera C, Wang J, Mosmiller E, Cerezo A, Maragakis NJ. Olfactory dysfunction in amyotrophic lateral sclerosis. Ann Clin Transl Neurol. 2018;5:976–81.
- Gunther R, Schrempf W, Hahner A, Hummel T, Wolz M, Storch A, et al. Impairment in respiratory function contributes to olfactory impairment in amyotrophic lateral sclerosis. Front Neurol. 2018;9:79.
- Bergmann M, Volpel M, Kuchelmeister K. Onuf’s nucleus is frequently involved in motor neuron disease/amyotrophic lateral sclerosis. J Neurol Sci. 1995;129:141–6.
- Carvalho M, Schwartz MS, Swash M. Involvement of the external anal sphincter in amyotrophic lateral sclerosis. Muscle Nerve. 1995;18:848–53.
- Gibbons CJ, Thornton EW, Young CA. The patient experience of fatigue in motor neurone disease. Front Psychol. 2013;4:788.
- Dhawan V, Healy DG, Pal S, Chaudhuri KR. Sleep-related problems of Parkinson’s disease. Age Ageing. 2006;35:220–8.
- Aarsland D, Brønnick K, Ehrt U, De Deyn PP, Tekin S, Emre M, et al. Neuropsychiatric symptoms in patients with Parkinson’s disease and dementia: frequency, profile and associated care giver stress. J Neurol Neurosurg Psychiatry. 2007;78:36–42.
- Doty RL. Olfactory dysfunction in Parkinson disease. Nat Rev Neurol. 2012;8:329–39.
- Suttrup I, Warnecke T. Dysphagia in Parkinson’s disease. Dysphagia 2016;31:24–32.
- Metta V, Leta V, Mrudula KR, Prashanth LK, Goyal V, Borgohain R, et al. Gastrointestinal dysfunction in Parkinson’s disease: molecular pathology and implications of gut microbiome, probiotics, and fecal microbiota transplantation. J Neurol. 2022;269:1154–63.
- Yeo L, Singh R, Gundeti M, Barua JM, Masood J. Urinary tract dysfunction in Parkinson’s disease: a review. Int Urol Nephrol. 2012;44:415–24.
- Rukavina K, Leta V, Sportelli C, Buhidma Y, Duty S, Malcangio M, et al. Pain in Parkinson’s disease: new concepts in pathogenesis and treatment. Curr Opin Neurol. 2019;32:579–88.