
Abstract
Objective
Most TDP-43 mouse models of ALS do not display cytoplasmic mislocalisation or protein aggregation of TDP-43 in spinal motor neurons in vivo. Thus, we investigated whether a combination of defective dynein with a TDP-43 mutation could trigger TDP-43 pathology.
Methods
Using immunohistochemical methods we examined the intracellular motor neuron pathology of the offspring of TDP-43WT and TDP-43M337V transgenic mice bred to heterozygous Loa mice, which carry an autosomal dominant mutation in dynein cytoplasmic 1 heavy chain 1 (Dync1h1).
Results
These mice did not exhibit TDP-43 mislocalisation in spinal motor neurons, but the expression of mutant dynein in combination with wildtype human TDP-43 resulted in p62 upregulation and TDP-43 aggregation, thus partially recapitulating the human disease.
Conclusions
These findings provide new insights into the possible relationship between dynein and TDP-43 and could prove useful in future studies looking to elucidate the mechanism behind the TDP-43 pathology observed in ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a form of motor neuron degeneration that affects the motor cortex, brainstem, and spinal cord, leading to the death of upper and lower motor neurons that control voluntary muscles. Over 50 mutations in TARDBP, the gene encoding TDP-43, have been identified in ALS (Citation1–6), and approximately 98% of all ALS cases show increased translocation of nuclear TDP-43 into the cytoplasm and its aggregation. The mechanisms by which these mutations and TDP-43 mislocalisation cause disease remain largely unknown. TDP-43 is an RNA binding protein involved in the regulation of gene transcription, mRNA splicing, stability, translation, and transport (Citation7, Citation8). A transgenic mouse model carrying a single copy of human TARDBP encoding the ALS-associated TDP-43M337V mutation (Citation3, Citation9) has provided insights into the mechanisms of TDP-43 pathology (Citation10). This mouse strain develops a progressive motor deficit and a loss of neuromuscular junction integrity beginning between 6-9 months of age, and a 50% decrease in survival by 2 years of age, but not cytoplasmic mislocalisation or protein aggregation of TDP-43 in spinal motor neurons in vivo (Citation10).
The delivery of misfolded proteins to proteasomes and lysosomes for degradation is highly dependent on the cytoplasmic dynein 1 complex (henceforth dynein) (Citation11, Citation12), as dynein inhibition or mutations lead to accumulation of protein aggregates (Citation13). While mutations in dynein have not been identified in ALS patients (Citation14, Citation15), polymorphisms in various genes related to dynein have been found in early-onset ALS patients (Citation16) and mutations in DCTN1 encoding the largest subunit of the dynein regulator and partner dynactin complex cause ALS (Citation17).
The “Legs at odd angles” (Loa) mutation in mouse dynein cytoplasmic 1 heavy chain 1 (Dync1h1) leads to a F580Y missense substitution causing the death of homozygous Loa mutants within a day after birth (Citation18, Citation19). In contrast, heterozygous Loa mice have a normal life span but display abnormal gait from approximately one month of age (Citation19, Citation20), reduced muscle tone, motor coordination, and hindlimb grip strength (Citation21), and defective retrograde axonal transport (Citation19, Citation22–26), likely as a result of impaired dynein-dynactin protein-protein interaction and motor processivity (Citation24, Citation27).
We hypothesized that a combination of mutant TDP-43 and defective dynein may be necessary for mice to display TDP-43 mislocalisation and aggregation in vivo. To test this and potentially create an improved mouse model to better reflect the intracellular pathology of the human disease we investigated the cellular pathology of the progeny from TARDBP hemizygous wildtype or hemizygous mutant transgenic mice on a heterozygous Loa background.
Materials and methods
Animals
Procedures involving animals were approved by the UK Home Office under the UK Animals Scientific Procedures Act 1986 and the University of Sussex Animal Welfare and Ethics Review Board. Mice were kept on a 12 h light/dark cycle (lights on at 7 am) in cages with up to five littermates, and free access to food and water. Ear biopsies were collected at 2 weeks of age for genotyping, and tail biopsies were collected postmortem for genotype confirmation. Transgenic mice carrying the human TARDBP gene (either wildtype or with the M337V mutation; JAX#029266) were bred with C57BL/6J mice (JAX#000664) to create in-house colonies. New mouse lines were generated by breeding Loa heterozygous mice (Citation19) to either TARDBP−/+ or TARDBP−/M337V transgenic mice (Citation10), to produce Dync1h1+/Loa;TARDBP−/+ and Dync1h1+/Loa;TARDBP−/M337V mice. N = 3 male adult mice per genotype (Dync1h1+/Loa; TARDBP−/−, Dync1h1+/Loa; TARDBP−/+, Dync1h1+/Loa; TARDBP−/M337V) were culled at 28 weeks by transcardial perfusion followed by decapitation, and littermates were used wherever possible.
Transcardial perfusion
Animals were terminally anesthetized with 450 mg/kg pentobarbital and transcardially perfused with artificial cerebrospinal fluid (aCSF) for 3 minutes. The vertebral column was removed and cut cross-sectionally through the intervertebral disks between vertebrae T9 and T10 and between vertebrae L6 and S1. The lumbar portion of the vertebral column (between vertebrae T10-L6) was drop-fixed in 4% formaldehyde (VWR, 9713.5000) for 24 hours, after which the spinal cord was dissected out and preserved in 30% sucrose (Fisher, S/8600/60) and 0.2% sodium azide (Sigma, S8032-100G) at 4 °C until cryo-sectioning.
Spinal cord cryo-sectioning and immunohistochemistry
Fixed spinal cords were mounted in optimal cutting temperature (OCT) compound (Agar Scientific, AGR1180) and sliced cross-sectionally in 30 μm-thick sections using a cryostat at −17 °C. Each section was kept in 200 μl of antifreeze solution (30% ethyleneglycol, Sigma-Aldrich, 324558; 30% glycerol, Fisher, G/0600/08, 30% dH2O; 10% 2X PBS) at 4 °C until immunostaining. One section per lumbar (L3-L6) segment per animal was immunostained. Sections were transferred to glass slides (Fisher, 10149870) and washed three times with PBS for 10 minutes per wash on an orbital shaker at 100 rpm. Antigen retrieval was performed with sodium citrate buffer, pH 6.0, pre-warmed to 80-95 °C for 30 minutes with gentle agitation. The slides were then washed three times with PBS containing 0.1% Triton X-100 (Sigma-Aldrich, 93443-100ML) to permeabilise the tissue for 10 minutes per wash at 100 rpm, followed by blocking with PBS containing 0.5% Triton X-100 and 5% horse serum (Fisher, 11520516) for 90 minutes at 100 rpm. Then, the slides were incubated overnight at 4 °C in a humidified chamber with PBS containing 0.1% Triton X-100, 1% horse serum, a 1:100 dilution of rabbit anti-TDP-43 primary antibody (Proteintech, 10782-2-AP), a 1:50 dilution of goat anti-ChAT primary antibody (Merck/Sigma, AB144P), and a 1:500 dilution of guinea pig anti-p62 primary antibody (Progen, GP62-C). The slides were washed three times with PBS for 10 minutes per wash at 100 rpm, then incubated for 3 hours in the dark with PBS containing 0.1% Triton X-100, a 1:500 dilution of Alexa Fluor 647-AffiniPure donkey anti-rabbit IgG secondary antibody (Jackson Immuno research, 711-605-152), a 1:500 dilution of Alexa Fluor 488 donkey anti-goat IgG secondary antibody (Life Technologies, A11055), and a 1:500 dilution of Rhodamine (TRITC)-AffiniPure donkey anti-guinea pig IgG secondary antibody (Jackson Immuno research, 706-025-148-JIR). The slides were finally washed three times with PBS for 10 minutes per wash at 100 rpm, with the addition of DAPI stain (Sigma, D9542) at 2 μg/ml final concentration during the second wash only, before sealing them with mounting medium (Fisher, 11539306). Omission of the primary or secondary antibodies resulted in no detectable staining.
Confocal microscopy and image analysis
Fluorescent images were acquired and analyzed blinded to genotype using a Leica SP8 confocal scanning electron microscope and captured using the LAS X version 3.4 software. Only one of the ventral horns per section was imaged at random. Z-stack images were acquired to provide full coverage of the cell volume and were converted to maximum projection images. Cholinergic spinal motor neurons were identified by their location in the ventral horn, morphology, and strong ChAT staining. The soma of ChAT+ cells and their nuclei (DAPI+) were outlined manually using the freehand selection tool in ImageJ, and regions of interest were created to represent the soma area, nucleus area, and cytoplasm area. Following background subtraction, the fluorescence intensity values of proteins of interest were measured. For analysis of punctate staining, the images were thresholded using the Minimum method (Citation28) and all cytoplasmic puncta with area ≥0.36 μm2 were measured.
Statistical analysis
Statistical analysis was undertaken in GraphPad Prism version 9, following the software’s recommended tests for each type of data set. Specific details are found in the figure legends. Outliers were identified using ROUT’s test with Q = 1% and were removed. The alpha significance threshold was set to 5%.
Results
Previous work indicates the absence of TDP-43 mislocalisation and aggregation in the lumbar spinal cord of the TARDBP−/+ and TARDBP−/M337V mice up to 2 years of age (Citation10). Therefore, we introduced the dynein Loa mutation in these models to examine whether the additional dysfunction of axonal transport would trigger TDP-43 proteinopathy. Lumbar spinal cord sections from segments L3-L6 of 28-week-old male mice were immunostained for TDP-43 with an antibody that detects both the mouse and human protein. We found significantly more TDP-43 in the soma of ChAT+ cells in mice expressing the wildtype TARDBP transgene, compared to non-transgenic controls, likely due to the presence of the extra copy of the gene (). There was also significantly increased TDP-43 in ChAT+ cells in mice expressing the M337V mutant transgene compared to non-transgenic controls, but this level was significantly lower compared to wildtype transgenic mice, indicating reduced TDP-43 expression in the presence of the M337V mutation (). Despite these differences in TDP-43 expression levels, there was no evidence of significant cytoplasmic mislocalisation in TDP-43 transgenic Loa mutants compared to non-transgenic Loa mutants (), suggesting that combined dynein and TDP-43 mutations do not trigger TDP-43 mislocalisation. Additionally, similar proportions of ChAT+ cells had cytoplasmic TDP-43 puncta in all genotypes (), but the number of puncta was significantly higher in Loa mutants expressing the wildtype transgene (). Conversely, the number of TDP-43 puncta in TARDBP−/M337V; Dync1h1+/Loa mice did not differ significantly from that seen in non-transgenic Loa mutants (). This suggests that expression of the mutant TDP-43 does not induce additional protein aggregation in Loa mutants, but that of the wildtype TDP-43 does.
Figure 1. TDP-43 fluorescence intensity, localization, and aggregation in ChAT+ cells in L3-L6 spinal cord segments. (A) Soma TDP-43 expression levels quantified by immunohistochemistry. Kruskal-Wallis test with Dunn’s post-hoc. (B) Cytoplasmic TDP-43 as a percentage of total soma expression. Kruskal-Wallis test with Dunn’s post-hoc. (C) Percentage of cells with cytoplasmic TDP-43 puncta. One-way ANOVA with Tukey’s post-hoc. (D) Number of cytoplasmic TDP-43 puncta per cell, normalized to cytoplasm area. Kruskal-Wallis test with Dunn’s post-hoc. (E) Representative immunohistochemistry images showing cytoplasmic TDP-43 puncta and predominantly nuclear TDP-43 expression in ChAT+ cells. Scale bar = 10 μm. Data is shown as mean ± SEM from 3 mice per genotype (N = 19 cells per mouse on average). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns = not significant.
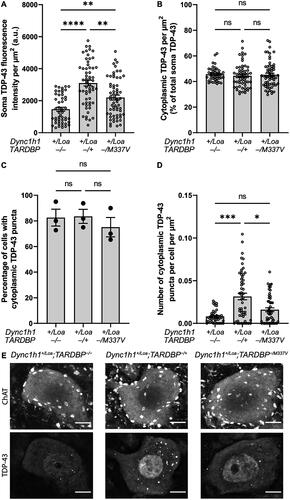
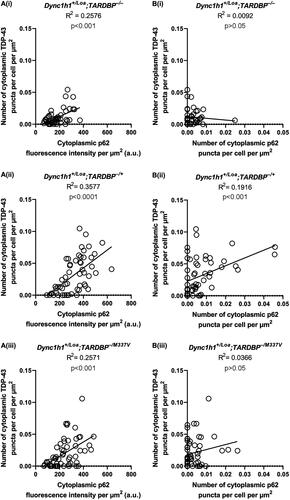
In all genotypes, we also found some ChAT+ cells with a primarily diffuse expression of cytoplasmic p62, whereas other cells had p62 puncta, which appeared predominantly around the perimeter of the soma (). The proportion of punctate cells () and the number of puncta per cell () were not statistically significant between genotypes. However, the overall cytoplasmic fluorescence intensity of p62 was statistically significant between the genotypes, with non-transgenic Loa and wildtype TARDBP transgenic Loa mice harboring the lowest and highest levels, respectively (). Collectively, these data suggest an impairment in autophagy caused by TDP-43 overexpression, which is however less apparent in Loa mice with the TARDBP−/M337V transgene, but still significantly more pronounced than in the non-transgenic controls. Surprisingly, there was zero spatial colocalisation between p62 puncta and TDP-43 puncta in all the cells examined (examples are shown in ), despite the presence of several ChAT+ cells with coincident cytoplasmic p62 puncta and TDP-43 puncta in all genotypes (). However, there was a statistically significant positive correlation between the number of TDP-43 puncta and the p62 expression level, indicating a relationship between TDP-43 aggregation and autophagy (). Nevertheless, the number of TDP-43 puncta was positively correlated with the number of p62 puncta only in Dync1h1+/Loa;TARDBP−/+ cells ().
Figure 2. P62 puncta number and p62 intensity in ChAT+ cells in L3-L6 spinal cord segments. (A) Representative immunohistochemistry images showing examples of punctate (left column) and diffuse (right column) p62 expression in ChAT+ cells. Scale bar = 10 μm. (B) Percentage of cells with cytoplasmic p62 puncta. One-way ANOVA with Tukey’s post-hoc. (C) Number of cytoplasmic p62 puncta per cell, normalized to cell area. Kruskal-Wallis test with Dunn’s post-hoc. (D) Cytoplasmic p62 expression in ChAT+ cells. Welch’s ANOVA with Dunnett’s T3 post-hoc. Data is shown as mean ± SEM from 3 mice per genotype (N = 19 cells per mouse on average). ***p < 0.001, ****p < 0.0001, ns = not significant.
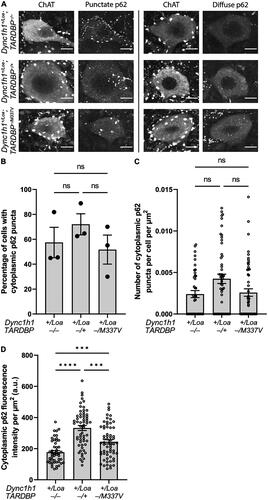
Figure 3. Colocalisation analysis of TDP-43 and p62 puncta in ChAT+ cells in L3-L6 spinal cord segments. (A) Representative immunohistochemistry (“staining”) and thresholded (“puncta only”) images showing the spatial relationship between TDP-43 (magenta) and p62 (cyan) puncta. Examples of TDP-43 puncta are indicated by yellow arrowheads, and examples of p62 puncta are indicated by white arrows. Scale bar = 10 μm. (B) Percentage of cells with coincident cytoplasmic p62 and TDP-43 puncta. One-way ANOVA with Tukey’s post-hoc. ns = not significant. Data is shown as mean ± SEM from 3 mice per genotype (N = 19 cells per mouse on average).
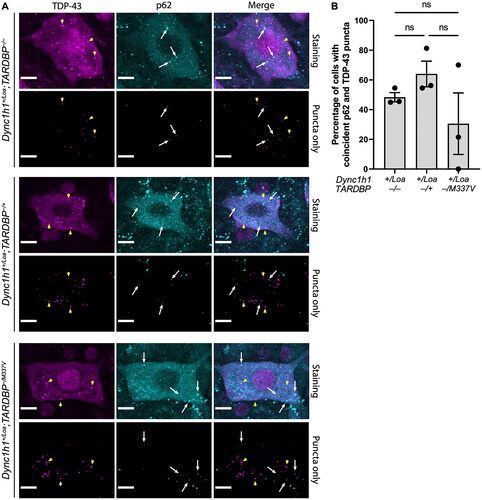
Figure 4. Correlation between TDP-43 and p62 puncta and expression levels in ChAT+ cells in L3-L6 spinal cord segments. (A) Correlations between p62 fluorescence intensity and TDP-43 puncta number. (B) Correlations between p62 puncta number and TDP-43 puncta number. Lines represent the simple linear regression fit and R2 values indicate the goodness of fit. Data is from 3 mice per genotype (N = 19 cells per mouse on average).
Discussion
Defective axonal transport in motor neurons is one of the earliest cellular pathologies in ALS and several genes linked to ALS have been associated with axonal transport (Citation23, Citation29, Citation30). Furthermore, the most significant risk factor for ALS is aging (Citation31), and evidence suggests that aging diminishes dynein function (Citation32, Citation33). Therefore, it is plausible that an aging-related reduction in dynein function may promote the protein aggregation seen in ALS, for example, by slowing the degradation of aggregate-prone proteins. Interestingly, mutant TDP-43 patient lines exhibit increased levels of microtubule-based transport proteins such as MAPT, TUBA4A, and DCTN1, which are found to be interacting with mutant TDP-43, potentially indicating a direct effect of TDP-43 proteinopathy on components of axonal transport (Citation34). Additionally, dynein light and heavy chains have been detected in TDP-43 aggregates purified from transfected neuroblastoma cells (Citation35), and there is evidence of a direct interaction between TDP-43 and the dynactin complex via its DCTN1 subunit, as well as a role for DCTN1 in regulating the nucleocytoplasmic transport and aggregation of TDP-43 (Citation36).
TDP-43 animal models used in ALS research generally do not exhibit TDP-43 cytoplasmic mislocalisation or protein aggregation pathology in vivo, despite displaying other disease-related molecular, neuromuscular, and behavioral phenotypes. Given the role of impaired axonal transport in ALS and the recent evidence suggesting an indirect interaction between TDP-43 and the dynein regulator dynactin (Citation36), we investigated whether mutant dynein in a transgenic TDP-43 mouse model would trigger TDP-43 proteinopathy in vivo, thus improving the model to better reflect the intracellular pathology of the human disease. Our results show increased levels of TDP-43 and its cytoplasmic aggregation in the spinal motor neurons of both Dync1h1+/Loa;TARDBP−/+ and Dync1h1+/Loa;TARDBP−/M337V male transgenic mice at 28 weeks of age, with the wildtype TARDBP transgene having a greater effect than the mutant one. While an increase in TDP-43 protein expression is expected in the presence of an extra TARDBP allele, the coincident presence of protein aggregates suggests that these may be formed due to too much TDP-43 protein expression in both transgenic genotypes. Previous studies did not observe TDP-43 aggregates in these transgenic mouse models, however, here we show that aberrant dynein function caused by the Loa mutation is sufficient to trigger TDP-43 aggregation in mice which also express the wildtype TDP-43 transgene. The absence of statistically significant TDP-43 aggregation in the mice expressing the mutant TDP-43 transgene may be due to the comparatively lower levels of TDP-43. A previous study showed impaired dynein-mediated retrograde axonal transport in the TARDBP−/M337V mice (Citation37). The impaired transport caused by mutant TDP-43 is however unlikely to be the cause of the difference in TDP-43 aggregation between the two genotypes in our study.
An impairment in dynein function leads to reduced trafficking of autophagosomes and fusion with lysosomes. This could explain the significantly higher level of p62 in Loa spinal motor neurons expressing either wildtype or mutant human TDP-43, signifying impaired autophagic flux. The less pronounced p62 levels in motor neurons with the M337V mutation follows the same pattern as the TDP-43 expression levels, indicating that autophagy is more compromised in cells expressing a higher amount of TDP-43 and exhibiting higher TDP-43 aggregation. Despite this, we found no colocalisation between TDP-43 and p62 puncta, an observation which differs from the recent evidence of colocalisation of p62 with TDP-43 inclusions in brain and spinal neurons from non-SOD1 familial and sporadic ALS and ALS-FTD patients (Citation38, Citation39). Nevertheless, our findings suggest that a combination of mutant dynein with overexpression of wildtype TDP-43 may impair the autophagic pathway, whereas the M337V mutation in TDP-43 either has less impact on autophagy or its expression is below the threshold for inducing this phenotype in these mice. Alternatively, while the increased p62 levels could initially be interpreted as a potential sign of impaired autophagy, we also consider that it could represent a more general response to cellular stress. Both these scenarios may be applicable, given the complex nature of cellular stress responses. An interesting observation is the presence of several p62 puncta outside of the cell boundaries. Extracellular release of p62 has previously been linked to inflammation, infection, and immunity (Citation40). It is difficult to delineate the relationship between p62 and TDP-43 in the disease. While a direct interaction between the two proteins has been observed (Citation41), studies have shown that p62 upregulation may either increase (Citation42) or decrease (Citation43) TDP-43 aggregation. Therefore, more research is needed to better understand the role of autophagy in the clearance of TDP-43 aggregates in ALS.
A limitation of this study is the use of only male mice, and therefore, the influence of sex on the interactions between TDP-43 and dynein mutations remains an open question. Furthermore, while the present study focused solely on cellular pathology, we acknowledge the importance of behavioral and survival analyses in understanding the full impact of the combined TARDBP and Loa mutations. Nevertheless, in our routine observations, the transgenic animals harboring the Loa mutation did not exhibit visible differences in gross motor behavior compared to those without the Loa mutation.
In summary, whilst expression of mutant dynein in wildtype or mutant TDP-43 transgenic mice did not result in TDP-43 mislocalisation, its presence resulted in statistically significant TDP-43 aggregation in spinal motor neurons harboring the wildtype TDP-43 transgene, thus partially recapitulating the human disease. These findings provide new insights into the possible relationship between dynein and TDP-43 and could prove useful in future studies looking to improve available transgenic mouse models of ALS for better presentation of ALS pathology and thus helping to fully elucidate the molecular mechanisms underpinning the TDP-43 pathology observed in ALS. Our study underlines the feasibility and importance of “second hit” or “double hit” models of disease progression. where the “first hit” is the presence of the TDP-43 mutation and the “second hit” is defective dynein. By exploring the combined effect of these factors, our model presents a more complex and nuanced representation of ALS pathology, allowing the opportunity to capture the multifactorial nature of ALS better. Consequently, our work provides a potential direction for future ALS research, which might look into combining genetic hits or focusing on the interplay between different pathogenic mechanisms in disease progression.
Acknowledgements
We would like to thank Dr Greig Joilin for critically reviewing the first draft of the manuscript.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Data availability statement
The data that support the findings of this study are available from the corresponding author, MH, upon reasonable request.
Additional information
Funding
References
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, et al. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–8.
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–4.
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7:409–16.
- Ratti A, Buratti E. Physiological functions and pathobiology of TDP-43 and FUS/TLS proteins. J Neurochem. 2016;138: 95–111.
- Ederle H, Dormann D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017;591:1489–507.
- Cohen TJ, Lee VMY, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;17:659–67.
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014;81:536–43.
- Rutherford NJ, Zhang Y-J, Baker M, Gass JM, Finch NA, Xu Y-F, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193.
- Gordon D, Dafinca R, Scaber J, Alegre-Abarrategui J, Farrimond L, Scott C, et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol Dis. 2019;121:148–62.
- Eschbach J, Dupuis L. Cytoplasmic dynein in neurodegeneration. Pharmacol Ther. 2011;130:348–63.
- Schiavo G, Greensmith L, Hafezparast M, Fisher EM. Cytoplasmic dynein heavy chain: the servant of many masters. Trends Neurosci. 2013;36:641–51.
- Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–6.
- Ahmad-Annuar A, Shah P, Hafezparast M, Hummerich H, Witherden AS, Morrison KE, et al. No association with common Caucasian genotypes in exons 8, 13 and 14 of the human cytoplasmic dynein heavy chain gene (DNCHC1) and familial motor neuron disorders. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:150–7.
- Shah PR, Ahmad‐Annuar A, Ahmadi KR, Russ C, Sapp PC, Robert Horvitz H, et al. No association of DYNC1H1 with sporadic ALS in a case‐control study of a northern European derived population: a tagging SNP approach. Amyotroph Lateral Scler. 2006;7:46–56.
- Steinberg KM, Yu B, Koboldt DC, Mardis ER, Pamphlett R. Exome sequencing of case-unaffected-parents trios reveals recessive and de novo genetic variants in sporadic ALS. Sci Rep. 2015;5:9124.
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur ELF, Tokito M, Mann E, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–6.
- Hoang HT, Schlager MA, Carter AP, Bullock SL. DYNC1H1 mutations associated with neurological diseases compromise processivity of dynein–dynactin–cargo adaptor complexes. Proc Natl Acad Sci U S A. 2017;114:E1597–E1606.
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–12.
- Chen X-J, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–24.
- Ilieva HS, Yamanaka K, Malkmus S, Kakinohana O, Yaksh T, Marsala M, et al. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc Natl Acad Sci U S A. 2008;105:12599–604.
- Perlson E, Jeong G-B, Ross JL, Dixit R, Wallace KE, Kalb RG, et al. A switch in retrograde signaling from survival to stress in rapid-onset neurodegeneration. J Neurosci. 2009;29:9903–17.
- Kieran D, Hafezparast M, Bohnert S, Dick JRT, Martin J, Schiavo G, et al. A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J Cell Biol. 2005;169:561–7.
- Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol. 2010;12:1228–34.
- Soppina V, Rai AK, Ramaiya AJ, Barak P, Mallik R. Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc Natl Acad Sci U S A. 2009;106:19381–6.
- Hendricks AG, Perlson E, Ross JL, Schroeder HW, 3rd, Tokito M, Holzbaur EL. Motor coordination via a tug-of-war mechanism drives bidirectional vesicle transport. Curr Biol. 2010;20:697–702.
- Deng W, Garrett C, Dombert B, Soura V, Banks G, Fisher EMC, et al. Neurodegenerative mutation in cytoplasmic dynein alters its organization and dynein-dynactin and dynein-kinesin interactions*. J Biol Chem. 2010;285:39922–34.
- Prewitt JMS, Mendelsohn ML. The analysis of cell images. Ann N Y Acad Sci. 1966;128:1035–53.
- Guo W, Stoklund Dittlau K, Van Den Bosch L. Axonal transport defects and neurodegeneration: molecular mechanisms and therapeutic implications. Semin Cell Dev Biol. 2020;99:133–50.
- De Vos KJ, Hafezparast M. Neurobiology of axonal transport defects in motor neuron diseases: opportunities for translational research? Neurobiol Dis. 2017;105:283–99.
- Pandya VA, Patani R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: a cellular perspective. Brain. 2020;143:1057–72.
- Kimura N, Imamura O, Ono F, Terao K. Aging attenuates dynactin-dynein interaction: down-regulation of dynein causes accumulation of endogenous tau and amyloid precursor protein in human neuroblastoma cells. J Neurosci Res. 2007;85:2909–16.
- Kimura N, Inoue M, Okabayashi S, Ono F, Negishi T. Dynein dysfunction induces endocytic pathology accompanied by an increase in Rab GTPases: a potential mechanism underlying age-dependent endocytic dysfunction. J Biol Chem. 2009;284:31291–302.
- Fazal R, Boeynaems S, Swijsen A, De Decker M, Fumagalli L, Moisse M, et al. HDAC6 inhibition restores TDP-43 pathology and axonal transport defects in human motor neurons with TARDBP mutations. Embo J 2021;40:e106177.
- Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21:228–39.
- Deshimaru M, Kinoshita-Kawada M, Kubota K, Watanabe T, Tanaka Y, Hirano S, et al. DCTN1 Binds to TDP-43 and Regulates TDP-43 Aggregation. Int J Mol Sci 2021;22.
- Sleigh JN, Tosolini AP, Gordon D, Devoy A, Fratta P, Fisher EMC, et al. Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Rep. 2020;30:3655–62.e2.
- Trist BG, Fifita JA, Hogan A, Grima N, Smith B, Troakes C, et al. Co-deposition of SOD1, TDP-43 and p62 proteinopathies in ALS: evidence for multifaceted pathways underlying neurodegeneration. Acta Neuropathol Commun. 2022;10:122.
- Hiji M, Takahashi T, Fukuba H, Yamashita H, Kohriyama T, Matsumoto M. White matter lesions in the brain with frontotemporal lobar degeneration with motor neuron disease: TDP-43-immunopositive inclusions co-localize with p62, but not ubiquitin. Acta Neuropathol. 2008;116:183–91.
- Zou B, Liu J, Klionsky DJ, Tang D, Kang R. Extracellular SQSTM1 as an inflammatory mediator. Autophagy 2020;16:2313–5.
- Tanji K, Zhang H-X, Mori F, Kakita A, Takahashi H, Wakabayashi K. p62/sequestosome 1 binds to TDP-43 in brains with frontotemporal lobar degeneration with TDP-43 inclusions. J Neurosci Res. 2012;90:2034–42.
- Foster AD, Flynn LL, Cluning C, Cheng F, Davidson JM, Lee A, et al. p62 overexpression induces TDP-43 cytoplasmic mislocalisation, aggregation and cleavage and neuronal death. Sci Rep. 2021;11:11474.
- Brady OA, Meng P, Zheng Y, Mao Y, Hu F. Regulation of TDP-43 aggregation by phosphorylation andp62/SQSTM1. J Neurochem. 2011;116:248–59.