
Abstract
Objective
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder resulting in upper and lower motor neuron loss. ALS often has a focal onset of weakness, which subsequently spreads to other body regions. Survival is limited to two to five years after disease onset, often due to respiratory failure. Cognitive impairment is present in approximately 30% to 50% of patients and in 10%–15% of patients, the clinical criteria of frontotemporal dementia (FTD) are met.
Methods
In this retrospective single-center ALS cohort study, we examined the occurrence of cognitive and behavioral impairment in relation to motor impairment at disease presentation and studied its impact on survival.
Results
The degree of lower motor neuron involvement was associated with a worse survival, but there was no effect for upper motor neuron involvement. Patients who were cognitively normal had a significantly better survival compared to patients with cognitive or behavioral impairment and to patients with comorbid FTD. There was no significant difference regarding survival between patients with FTD and patients with cognitive or behavioral impairment.
Conclusions
The extent of motor and extramotor involvement in patients with ALS at disease presentation holds complementary prognostic information.
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder resulting in upper and lower motor neuron loss. ALS usually has a focal onset of muscle weakness in one limb or in the bulbar region, but weakness typically spreads to other body regions with disease progression. The survival is limited to two to five years after disease onset, mostly due to respiratory failure (Citation1,Citation2). In about 30% of cases there is a bulbar onset of disease, with patients presenting with dysphagia or dysarthria (or less common dysphonia) (Citation1). Cognitive or behavioral impairment can be present in up to half of ALS patients and in 10%–15% of patients, the clinical criteria of frontotemporal dementia (FTD) are met (Citation1,Citation3).
Most cognitive domains can be affected, although predominantly impairment in verbal fluency, language, and executive dysfunction (i.e. ALS-Specific changes) is being reported (Citation4). Letter fluency impairment is most frequently observed (30.4%), followed by executive dysfunction (22.5%) and language dysfunction (21.3%) (Citation5). Nevertheless, psychomotor speed, visuoconstructive functions and Mini Mental State Examination (MMSE) also show a medium or large effect size in ALS patients (Citation4). The Edinburgh Cognitive and Behavioral ALS Screen (ECAS) is frequently being used to assess cognitive and behavioral problems in ALS patients and is a valid and sensitive assessment for patients with behavioral variant FTD (bv-FTD) (Citation4,Citation6,Citation7). It enables a correction for the presence of bulbar and spinal dysfunction, by correcting for slowing of speech and writing (i.e. verbal fluency index) (Citation6).
A validated prediction model for survival so far consists of the following eight predictors: bulbar versus non-bulbar onset, definite versus probable or possible ALS, diagnostic delay, and age of onset, ALS-FRS-R progression rate, forced vital capacity, presence of frontotemporal dementia, and presence of a C9orf72 repeat expansion (Citation8).
The number of regions with lower motor neuron involvement additionally shows a strong association with survival (Citation9). Moreover, the extent of extramotor impairment has also been linked to the outcome in ALS patients, as patient with concomitant FTD have a shorter survival (Citation10–13). Even though it is known that cognitive/behavioral changes in ALS patients are common, this domain is currently still not included in diagnostic criteria or in ALS staging systems.
Studies that evaluate whether combined profiles of extramotor and motor changes can be used to predict survival, are lacking (Citation9,Citation14–17).
The relation between extramotor and motor impairment has been studied in a few studies, suggesting that frontotemporal involvement is more common in bulbar onset ALS (Citation18,Citation19). Patients who suffered from worse cognition also progressed faster as measured by the ALS functional rating scale –revised (ALSFRS-R) (Citation20). How the degree and severity of motor involvement is related to concomitant extramotor involvement, however, remains to be investigated (Citation8).
In this study, we aimed to grade the extent of upper motor neuron (UMN) and lower motor neuron (LMN) involvement and the extent of frontotemporal involvement at disease presentation and to study the relation with disease outcome.
Methods
In this retrospective single-center cohort study, we examined cognitive and behavioral impairment in ALS patients and we investigated the extent of extramotor and motor involvement in relation to survival. Patients were eligible for inclusion in the study if they were diagnosed with definite ALS, probable ALS and probable laboratory-supported ALS and possible ALS by the revised El Escorial criteria, had undergone both nerve conduction and EMG analysis as well as neuropsychological testing by ECAS and attended the neuromuscular reference center of UZ Leuven with a date of diagnosis ranging from 2011 to October 2021.
All patients in our database of whom adequate clinical data and EMG analysis were available, were evaluated for inclusion within the given timeframe. Of this cohort, all patients who had undergone ECAS testing were included in this study and patients with PLS were excluded.
ECAS assessment was performed in Dutch. Normative data have been established in Dutch, with deficits in line with the established cognitive profile of ALS (Citation21). The following demographic information was also collected for the present study: age at disease onset, sex, diagnostic delay, site of symptom onset, date of death, total regions with upper and lower motor neuron symptoms at disease presentation based on clinical examination, El Escorial and Awaji criteria.
Extramotor involvement was defined as cognitive and/or behavioral impairment in patients with ALS. Cognitive status was classified according to the diagnostic criteria published by Strong et al into the following categories: ALS-Cn (cognitive normal); ALS-Bi (behavioral impairment); ALS-Ci (cognitive impairment); ALS-Cbi (cognitive and behavioral impairment); ALS-FTD (fulfilling criteria for FTD) (Citation22,Citation23). ALS-Bi requires 2 of 6 criteria for behavioral variant FTD to be fulfilled or the presence of apathy. ALS-Ci is classified as evidence of either executive dysfunction or language dysfunction. For the diagnosis of ALS-FTD, at least 3 symptoms associated with bvFTD must be present, 2 changes together with psychotic symptoms or fulfilled criteria for primary progressive aphasia (Citation1,Citation5,Citation24).
The primary endpoint was death with a significance level of 0.05. UMN groups were categorized from 0 regions affected to 3 or more. Lower motor neuron regions were deemed affected when either clinical examination, Awaji or El Escorial criteria were met. LMN groups were categorized from 1 to 3 or more regions affected, excluding patient with primary lateral sclerosis from the analysis. ECAS categories were grouped as (Citation1) ALS-Cn (Citation2), ALS patients with cognitive or behavioral impairment not meeting the criteria for concomitant FTD (ALS-Ci/ALS-Cbi/ALS-Bi) and (Citation3) ALS-FTD. provides an overview of the different patient groups in this study. We used a multivariable Cox proportional hazards regression model to estimate the effect of cognitive status, LMN and UMN involvement, as well as a combination of both motor and extramotor involvement on survival.
Table 1. Definition of the different patient groups in this study.
Ethical consent for this study was achieved via the ethics committee of University Hospitals Leuven (S50354) and informed consent forms were obtained for all patients included in the study.
Results
Demographic data
In total, 179 patients were included in this study. Sex at birth, age of onset, diagnostic delay, site of onset and median ECAS score are reported in . In line with previous studies, half (n = 85; 47.5%) of ALS patients were cognitively normal, and half suffered from either behavioral or cognitive impairment (n = 70; 39.1%) or FTD (n = 24; 13.4%).
Table 2. Demographic data for ALS patients.
Age of onset was lowest in the cognitively normal group, with a 8.9-year difference compared to the FTD group. Median ECAS score was 96.5 for all participants, with a score of 110.5 for the ALS-Cn group and 75.3 for the ALS-FTD group, respectively. Bulbar onset ALS occurred in 29.1% of patients. The frequency of bulbar onset ALS was 20.0% in the ALS-Cn cohort versus 41.7% in the ALS-FTD cohort.
What is the effect of LMN, UMN and extramotor involvement on survival?
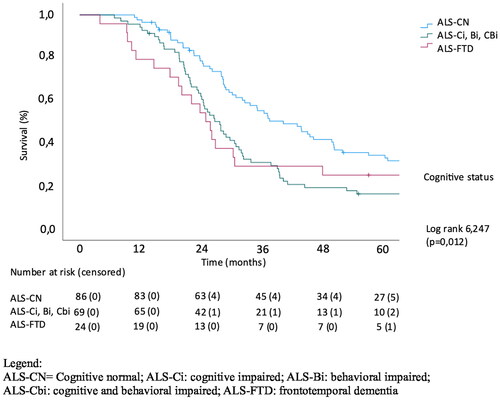
The effect of extramotor involvement, being defined as cognitive and/or behavioral impairment in patients with ALS as well as LMN and UMN involvement on survival was evaluated. In a univariate analysis of effect of the number of regions with UMN involvement, there was no significant effect on survival (p = 0.905). The number of LMN regions affected was significantly related to survival (p = 0.008) with a median survival of 32.9 months in the patients with LMN involvement in 1 region (LMN group 1) versus 29.1 months in patients with LMN involvement in 3 regions (LMN group 3). Cognitive involvement also significantly correlated with survival (p = 0.012), with median survival being 42.6, 28.1 and 25.3 months in cognitively normal patients, patients with cognitive and/or behavioral impairment and with concomitant FTD, respectively, (Log Rank Chi square 6.43). Kaplan-Meier analysis of the effect of the extent of LMN, UMN and cognitive/behavioral involvement on survival is shown in .
Figure 1. Univariate analysis of effect of LMN involvement on survival.
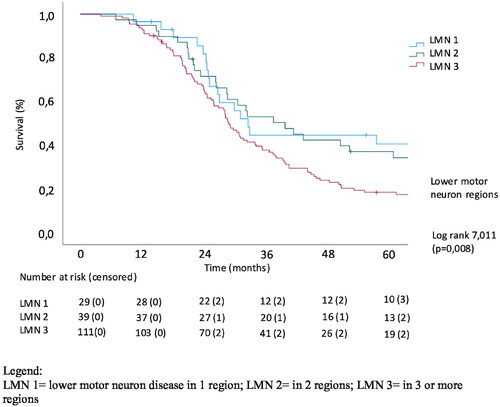
Figure 2. Univariate analysis of effect of UMN involvement on survival.
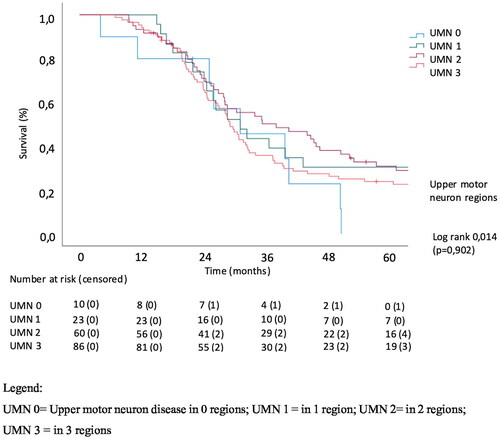
Figure 3. Univariate analysis of effect of extramotor involvement on survival.
What is the effect of LMN and extramotor involvement on survival in a multivariate analysis?
Next, a multivariable analysis was performed, including age of onset, site of onset, sex at birth and diagnostic delay.
Diagnostic delay and age of onset were significant predictors of survival in our multivariable Cox analysis, whereas site of onset and sex at birth did not reach statistical significance. As expected, the number of UMN regions was not an independent predictor of survival. Subsequently, a multivariable analysis with diagnostic delay, age of onset, number of LMN regions and cognitive/behavioral impairment was performed. LMN group 3 was used as the reference group since this was the group with the largest numbers of patients. depicts the hazard ratios (HR), p values and confidence intervals for the different covariates. Lower motor neuron involvement in only 1 region had a hazard ratio of 0.524 versus 3 regions affected (p = 0.012; 95% confidence interval (CI) 0.317–0.866). ECAS was also significantly associated with survival, with ALS-Ci, ALS-Cbi, ALS-Bi and ALS-FTD having an HR of 1.711 and 1.241, respectively, (p = 0.004; 95% CI 1.187–2.467 for ALS-Ci, ALS-Cbi, ALS-Bi and p = 0.396; 95% CI 0.753–2.046 for ALS-FTD). This shows that the extent of extramotor involvement has a significant impact on survival in patients with ALS. Diagnostic delay had a hazard ratio of 0.997 (p < 0.001) and age of onset a HR of 1.034 (p < 0.001).
Table 3. Multivariable Cox analysis of effect of ECAS, LMN, age of onset, and diagnostic delay on survival.
What is the combined effect of LMN and extramotor involvement on survival?
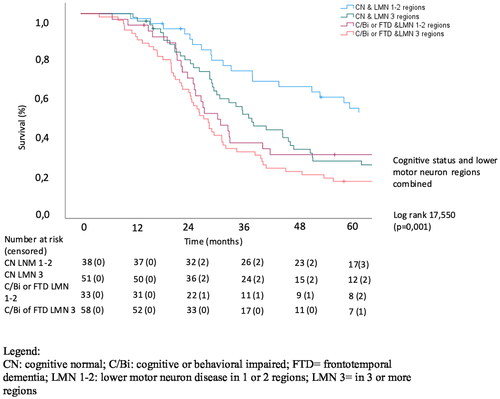
Finally, we wanted to analyze the combined effect of lower motor neuron involvement and extramotor involvement on the survival of patients with ALS. We therefore performed a Kaplan Meier analysis in which patient groups were dichotomized in either having a low or high burden of lower motor neuron disease and cognitive or behavioral impairment. Lower motor neuron disease was scored either as being present in 1 to 2 regions or 3 or more regions and cognition was scored as either cognitively normal or cognitive/behavioral impaired and FTD. This classification showed a strong correlation with survival (p = 0.001; Log Rank Chi square 17.550) with median survival being 61.3 months in the CN & LMN 1-2 regions group versus 26.1 months in the Ci/Bi/Cbi or FTD & LMN 3 regions group. Cognitive and/or behavioral impairment combined with a low burden of LMN involvement (ALS-Ci/Bi/Cbi or ALS-FTD & LMN 1-2 regions group) had a median survival of 27.3 months, which is lower than patients with a high burden of LMN involvement while being cognitively normal (ALS-CN & LMN 3 regions group), with a median survival of 35.7 months. Of note, the differences in survival between the ALS-Ci/Bi/Cbi or ALS-FTD & LMN 1-2 regions group and Ci/Bi/Cbi or FTD & LMN 3 regions group were rather small in this study. Kaplan-Meier curves of the effect of the combined effect of LMN and cognitive/behavioral involvement on survival is shown in .
Figure 4. Analysis of the combined effect of LMN and extramotor involvement on survival.
Discussion
This paper evaluates the link between extra-motor changes, upper and lower motor neuron involvement at diagnosis and survival in ALS patients. We established a significant effect for the number of regions with lower motor neuron involvement on survival and for cognitive/behavioral impairment and FTD on survival. In line with previous studies, the univariable analysis of effect of the number of regions with upper motor neuron involvement on survival, revealed no significant effect. The number of regions affected by LMN involvement was significantly related to survival in the Kaplan-Meier analysis. Mean survival also declined with each subsequent ECAS group (i.e. ALS-CN, ALS-Ci/ALS-Bi/ALS-CBi, ALS-FTD). In the multivariable analysis, diagnostic delay, site of onset, number of LMN regions and cognitive/behavioral impairment and FTD were significantly related to survival.
Both cognitive/behavioral impairment and FTD were associated with a decreased survival, yet for the ALS-FTD group we were not able to find a significant effect, possibly due to the small sample size. The percentage of bulbar onset rose with increasing cognitive/behavioral changes, but site of onset was not significantly associated with survival.
In line with our findings, other studies have found ALS-FTD patients to have a worse prognosis and a shorter survival than patients with FTD without concomitant motor neuron symptoms and patients with ALS without cognitive/behavioral changes (Citation24). Time of onset of cognitive impairment also holds prognostic value, as a previous study revealed that ALS-FTD patients with an initial motor presentation had a significantly shorter median survival than ALS-FTD patients with an initial cognitive presentation (Citation25).
Our study aimed to evaluate the combined effect of lower motor neuron disease and cognition on survival. Classification according to a high or low burden of disease regarding LMN involvement and cognitive/behavioral changes resulted in a strongly significant result (p = 0.001; Log Rank Chi square 17.550). It also showed that patients who were cognitively normal, but presented with a high burden of LMN involvement had a better survival than patients with a lower burden of disease regarding LMN involvement, while having cognitive/behavioral impairment or FTD. Clinicians can use both the extent of lower motor neuron involvement as well as extramotor involvement to evaluate the life expectancy of patients with ALS.
Non-adherence to tube feeding and noninvasive ventilation can possibly explain a difference in survival, as this was the case in the study by Caga et al in 31% and 38% for cognitively normal ALS patients, compared to 72% and 75% for ALS-FTD patients (Citation26). Both respiratory insufficiency as well as nutritional deficiency can also have secondary confounding effect on cognition (Citation19). The presence of neurobehavioral dysfunction in ALS has also shown to reduce the efficacy of life-prolonging therapies such as noninvasive ventilation and percutaneous gastrostomy feeding (Citation27).
There are certain limitations for this article. Firstly, only patients for whom clinical data, EMG analysis and ECAS testing was available, could be included for his study, possibly introducing a selection bias. Patients with cognitive and behavioral problems might have refused additional testing and for other patients the burden of additional neuropsychological testing aside from the regular diagnostic work-up can be too time-consuming or exhausting. Secondly, data was collected from a single center cohort. Future studies could focus on the validation of our findings in a multi-centered approach.
Conclusion
The degree of lower motor neuron involvement was associated with a worse survival and patients who were cognitively normal had a significantly better survival compared to patients with concomitant cognitive and/or behavioral impairment or comorbid FTD. The effect of cognitive impairment on survival was stronger in patients with lower LMN involvement. These results therefore emphasize the need for tools and systems for disease staging at diagnosis which combine motor neuron involvement and extra-motor involvement, since the combination holds complementary prognostic information.
Acknowledgements
This study was supported by a TBM grant from FWO-Vlaanderen (n° T003519N). PVD holds a senior clinical investigatorship of FWO-Vlaanderen (G077121N) and is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the ALS Liga België and the KU Leuven funds “Een Hart voor ALS,” “Laeversfonds voor ALS Onderzoek,” and the “Valéry Perrier Race against ALS Fund.”
Declaration of interest statement
None.
References
- Masrori P, Van Damme P. Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol. 2020;27:1918–29.
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–55.
- Canosa A, Pagani M, Cistaro A, Montuschi A, Iazzolino B, Fania P, et al. 18F-FDG-PET correlates of cognitive impairment in ALS. Neurology 2016;86:44–9.
- Beeldman E, Raaphorst J, Twennaar MK, De Visser M, Schmand BA, De Haan RJ. The cognitive profile of ALS: a systematic review and meta-analysis update. J Neurol Neurosurg Psychiatry. 2016;87:611–9.
- Costello E, Rooney J, Pinto-Grau M, Burke T, Elamin M, Bede P, et al. Cognitive reserve in amyotrophic lateral sclerosis (ALS): a population-based longitudinal study. J Neurol Neurosurg Psychiatry. 2021;92:460–5.
- Beeldman E, Jaeger B, Raaphorst J, Seelen M, Veldink J, Van Den Berg L, et al. The verbal fluency index: Dutch normative data for cognitive testing in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:388–91.
- De Icaza Valenzuela MM, Bak TH, Thompson HE, Colville S, Pal S, Abrahams S. Validation of The Edinburgh cognitive and behavioural ALS screen (ECAS) in behavioural variant frontotemporal dementia and Alzheimer’s disease. Int J Geriatr Psychiatry. 2021;36:1576–87.
- Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17:423–33.
- Reniers W, Schrooten M, Claeys KG, Tilkin P, D'Hondt A, Van Reijen D, et al. Prognostic value of clinical and electrodiagnostic parameters at time of diagnosis in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:341–50.
- Xu Z, Alruwaili ARS, Henderson RD, McCombe PA. Screening for cognitive and behavioural impairment in amyotrophic lateral sclerosis: frequency of abnormality and effect on survival. J Neurol Sci. 2017;376:16–23.
- Ye S, Jin P, Chen L, Zhang N, Fan D. Prognosis of amyotrophic lateral sclerosis with cognitive and behavioural changes based on a sixty-month longitudinal follow-up. PLoS One. 2021;16:e0253279.
- Nguyen C, Caga J, Mahoney CJ, Kiernan MC, Huynh W. Behavioural changes predict poorer survival in amyotrophic lateral sclerosis. Brain Cogn 2021;150:105710.
- Hu WT, Shelnutt M, Wilson A, Yarab N, Kelly C, Grossman M, et al. Behavior matters-cognitive predictors of survival in amyotrophic lateral sclerosis. PLoS One. 2013;8:e57584.
- Devine MS, Ballard E, O'Rourke P, Kiernan MC, Mccombe PA, Henderson RD. Targeted assessment of lower motor neuron burden is associated with survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener. 2016;17:184–90.
- Fujimura-Kiyono C, Kimura F, Ishida S, Nakajima H, Hosokawa T, Sugino M, et al. Onset and spreading patterns of lower motor neuron involvements predict survival in sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82:1244–9.
- Zakharova MN, Abramova AA. Lower and upper motor neuron involvement and their impact on disease prognosis in amyotrophic lateral sclerosis. Neural Regen Res. 2022;17:65–73.
- Kjældgaard AL, Pilely K, Olsen KS, Jessen AH, Lauritsen AØ, Pedersen SW, et al. Prediction of survival in amyotrophic lateral sclerosis: a nationwide, Danish cohort study. BMC Neurol. 2021;21:164.
- Chiò A, Moglia C, Canosa A, Manera U, Vasta R, Brunetti M, et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019;93:E984–e994.
- Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology 2018;91:E1370–e1380.
- Canosa A, Moglia C, Manera U, Vasta R, Torrieri MC, Arena V, et al. Metabolic brain changes across different levels of cognitive impairment in ALS: a 18 F-FDG-PET study. J Neurol Neurosurg Psychiatry 2021;92:357–63.
- Bakker LA, Schröder CD, Spreij LA, Verhaegen M, De Vocht J, Van Damme P, et al. Derivation of norms for the Dutch version of the Edinburgh cognitive and behavioral ALS screen. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:19–27.
- Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis—frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:153–74.
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–77.
- Rusina R, Vandenberghe R, Bruffaerts R. Cognitive and behavioral manifestations in ALS: beyond motor system involvement. Diagnostics 2021;11:624.
- Ahmed RM, Devenney EM, Strikwerda-Brown C, Hodges JR, Piguet O, Kiernan MC. Phenotypic variability in ALS-FTD and effect on survival. Neurology 2020;94:E2005–e2013.
- Caga J, Hsieh S, Lillo P, Dudley K, Mioshi E. The impact of cognitive and behavioral symptoms on ALS patients and their caregivers. Front Neurol. 2019;10:192.
- Chio A, Ilardi A, Cammarosano S, Moglia C, Montuschi A, Calvo A. Neurobehavioral dysfunction in ALS has a negative effect on outcome and use of PEG and NIV. Neurology 2012;78:1085–9.