
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
To determine the average time from Amyotrophic Lateral Sclerosis (ALS) symptom onset to 11 pre-defined milestones, overall and according to ALS progression rate and geographic location.
Methods
Data were drawn from the Adelphi Real World ALS Disease-Specific ProgrammeTM, a point-in-time survey of neurologists caring for people living with ALS (pALS) conducted in France, Germany, Italy, Spain, the United Kingdom and the United States from 2020–2021. ALS progression rate was calculated using time since symptom onset and ALS Functional Rating Scale Revised score.
Results
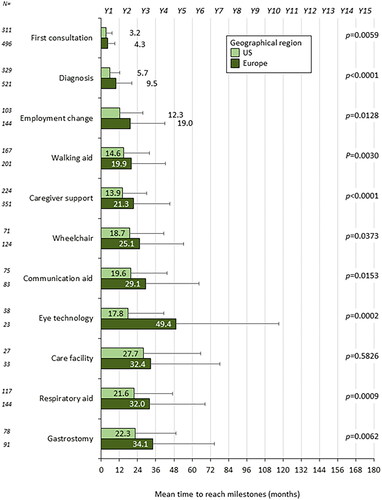
Survey results were available for N = 1003 pALS (progression rate for N = 867). Mean time from symptom onset was 3.8 months to first consultation, 8.0 months to diagnosis, 16.2 months to employment change (part-time/sick leave/retirement/unemployment), 17.5 months to use of a walking aid, 18.5 months to first occurrence of caregiver support, 22.8 months to use of a wheelchair, 24.6 months to use of a communication aid, 27.3 months to use of a respiratory aid, 28.6 months to use of gastrostomy feeding, 29.7 months to use of eye gaze technology and 30.3 months to entering a care facility. Multivariate analysis indicated significant effects of fast (versus slow) progression rate on time to reach all 11 milestones, as well as US (versus European) location, age, body mass index and bulbar onset (versus other) on time to reach milestones.
Conclusions
pALS rapidly reached clinical and disease-related milestones within 30 months from symptom onset. Milestones were reached significantly faster by pALS with fast versus slow progression. Geographic differences were observed.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive neuromuscular disorder characterized by the degeneration of motor neurons in the cortex, bulbar and spinal regions (Citation1,Citation2). Drugs currently approved by the United States (US) Food and Drug Administration for the treatment of ALS are riluzole, edaravone, sodium phenylbutyrate/taurursodiol and tofersen. ALS is often characterized by focal skeletal muscle atrophy and weakness but progresses to involve most muscles, and death due to neuromuscular respiratory failure generally occurs within 3–5 years of symptom onset (Citation1–3). Decline is overall linear and people living with ALS (pALS) have an intrinsic disease progression rate (Citation4).
Loss of function associated with disease progression will over time lead to pALS reaching defined clinical milestones (Citation5). Such milestones provide important prognostic information and have been employed in the development of ALS staging systems used in clinical practice. The ALS Functional Rating Scale-Revised (ALSFRS-R) includes 12 items assessing bulbar function, fine and gross motor skills, and respiratory functions (Citation6), the King’s clinical staging system is based on the number of affected regions and the requirement for gastrostomy or noninvasive ventilation (Citation5,Citation7,Citation8) and Milan Torino Staging system stages relate to loss of independence in four domains (Citation8,Citation9).
The ALS phenotype is, however, highly heterogeneous. There is a wide spectrum of clinical presentations as well as variation in age at onset and progression rate (Citation10–13). Although death usually occurs ≤3 years from symptom onset, 5–10% of pALS live >10 years (Citation4,Citation9,Citation14,Citation15). Factors associated with worse prognosis include older age at onset, bulbar onset disease, frontotemporal dysfunction, faster rate of decline in the ALSFRS-R, lower forced vital capacity, shorter diagnostic delay, definite ALS by El Escorial classification, and the C9orf72 expansion mutation (Citation16).
In order to better inform and empower pALS and their physicians and to assist in the planning and delivery of optimal care, it is important to empirically assess the time from symptom onset to a wide range of relevant milestones – both clinical and wider disease-related – using a representative, multinational dataset. These milestones may impact pALS and their families/caregivers and are associated with increasing healthcare resource utilization (Citation17).
The time between symptom onset and (a) first physician consultation and (b) ALS diagnosis (i.e. diagnostic delay) have been studied previously and determined as 3–6 months and 10–16 months, respectively (Citation18). Diagnostic delay differed according to progression rate (7.8, 15.4 and 17.8 months for pALS with fast/intermediate/slow progression, respectively(Citation19) or 6.1 versus 81.7 months (Citation14])or 16.1 versus 19.2 months (Citation20) for those with slow/fast progression, respectively). However, studies of the time to reach other milestones are less common and results vary widely. For example, utilization of a walker or wheelchair occurred 4.9 months and 10.9–46.7 months post-diagnosis, respectively (Citation17,Citation21); communication-related milestones (such as loss of intelligible verbal communication or utilization of eye-tracking devices for communication) occurred between 10 months post-diagnosis and 6 years post-symptom onset (Citation19,Citation20, Citation22); respiratory aids were initiated 17.0–31.5 months post-symptom onset or 10.0–34.6 months post-diagnosis (Citation5,Citation17,Citation19,Citation21,Citation23,Citation24); gastrostomy feeding was initiated 17.0–27.3 months post-symptom onset or 7.3–31.1 months post-diagnosis (Citation5,Citation21,Citation23–27) and differed according to progression rate (Citation25); and admission into a hospice occurred 16.6 months post-diagnosis (Citation17).
The study objective was to analyze data from the Adelphi Real World ALS Disease-Specific ProgrammeTM (DSPTM) cross-sectional survey with retrospective chart review to ascertain the average time from symptom onset to key milestones, and to compare groups of pALS with different rates of disease progression.
Materials and methods
Study design
DSPsTM are large, multinational, cross-sectional surveys with retrospective chart review conducted in clinical settings (but outside the context of clinical trials), describing current disease management and disease burden from the viewpoint of pALS and their physicians (neurologists). The ALS DSP™ was conducted in France, Germany, Italy, Spain, the United Kingdom (UK) and the US from July 2020 until March 2021.
A complete description of the DSP™ methodology employed has been previously published and validated (Citation28–30); details of recruitment, data collection/aggregation, and ethical approval are provided in Supplementary Appendix 1. Recruited physicians were based in a range of settings, including ALS centers and other clinical locations.
Briefly, data were collected in two stages following informed consent: (Citation31) an online physician-completed chart review about the next 1–10 consecutive consulting pALS that met inclusion criteria (completed via review of available clinical records as well as the judgment and diagnostic skills of the physician, thus reflecting usual clinical practice); and (Citation1) a voluntary self-completion pen and paper form for pALS (results not presented). Physicians were personally responsible for treatment decisions and management of pALS and consulted with ≥2 pALS per month; they were financially incentivized for survey completion. Eligible pALS were aged ≥18 years with a confirmed diagnosis who visited their neurologist for routine care.
Ethical approval
The survey was reviewed by the Western Institutional Review Board and granted an exemption (protocol number: AG8802) as data were collected in such a way that individuals could not be identified; all data were aggregated and de-identified before receipt. Data collection was in line with relevant legislation and guidelines. Online survey components used Forsta software, and all data were stored in SPSS Statistics format.
Rate of ALS progression
The ALSFRS-R is a validated instrument for monitoring disability progression in pALS (Citation6). ALSFRS-R score and time since symptom onset were employed to calculate ‘points of score lost per month’ or rate of progression:
Based upon the findings for this study population, disease progression was divided into tertiles of slow (≤0.36 points/month), intermediate (>0.36 – <0.77) and fast (≥0.77) rates.
Physician-reported milestones
Following literature review and discussion with physicians, pALS and caregivers, 11 disease milestones were selected: (Citation31) date when person first saw a healthcare professional with concerns over symptoms later attributed to ALS, (Citation1) date of ALS diagnosis, (Citation2) change in employment: working part time, on long term sick leave, retired or unemployed, (Citation3) first use of a walking aid: walking stick/cane, walking frame or wheeled walker (Citation4) first occurrence of caregiver support: professional or nonprofessional (partner/spouse, son/daughter, other relative[s], friend[s], or other), (Citation5) first use of a wheelchair, (Citation6) first use of a communication aid: voice amplification system, text-to-speech system, or communication boards, (Citation7) first use of a respiratory aid: noninvasive ventilation (NIV; continuous positive airway pressure or bi-level positive airway pressure) or tracheostomy (Citation8) initiation of gastrostomy feeding, (Citation9) first use of eye gaze technology/eye gesture communication, and (Citation10) pALS became a resident of a care facility: hospice (inpatient or home hospice), nursing home, residential home or assisted living. residence. Aggregate mean time (months) from symptom onset to each milestone was reported.
Statistical analysis
Results were reported overall, stratified by rate of disease progression and region (US versus Europe). Sample size, mean and standard deviation (numeric variables) or sample size and number/percent (categorical variables) were calculated and compared using t-test, Fishers Exact test, Chi Squared test, or Analysis of Variance. Data were descriptive, with no formal hypothesis testing or multiple comparison controls conducted.
Cox regressions (Citation27) were performed to identify independent predictors of time to each milestone; risk factors included were location (US versus Europe), ALS progression rate (fast versus slow and intermediate versus slow), age, sex (female versus male), body mass index (BMI) and region of onset (bulbar versus non-bulbar).
Missing data were not imputed; the number of pALS included could vary between variables and was reported for each analysis.
All statistical analyses were performed using STATA v17 (StataCorp. 2021. Stata Statistical Software: Release 17. College Station, TX: StataCorp LLC).
Results
People living with ALS
For 867 pALS, progression rate was calculated, tertiles determined and patients classified as having fast (34.0%), intermediate (33.1%) or slow (32.9%) progression (). Of these 867 individuals, 114 were from France, 65 from Germany, 119 from Italy, 153 from Spain, 76 from the UK, and 340 from the US.
Table 1. Demographic and clinical characteristics of people with ALS, overall and according to disease progression category.
Table 2. Time from symptom onset to milestones according to progression rate (all people with ALS).
Table 3. Time from symptom onset to milestones according to progression rate (people with ALS in the US).
Table 4. Time from symptom onset to milestones according to progression rate (people with ALS in Europe).
A faster rate of ALS progression was significantly associated with older patient age, lower ALSFRS-R score, shorter times since symptom onset and diagnosis (). Type of treatment currently received also varied according to progression rate ().
Characteristics also differed significantly between pALS in the US and Europe; European pALS were older, had a lower BMI, were more likely to be of white/Caucasian ethnicity, had a longer time since symptom onset and diagnosis, and had a lesser increase in ALSFRS-R points/month (). Type of treatment currently received also varied according to location ().
Time from ALS symptom onset to milestones: overall population
On average, within 30 months of symptom onset pALS had reached the milestones of wheelchair use (indicating loss of mobility), use of a respiratory aid (indicating loss of respiratory function) and gastrostomy (indicating loss of nutritional function) (; ).
Figure 1. Time from symptom onset to milestones for all people with ALS.
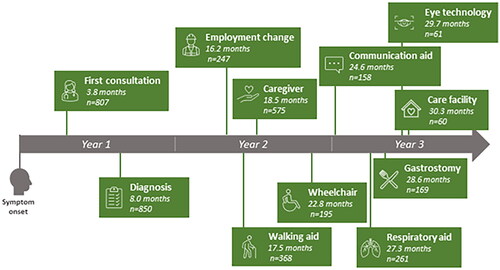
Figure 2. Time from symptom onset to milestones according to progression rate. Y: year. Error bars show 95% confidence intervals
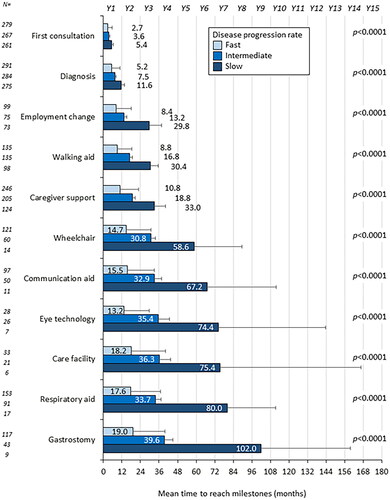
Figure 3. Time from symptom onset to milestones according to region (Europe versus US). US: United States of America; Y: year. Error bars show 95% confidence interval
Time from ALS symptom onset to milestones: disease progression rate
Cox regression results showed a significant effect of fast (versus slow) progression rate on the time to reach all 11 milestones, and of intermediate (versus slow) progression rate on time to reach 9/11 milestones (Supplementary Tables 2–12). For example, among pALS with fast progression, ‘diagnosis’ was reached by 5.2 months, compared with 7.5 months in the intermediate category and 11.6 months in the slow category (; –5). The latest milestone reached was ‘gastrostomy’; pALS with fast progression reached this by 19.0 months, compared with 39.6 and 102.0 months for those in intermediate and slow categories, respectively (; ).
Time from ALS symptom onset to milestones: Europe versus US
pALS in the US and Europe differed in terms of classification into fast (US 41.8%, Europe 29.0%), intermediate (US 28.5%, Europe 36.1%) or slow (US 29.7%, Europe 34.9%) progression rate categories (Supplementary Table 1). When comparing those with fast progression only, there were numeric differences (statistical comparisons not performed) between US and European pALS in average progression rate (US 1.49 points/month, Europe 1.41 points/month), age (US 61.9 years, Europe 64.6 years) and gender (US 70.4% male, Europe 57.5% male) (Supplementary Table 1).
Multivariate analysis indicated that pALS in the US (versus Europe) had significant reduction in time to reach six milestones: ‘first consultation’, ‘diagnosis’, ‘eye gaze technology’, ‘gastrostomy’, ‘respiratory aid’ and ‘caregiver’ (Supplementary Tables 2–12). The time difference between pALS in the two regions ranged from 1.1 months for first consultation to 31.6 months for eye gaze technology ().
Time from ALS symptom onset to milestones: other factors
Results of multivariate analysis indicated that bulbar site of onset (versus other site) significantly reduced time to reach four of the milestones (Supplementary Tables 2–12). Effects by age and body mass index were observed for a small number of milestones (three and one, respectively) though hazard ratios were approaching 1 (Supplementary Tables 2–12).
Discussion
The findings of this large, multinational, cross-sectional survey with retrospective chart review demonstrate that pALS rapidly reach various disease-related and clinically important milestones. The rate of disease progression was heterogeneous. pALS with a fast progression rate reached each of 11 milestones significantly sooner than those with a slow rate, from the first consultation and diagnosis through to the initiation of gastrostomy feeding. Reaching these milestones would generally suggest an individual had experienced substantial loss of mobility, respiratory and nutritional function. Six milestones were reached significantly sooner by pALS in the US versus Europe and four were reached significantly sooner by pALS with bulbar versus other site of onset.
The average diagnostic delay of 8.0 months observed in this study was shorter than has been reported previously (Citation18), though within the range observed in a single-center study at Massachusetts General Hospital (Citation32). The 9.5-month delay observed in European countries was more aligned. Previous studies in which diagnostic delay was described for pALS with different progression rates have also generally observed longer delays in each subgroup, compared with this study (Citation13,Citation33,Citation34).
After diagnosis and treatment initiation, information about when pALS may expect to reach particular clinical inflection points in their disease progression has practical value in terms of management planning for pALS, their families and the multi-disciplinary care team. In addition, milestones relating to swallowing, walking, self-care, communicating and breathing are associated with decreased quality of life as well as increased healthcare resource use and costs (Citation8,Citation9) and have important prognostic value (Citation5). In this study, mobility, communication and respiratory aids were utilized sooner by pALS with a faster progression rate (fast or intermediate versus slow, except for intermediate versus slow time to wheelchair). Overall, wheelchairs were utilized at a broadly similar time point to a previous US study (Citation17) but substantially earlier than in Italy (Citation21). Communication aids were utilized later compared with previous studies in Italy (Citation19) and Finland (Citation22), but substantially sooner than in a different Italian study (Citation20) (though all pALS in this study were essentially locked in and 70% had a tracheostomy and were mechanically ventilated [ALS-related], suggesting that this was provided when the person had reached a point of marked disability). This suggests that there may be variability as to when health systems provide eye-tracking devices, which are relatively expensive. Respiratory aids were initiated at a comparable time point to previous reports (Citation5,Citation17,Citation21,Citation23,Citation24). Time to gastrostomy feeding initiation was within the range reported previously (Citation5,Citation19,Citation21,Citation23–26). Given that European recommendations around NIV initiation in ALS are less stringent, one might expect that this would commence earlier compared to the US where most insurers follow stricter Medicare guidelines (Citation35–37). Time to gastrostomy was comparable with previous studies for the fast progression group only (Citation25). Finally, timing of admission into a care facility was approximately aligned with a US study (Citation17).
Taken together, the average loss of mobility, respiratory and nutritional function 30 months post-symptom onset in this study demonstrates the rapid progression of ALS. This also aligns with average survival of 36 months from symptom onset (Citation1–3).
Several differences between pALS in Europe and the US should also be considered. Mean BMI was lower in Europe, in line with known differences (obesity rates are 16% in the European Union (Citation38) and 42% in the US (Citation39). pALS often experience weight loss before onset of other symptoms and further loss of weight as the disease progresses; lower BMI earlier in life may increase ALS risk and decrease survival (Citation40–46). Secondly, a greater percentage of European participants were white/Caucasian, compared with the US, though this difference was broadly consistent with observed figures from available national census data (Citation47,Citation48). Thirdly, edaravone was approved in the US but not Europe at the time of the survey. Finally, pALS in Europe were ∼2 years older (again, broadly consistent with national census data (Citation49,Citation50) and exhibited a longer time since symptom onset (∼8 months) versus pALS in the US. Time since diagnosis was ∼4 months longer versus pALS in the US, which may relate to the longer diagnostic delay. However, ALSFRS-R scores were similar, indicating similar severity. In addition, the US cohort comprised a higher proportion of pALS with a fast progression rate versus the European cohort, and in the US pALS with a fast rate were younger and had a higher proportion of males.
Demographics and clinical characteristics also differed significantly between progression groups. This included site of onset, as documented previously (Citation4), and age. Different therapeutic strategies may be appropriate for pALS with differing progression rates. Current treatment differed in this study (no ALS-approved treatment in 15.4%, 16.4% and 27.5% of people in slow/intermediate/fast progression groups, respectively). This may be due to differences in treatment approach, or people in later stages may be less likely to continue receiving treatment due to extensive function loss.
It has been reported that ALS management, from diagnosis to prognosis, is currently suboptimal (Citation51). It is recommended that people with symptoms suggestive of ALS are assessed as soon as possible and early diagnosis pursued (Citation36). pALS and relatives/carers should receive regular support from a multidisciplinary care team, symptom management should be attempted, and devices and aids introduced at appropriate times to improve survival and quality of life (Citation36,Citation52). Furthermore, effective therapies that slow progression would potentially reduce the burden of ALS by delaying the time to reach some milestones, which could help with planning to optimize multidisciplinary care.
The study has several limitations. First, the DSPTM is not based on a true random sample of physicians or pALS; participation is influenced by willingness to complete the survey. Minimal criteria governed physician selection and identification of pALS was based on physician judgment (reflecting usual classification of patients within a clinical context). Given the rarity of ALS, this was unlikely to be an issue. Secondly, the study design prevents conclusions about causal relationships. Thirdly, recall bias may affect physician responses (a common limitation of surveys), though data were collected during clinic appointments when physicians would be expected to have access to medical records. Finally, as missing data were not imputed, the number of pALS included could differ for assessed variables.
In conclusion, this multinational survey of physicians treating 867 pALS found that they rapidly reached various milestones, on average losing mobility, respiratory function and nutritional function within 30 months from symptom onset. Those with fast progression rates reached all key milestones significantly sooner than those with slow rates.
Data deposition
All data, i.e. methodology, materials, data and data analysis, that support the findings of this survey are the intellectual property of Adelphi Real World. All requests for access should be addressed directly to Jennifer Mellor at [email protected].
Table 5. Time from symptom onset to milestones according to region (US or Europe).
Supplemental Material
Download MS Word (30.5 KB)Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
PG, JB and SAR own stock in and are employees of Cytokinetics Inc.
JM, JW, LE, NB, HI, OT and GC are employees of Adelphi Real World
Data collection was undertaken by Adelphi Real World as part of an independent survey, entitled the Adelphi ALS Disease Specific Programme™, sponsored by multiple pharmaceutical companies, one of which was Cytokinetics Inc. The study described here using data from the Adelphi ALS Disease Specific Programme was funded by Cytokinetics Inc.
Medical writing support under the guidance of the authors was provided by Bethan Hahn, PhD, on behalf of Adelphi Real World and Cytokinetics, in accordance with Good Publication Practice guidelines (Citation31).
References
- Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–28.
- Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;77:162–72.
- Qureshi M, Schoenfeld DA, Paliwal Y, Shui A, Cudkowicz ME. The natural history of ALS is changing: improved survival. Amyotroph Lateral Scler. 2009;10:324–31.
- Chiò A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler. 2009;10:310–23.
- Roche JC, Rojas-Garcia R, Scott KM, Scotton W, Ellis CE, Burman R, et al. A proposed staging system for amyotrophic lateral sclerosis. Brain. 2012;135:847–52.
- Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (Phase III). J Neurol Sci. 1999;169:13–21.
- Balendra R, Al Khleifat A, Fang T, Al-Chalabi A. A standard operating procedure for King’s ALS clinical staging. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:159–64.
- Corcia P, Beltran S, Lautrette G, Bakkouche S, Couratier P. Staging amyotrophic lateral sclerosis: a new focus on progression. Rev Neurol. 2019;175:277–82.
- Chiò A, Hammond ER, Mora G, Bonito V, Filippini G. Development and evaluation of a clinical staging system for amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2015;86:38–44.
- Kimura F, Fujimura C, Ishida S, Nakajima H, Furutama D, Uehara H, et al. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006;66:265–7.
- Qureshi MM, Hayden D, Urbinelli L, Ferrante K, Newhall K, Myers D, et al. Analysis of factors that modify susceptibility and rate of progression in amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler. 2006;7:173–82.
- Takeda T, Kitagawa K, Arai K. Phenotypic variability and its pathological basis in amyotrophic lateral sclerosis. Neuropathology. 2020;40:40–56.
- Witzel S, Wagner M, Zhao C, Kandler K, Graf E, Berutti R, et al. Fast versus slow disease progression in amyotrophic lateral sclerosis–clinical and genetic factors at the edges of the survival spectrum. Neurobiol Aging. 2022;119:117–26.
- Hardiman O, Al-Chalabi A, Brayne C, Beghi E, van den Berg LH, Chio A, et al. The changing picture of amyotrophic lateral sclerosis: lessons from European registers. J Neurol Neurosurg Psychiatry. 2017;88:557–63.
- Rosenbohm A, Peter RS, Erhardt S, Lulé D, Rothenbacher D, Ludolph AC, et al. Epidemiology of amyotrophic lateral sclerosis in Southern Germany. J Neurol. 2017;264:749–57.
- Westeneng HJ, Debray TPA, Visser AE, van Eijk RPA, Rooney JPK, Calvo A, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17:423–33.
- Meng L, Bian A, Jordan S, Wolff A, Shefner JM, Andrews J. Profile of medical care costs in patients with amyotrophic lateral sclerosis in the Medicare programme and under commercial insurance. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:134–42.
- Richards D, Morren JA, Pioro EP. Time to diagnosis and factors affecting diagnostic delay in amyotrophic lateral sclerosis. J Neurol Sci. 2020;417:117054.
- Vacchiano V, Di Stasi V, Rizzo G, Giannoccaro MP, Donadio V, Bartolomei I, et al. Prognostic value of EMG genioglossus involvement in amyotrophic lateral sclerosis. Clin Neurophysiol. 2021;132:2416–21.
- Spataro R, Ciriacono M, Manno C, La Bella V. The eye-tracking computer device for communication in amyotrophic lateral sclerosis. Acta Neurol Scand. 2014;130:40–5.
- Beghi E, Millul A, Logroscino G, Vitelli E, Micheli A,. Outcome measures and prognostic indicators in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:163–7.
- Makkonen T, Ruottinen H, Puhto R, Helminen M, Palmio J. Speech deterioration in amyotrophic lateral sclerosis (ALS) after manifestation of bulbar symptoms. Int J Lang Commun Disord. 2018;53:385–92.
- Fileccia E, De Pasqua S, Rizzo G, Di Stasi V, Vacchiano V, Avoni P, et al. Denervation findings on EMG in amyotrophic lateral sclerosis and correlation with prognostic milestones: Data from a retrospective study. Clin Neurophysiol. 2020;131:2017–22.
- Talman P, Duong T, Vucic S, Mathers S, Venkatesh S, Henderson R, et al. Identification and outcomes of clinical phenotypes in amyotrophic lateral sclerosis/motor neuron disease: Australian national motor neuron disease observational cohort. BMJ Open. 2016;6:e012054.
- Mariani L, Ruoppolo G, Cilfone A, Cocchi C, Preziosi Standoli J, Longo L, et al. Progression of oropharyngeal dysphagia in amyotrophic lateral sclerosis: a retrospective cohort study. Dysphagia 2022;37:868–78.
- Vergonjeanne M, Fayemendy P, Marin B, Penoty M, Lautrette G, Sourisseau H, et al. Predictive factors for gastrostomy at time of diagnosis and impact on survival in patients with amyotrophic lateral sclerosis. Clin Nutr. 2020;39:3112–8.
- Cox DR. Regression models and life-tables. JR StatSoc: Series B (Methodol) 1972;34:187–202.
- Anderson P, Benford M, Harris N, Karavali M, Piercy J. Real-world physician and patient behaviour across countries: disease-specific programmes - a means to understand. Curr Med Res Opin. 2008;24:3063–72.
- Babineaux SM, Curtis B, Holbrook T, Milligan G, Piercy J. Evidence for validity of a national physician and patient-reported, cross-sectional survey in China and UK: the disease specific programme. BMJ Open. 2016;66:e010352.
- Higgins V, Piercy J, Roughley A, Milligan G, Leith A, Siddall J, et al. Trends in medication use in patients with type 2 diabetes mellitus: a long-term view of real-world treatment between 2000 and 2015. Diabetes Metab Syndr Obes. 2016;9:371–80.
- DeTora LM, Lane T, Sykes A, DiBiasi F, Toroser D, Citrome L. Good publication practice (GPP) guidelines for company-sponsored biomedical research: 2022 update. Ann Intern Med. 2023;176:eL220490.
- Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A, et al. Diagnostic timelines and delays in diagnosing amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:453–6.
- Lu CH, Petzold A, Topping J, Allen K, Macdonald-Wallis C, Clarke J, et al. Plasma neurofilament heavy chain levels and disease progression in amyotrophic lateral sclerosis: insights from a longitudinal study. J Neurol Neurosurg Psychiatry. 2015;86:565–73.
- Rong P, Yunusova Y, Eshghi M, Rowe HP, Green JR. A speech measure for early stratification of fast and slow progressors of bulbar amyotrophic lateral sclerosis: lip movement jitter. Amyotroph Lateral Scler Frontotemporal Degener. 2020;21:34–41.
- National institute for Health and Care Excellence. Motor neurone disease: assessment and management [Internet]. 2023 [cited 2020 Jan 15]. Available from: https://www.nice.org.uk/guidance/ng42
- Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, et al. EFNS guidelines on the clinical management of amyotrophic lateral sclerosis (MALS)–revised report of an EFNS task force. Eur J Neurol. 2012;19:360–75.
- ResMed. Respiratory assist device (RAD) coverage guidelines [Internet]. 2019 [cited 2020 Jan 15]. Available from: https://www.resmed.com/us/dam/documents/articles/1010293_RAD_Guidelines.pdf
- Eurostat Statistical Office of the European Union. Body mass index (BMI) by sex, age and educational attainment level [Internet]. 2022 [cited 2023 Jun 13]. Available from: https://ec.europa.eu/eurostat/databrowser/view/HLTH_EHIS_BM1E__custom_1162105/bookmark/table?lang=en&bookmarkId=6f21bd9e-cbe6-4467-9821-2050435af363
- Centers for Disease Control and Prevention. Adult Obesity Facts [Internet]. 2022 [cited 2023 Jun 13]. Available from: https://www.cdc.gov/obesity/data/adult.html
- Moglia C, Calvo A, Grassano M, Canosa A, Manera U, D'Ovidio F, et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population-based cohort. J Neurol Neurosurg Psychiatry. 2019;90:666–73.
- Peter RS, Rosenbohm A, Dupuis L, Brehme T, Kassubek J, Rothenbacher D, et al. Life course body mass index and risk and prognosis of amyotrophic lateral sclerosis: results from the ALS registry Swabia. Eur J Epidemiol. 2017;32:901–8.
- O'Reilly ÉJ, Wang M, Adami H-O, Alonso A, Bernstein L, van den Brandt P, et al. Prediagnostic body size and risk of amyotrophic lateral sclerosis death in 10 studies. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19:396–06.
- Gallo V, Wark PA, Jenab M, Pearce N, Brayne C, Vermeulen R, et al. Prediagnostic body fat and risk of death from amyotrophic lateral sclerosis: the EPIC cohort. Neurology 2013;80:829–38.
- Mariosa D, Beard JD, Umbach DM, Bellocco R, Keller J, Peters TL, et al. Body mass index and amyotrophic lateral sclerosis: a study of US Military Veterans. Am J Epidemiol. 2017;85:362–71.
- Goutman SA, Boss J, Iyer G, Habra H, Savelieff MG, Karnovsky A, et al. Body mass index associates with amyotrophic lateral sclerosis survival and metabolomic profiles. Muscle Nerve. 2023;67:208–16.
- Dardiotis E, Siokas V, Sokratous M, Tsouris Z, Aloizou AM, Florou D, et al. Body mass index and survival from amyotrophic lateral sclerosis: a meta-analysis. Neurol Clin Pract. 2018;8:437–44.
- Office for National Statistics. Ethnicity Facts and Figures: Population of England and Wales [Internet]. 2022 [cited 2023 Nov 6]. Available from: https://www.ethnicity-facts-figures.service.gov.uk/uk-population-by-ethnicity/national-and-regional-populations/population-of-england-and-wales/latest#by-ethnicity
- United States Census Bureau. Racial and Ethnic Diversity in the United States: 2010 Census and 2020 Census [Internet]. 2022 [cited 2023 Nov 6]. Available from: https://www.census.gov/library/visualizations/interactive/racial-and-ethnic-diversity-in-the-united-states-2010-and-2020-census.html
- Eurostat Statistical Office of the European Union. Population Structure and Ageing [Internet]. 2023 [cited 2023 Nov 6]. Available from: https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Population_structure_and_ageing&stable=1#Median_age_is_highest_in_Italy_and_lowest_in_Cyprus
- United States Census Bureau. America Is Getting Older [Internet]. 2023 [cited 2023 Nov 6]. Available from: https://www.census.gov/newsroom/press-releases/2023/population-estimates-characteristics.html
- Feldman EL, Goutman SA, Petri S, Mazzini L, Savelieff MG, Shaw PJ, et al. Amyotrophic lateral sclerosis. Lancet. 2022;400:1363–80.
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the. Neurology 2009;73:1227–33.