
Abstract
Based on our previous work on the PLGA nanoparticles modified with biotinylated chitosan (Bio-CS-PLGA NPs), we further studied the stability, toxicity, pharmacokinetics, and in vivo efficacy. The safety of NPs was studied through single-dose toxicity test in mice, and the result showed that NPs were well tolerated at the dose of 300 mg/kg. Compared with the free EPB group, the NPs group exhibited higher plasma drug concentration, longer half-life time. EPB-loaded NPs significantly inhibited the tumor growth compared to free EPB. All results suggested that Bio-CS-PLGA NPs were stable, safe, and showed a promising potential on targeted drug delivery.
Introduction
Nanotechnology has expanded widely over the last decades not only to involve application areas for information technology and electronics, but also pharmaceuticals, bringing a whole new perspective into health and medicine (Farokhzad and Langer Citation2009). There has been tremendous interest in using nanoparticles (NPs) in drug delivery systems for transport and targeting of specific compartments, normally inaccessible to some pharmaceutics (Paolo et al. Citation2013; Sedigheh et al. Citation2016).
Adverse effects of pharmaceuticals can be attributed to their lack of targeting specificity (Nel et al. Citation2006). Improving this parameter by implementing a nanocarrier could decrease the risk of unwanted adverse effects (Adny et al. Citation2016; Fatemeh et al. Citation2016; Vander et al. Citation2013), where an intravenous (i.v.) injection of the cancer drug doxorubicin in rats showed a reduced cardio- and testicular toxicity when being encapsulated by NPs (Gabizon et al. Citation2003; Guo et al. Citation2014; Laura et al. Citation2016; Pereverzeva et al. Citation2008).
Active targeting to cancer cells is a widely acceptable strategy for drug delivery, in which ligand conjugates can directly interact with complementary moieties on the surface of target cancer cells (Alexander-Bryant et al. Citation2013). Polylactic-co-glycolic acid (PLGA) is FDA-approved biodegradable polymer. Various kinds of NPs prepared from PLGA have been investigated and showed great potential as carriers for cancer therapy (Giarra et al. Citation2016). However, the main drawback of PLGA NPs is their lack of suitable functional groups for efficient covalent conjugation with bioactive ligands (Zhu et al. Citation2014). Chitosan NPs have many advantages such as bioadhesive property and prolonged drug release properties (Walter et al. Citation2016). In our previous reports, we prepared biotinylated chitosan (Bio-CS)-based surface modified PLGA NPs to be used as drug carriers (Chen et al. Citation2009, Citation2014), and also found that using Bio-CS conjugate as a surface-modified material could realize the distribution of functional biotin groups on the surface of PLGA NPs. In MCF-7 tumor-bearing nude mice, Bio-CS-PLGA NPs were mainly distributed in the tumor after 24 h post administration (Chen et al. Citation2016). But the biological effects of Bio-CS-PLGA NPs in vivo have not been investigated deeply up to date. There is a potential risk when using nanomaterials, a consideration supported by findings of toxicological effects of e.g., metal oxides, carbon nanotubes, or NPs that may promote inflammation, cytotoxicity, and allergic sensitization (Kristina et al. Citation2015; Vega-Villa et al. Citation2008). Herein, in the present study, we studied the stability and in vivo toxicity of Bio-CS-PLGA NPs. Due to limited information available in terms of potential toxicity, we conducted study of systemic NPs administration by i.v. injection to mimic the delivery route of a drug carrier. We assessed the toxicological endpoints by histopathology, hematology, and clinical chemistry parameters. Moreover, epirubicin (EPB) was loaded into Bio-CS-PLGA NPs and its pharmacokinetics was also assessed in rats to compare to the free drug. We also established a xenograft model by implanting MCF-7 cells subcutaneously into nude mice and further evaluated the in vivo antitumor activity of EPB-loaded Bio-CS-PLGA NPs using free EPB as the controls.
Materials and methods
Materials
PLGA (MW 10,000–30,000, the ratio of lactic to glycolic acid in the copolymer is 50:50, with uncapped end-group) was purchased from Shandong Institute of Medical Instruments. CS (low molecular weight, >80% deacetylation, Brookfield viscosity 20,000 cps) and polyvinyl alcohol (PVA, wt 30,000–70,000) was purchased from Sigma-Aldrich (St. Louis, Mo). Biotin was purchased from Sigma-Aldrich. Ultrapure water (Millipore, Bedford, MA) was used throughout the experiment. All other chemical reagents were of analytical or HPLC grade and obtained from commercial sources. ICR mice and Wistar rats were purchased from the Institute of Radiology, Chinese Academy of Medical Science. All animal experiments were performed in compliance with the Institutional Animal Care and Use Committee (IACUC) guidelines.
Synthesis of biotinylated CS (Bio-CS)
Bio-CS was synthesized according to our previous published work (Chen et al. Citation2016). In brief, biotin (0.5 mol), dicyclohexylcarbodiimide (DCC, 0.5 mol), and N-hydroxysuccinimide (NHS, 0.6 mol) were mixed in 20 mL DMF followed by stirring for 18 h. The reaction mixture was filtered and the excess of diethyl ether was then added to get the precipitate of NHS-biotin. The thus-obtained crude product of NHS-biotin was purified by the recrystallization in 2-propanol (70 °C). To conjugate biotin with CS, 10 mL 1% CS in acetic acid solution was added dropwise into certain amount of NHS-Bio in DMF, and the mixture was then continuously stirred for 20 h at room temperature. After that, the mixture was dialyzed against deionized water using dialysis tubing with Mw cut-off of 8000 Da (Spectrum Laboratories, Rancho Dominguez, CA, USA) for 48 h with 6-water exchanges, and then freeze-dried to obtain Bio-CS.
Preparation of biotinylated chitosan surface modified PLGA nanoparticle
PLGA NPs were prepared by the modified solvent displacement method as described previously (Chen et al. Citation2009, Citation2014). To activate the carboxylic groups on the surfaces of PLGA NPs, 100 mg of PLGA NPs powder was dispersed in 5 mL of PBS (pH5.0) containing EDC (40 mg) and NHS (20 mg). Bio-CS (60 mg) was then added to this suspension and the resulting mixture was stirred for 24 h at ambient temperature. Upon completion of the reaction, the excess coupling reagent was removed by centrifugation at 15,000 rpm for 10 min to get the powders of Bio-CS-PLGA NPs. Furthermore, blank Bio-CS-PLGA NPs were also prepared at the same time.
Characterization of the NPs
Surface morphologies of NPs were visualized by a field emission scanning electron microscopy (SEM, Jeol, JSM-6700F, Akishima, Tokyo, Japan) system at an accelerating voltage of 5 kV. Before imaging, the freeze-dried samples were coated by platinum for 4 min. Atomic force microscopy (AFM) images were collected on a Nanoscope V atomic force microscope (multimode 8, Veeo, USA).
The particle size and size distribution were measured by 90 Plus Particles Size Analyzer (Nano ZS 90, Malvern Instruments Ltd., Malvern, UK) based on the laser light scattering technique. Before measurement, the freshly prepared NPs were appropriately diluted and the final concentrations were about 0.5 mg/mL. The zeta potential was determined by Zeta Potential Analyzer in deionized water. The mean particle size and mean zeta potential values were calculated from five measurements per sample. Drug loading content and encapsulation efficiency were determined as described previously (Chen et al. Citation2009, Citation2014).
The in vitro releases of EPB from above NPs were studied using dynamic dialysis method in media at physiological pH (PBS, pH 7.4) and in acidic media (PBS, pH 5.5), respectively. Briefly, 20 mg of EPB-loaded NPs was suspended in 5 mL of PBS and vibrated in a water-bath shaker at 75 rpm at 37 °C. At designated time intervals, all samples were centrifuged at 15,000 rpm for 10 min, the supernatants were taken out for the detection of EPB content and 5 mL of fresh release media were then added at the same time. The EPB release amounts were determined by HPLC analysis as described (Chen et al. Citation2014, Citation2016). For the evaluation of the release kinetics, the release data were fitted into first-order, zero-order, and Higuchi models. The subsequent selection of the optimum model was based on the comparison of the relevant correlation coefficients.
Storage stability of Bio-CS-PLGA NPs
Bio-CS-PLGA NPs samples were stored in normal saline at 4 °C for a period of 8 weeks. Particle sizes and zeta potentials of Bio-CS-PLGA NPs suspensions were measured once a week. Furthermore, the surface morphologies of the NPs were visualized by SEM after 4 weeks and 8 weeks, respectively. In order to study the stability of Bio-CS-PLGA NPs powder, the NPs lyophilized powder were stored for 12 months at 4 °C, particle sizes and zeta potentials of Bio-CS-PLGA NPs suspensions were measured once a months.
In vivo acute toxicity
The acute toxicity of Bio-CS-PLGA NPs was evaluated in vivo according to the previous reported method (Chen et al. Citation2016; Kristina et al. Citation2015; Vega-Villa et al. Citation2008). Adult male and female ICR mice (20–24 g) were randomly divided equally into two groups, each with 12 mice. The experimental group received a single i.v. dose of blank Bio-CS-PLGA NPs (300 mg/kg), the control group was treated with a single i.v. dose of same amount of normal saline to make a comparison keeping similar environment. All animals were fed with normal diet, and water was provided ad libitum. Animals were observed carefully for the onset of any signs of toxicity and monitored for changes in food intake and body weight (BW) at 1, 8, and 15 days. Before being sacrificed at 15 days, we assessed the hematology and clinical biochemical parameters by hematology analyzer (MEK-6318K, Nihon Kohden, Japan) and biochemical analyzer (microlab300, Vital Scientific, the Netherlands) at 15 days.
After being sacrificed at 15 days, internal organs of each animal were harvested and observed grossly. The weights of brain, liver, spleen, kidneys, adrenal glands, lungs, and thymus were recorded and individual organ to BW ratios were calculated. All gross lesions were retained and examined. The histological changes, such as acute-chronic inflammatory symptoms, fibroblastic proliferation, and any other inflammation symptoms were observed. For further histological examinations, specimens of major organs such as heart, liver, spleen, lung, and kidney were fixed in 10% phosphate-buffered formalin, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E).
In vivo pharmacokinetics
In vivo pharmacokinetic study was conducted by the routine method according to the papers (Moghimi et al. Citation2001; Tang et al. Citation2010; Xiong et al. Citation2012; Yu et al. Citation2015). The drug content of the Bio-CS-PLGA NPs was determined according to our previous study and drug-loaded NPs with 3.46% was selected to carry out the pharmacokinetics studies. Female Wistar rats of 180–250 g and 4–6 weeks old were held in an air-conditioned facility, provided with standard food and filtered water. Animals were randomly assigned to two groups, each with six rats, which received an i.v. injection via the tail vein of free EPB and the Bio-CS-PLGA NPs solution in saline at 10 mg/kg equivalent dose, respectively. All animals were observed for mortality, general condition, and potential clinical signs.
The blood samples were collected with heparinized tube at 0 (pre-dose), 0.0167, 0.0833, 0.167, 0.5, 1, 2, 4, 8, 12, 24, and 48 h post-treatment. Plasma samples were harvested by centrifugation at 3000 rpm for 15 min and stored at −20 °C until analysis. Liquid–liquid extraction was performed prior to the HPLC analysis. Briefly, the plasma (100 μL) was mixed with dichloromethane–methanol (4:1, v/v) on a vortex-mixer for 3 min to extract the drug. Upon centrifugation at 10,000 rpm for 15 min, the upper aqueous layer was removed by aspiration and the organic layer was transferred to a tube and evaporated under nitrogen at 50 °C. The residue was dissolved in 100 μL of anhydrous methanol by vortex. For the HPLC analysis, the C-18 column was used and the mobile phase (0.02 M KH2PO4/CH3CN/CH3OH =49:17:34 v/v/v) was delivered at a rate of 1 mL/min. Sample (20 μL) was injected and the column effluent was detected with a UV detector at 232 nm. The main pharmacokinetic parameters were calculated by DAS 1.0 (Anhui, China) program. Bioavailability (BA) is a measurement of the rate and extent of a therapeutically active drug that reaches the systemic circulation and is available at the site of action. When a medication is administered intravenously, its bioavailability is 100%. When the standard consists of intravenously administered drug, this is known as relative bioavailability (BAR). The BAR of Bio-CS-PLGA NPs after administration was calculated using the following formula (Florescu et al. Citation2014; Tang et al. Citation2010).
where AUC is the area under the curve.
In vivo antitumor study
Female BALB/c nude mice were inoculated subcutaneously with 200 mL cell suspensions containing 1 × 106 MCF-7 cells. When the tumors grew up to 300–400 mm3 in average, the mice were randomly divided into three groups, respectively treated with physiological saline (control), free EPB, and Bio-CS-PLGA NPs. The treatments with EPB dose of 3 mg/kg were injected through the tail vein every 3 days for a total of 5 times. After 21 days, all animals were killed and tumors were excised for size determination according to the following formula.
Here, L and W are the length and the width of the tumor measured by a caliper, respectively.
Statistical analysis
Statistical analysis was conducted by using Student’s t-test with P < 0.05 as a significant difference. All data reported were mean value ± SD, unless otherwise noted.
Results and discussion
The preparation and characterization ofBio-CS-PLGA NPs
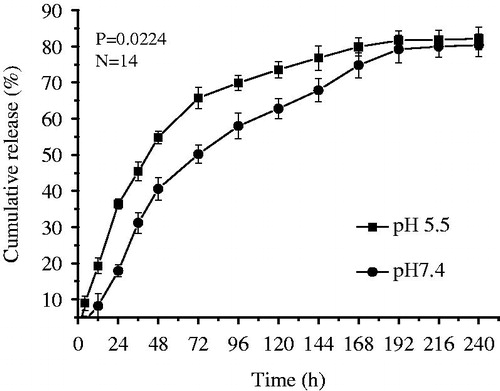
Bio-CS was synthesized and the DS of biotin group in its molecule was about 31%, which was determined by the 1H NMR and ICP methods in our previous study (Chen et al. Citation2016). The SEM images are shown in . Bio-CS-PLGA NPs had the regularly spherical shapes with the relatively uniform distributions. A close examination of AFM further clarified the morphology of the NPs. The height images show round shapes of all samples and the horizontal distance is similar to the diameters observed by AFM (). The mean diameter of Bio-CS-PLGA NPs was 218.4 ± 21.0 nm with a polydispersity index of 0.179 ± 0.032, indicating a relatively narrow size distribution. The zeta potential of Bio-CS-PLGA NPs was 24.15 ± 1.41 mV. In acidic media (PBS, pH 5.5), the mean diameter and zeta potential of Bio-CS-PLGA NPs was 247.5 ± 24.2 ± nm and 32.43 ± 3.22 mV, respectively. The EPB-loading content and encapsulation efficiency in PLGA NPs were 3.45% and 71.4 ± 3.6%, respectively. EPB in vitro releases from the NPs were investigated in PBS solutions with pH values of 7.4 and 5.5, respectively. The release profiles are shown in . A sustained drug release pattern was observed, and the amount of EPB released in acidic media (pH 5.5) was higher than in physiological media (pH 7.4). For example, EPB release amount was only 18.2% at pH 7.4 over a 24-h period, but reached up to 35.5% at pH 5.5, respectively. It is well known that the release of a drug from polymeric NPs is often modulated by different physicochemical parameters of the medium (Laura et al. Citation2013). In acidic media, Bio-CS layers surrounding PLGA NPs, whose amino groups are mostly protonated, dissolve in water, and therefore do not slow down drug diffusion from PLGA NPs core. It was clear that about 80% of the loaded EPB was released after 10 days, indicating a large proportion of EPB could be released from these NPs. All of the profiles were characterized by a rapid procedure of initial drug releases followed by a continuous period of slow drug releases after 48 h.
Figure 1. SEM images and size distribution of Bio-CS-PLGA NPs (a: Bio-CS-PLGA NPs, b: degraded for 4 week; c: degraded for 8 weeks).

Figure 2. AFM image of Bio-CS-PLGA NPs deposited on silicon dioxide at 300 K.

Figure 3. Cumulative release profiles of EPB from Bio-CS-PLGA NPs.
Storage stability of NPs
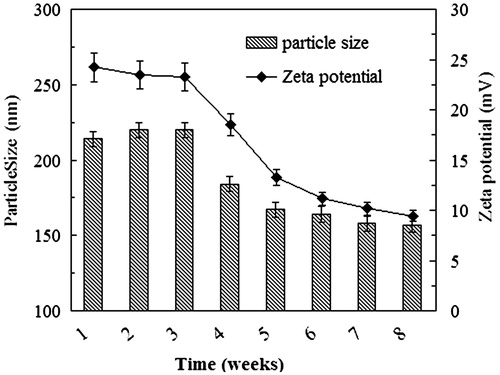
The NP suspensions were stored in normal saline at 4 °C for a period of 8 weeks. During the first 3 weeks, the physicochemical properties such as the size and the zeta potential were effectively unchanged and no sediment was observed (), indicating that the particle system was highly stable over this period of time. Storage beyond 3 weeks, however, led to reductions in the particle size and the absolute value of the zeta potential. Furthermore, the SEM images revealed that the Bio-CS-PLGA NPs tended toward aggregation and disintegration during the fourth week of storage under these conditions ()).
Figure 4. The changes of the size and the zeta potential of the NPs against the storage time.
Stability is an important facet of preparation and a necessary step in the development process, it will indicate potentiality of clinical application and industry production. For the stability investigations, the NP lyophilized powder was stored at 4 °C for a period of 12 months. During this period, the physicochemical properties such as the size, the zeta potential, and drug loading were effectively unchanged. Therefore, it could be concluded that NP powder was stable for at least 12 months at 4 °C.
The acute toxicity of Bio-CS-PLGA NPs in mice
To demonstrate the safety of Bio-CS-PLGA NPs as a drug carrier, the acute toxicity in vivo experiment was studied. All mice were healthy throughout the experiment period, as judged from the BW data, food intake data, and pathological changes of mice during the experiment. No significant differences between the Bio-CS-PLGA NPs group and the control group in clinical signs, e.g., diarrhea, fever, and other systemic symptoms, and no mortality occurred throughout the entire study course. As shown in and , the data of the BW and food intake for both male and female mice between the two studied groups are also not shown significant difference. There was no effect on BW in animals given NPs as compared to controls (data not shown). The data of hematological indexes and biochemical indexes for both male and female mice between the two studied groups did not show significant difference (). Animals were sacrificed for histopathological study on 15th days. shows microscopic examination of the representative histopathological of major organs including heart, liver, spleen, lung, and kidney sections stained with H&E. No significant differences were observed between the Bio-CS-PLGA NPs group and the control group in histological analysis. All the above results indicated that no apparent acute toxicity of the Bio-CS-PLGA NPs was found in the experimental animals after i.v. at a dose of 300 mg/kg. Thus, histopathological studies confirmed that Bio-CS-PLGA NPs are safe and biocompatible for use in drug delivery systems.
Figure 5. Representative photomicrographs of the heart, liver, spleen, lung, and kidney sections (H&E staining) of mice of control group (a) and treated with test Bio-CS-PLGA NPs (b).
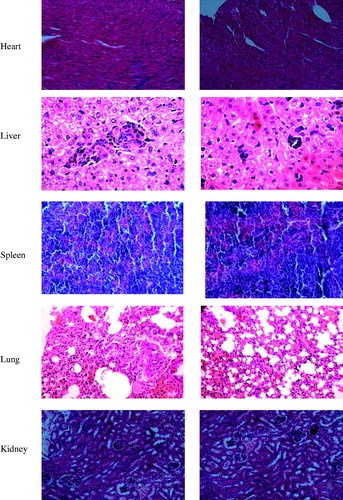
Table 1. Body weight of Bio-CS-PLGA NPs i.v. injected in mice at 300 mg/kg dose (xo ± s, g).
Table 2. Food intake of Bio-CS-PLGA NPs i.v. injected in mice at 300 mg/kg dose (x(± s), g).
Table 3. Hematological indexes and biochemical indexes.
In vivo pharmacokinetics
The plasma levels of EPB were determined following a single i.v. injection of EPB or Bio-CS-PLGA NPs (10 mg/kg EPB equiv.) in female Wistar rats. The plasma levels over 48 h are shown in and the pharmacokinetic parameters are summarized in . The peak concentration of total EPB in plasma was 13.10 mg/L at 1 min after injection and then decreased to nearly undetectable levels after 24 h. The maximum concentration (Cmax) of drug-loaded Bio-CS-PLGA NPs at 5 min was 9.80 mg/L, which was obviously lower than that of EPB because of sustained release from NPs. According to the previous report, free EPB rapidly disappeared from the circulation due to its short half-life. In this study, the terminal elimination half-life of free EPB was 6.753eh (), which was consistent with that of 7.3 h of doxorubucin. In contrast, Bio-CS-PLGA NPs showed a much longer circulation time and its elimination half-life time was 16.642 h, 2.464 times that of free EPB. The clearance of EPB loaded in NPs was 0.117 L/h, which is 3.119 times lower than free EPB. Moreover, the mean residence time (MRT) of Bio-CS-PLGA NPs in the plasma was 19.067 h, 21.25 times that of free EPB. Altogether, Bio-CS-PLGA NPs had the longer t1/2 and MRT, lower Vd and CL than the free drug in rats. At the same time, the AUC of EPB and Bio-CS-PLGA NPs from 0 to 24 h were 42,356.412 and 92,124.110 μg h/L, respectively. The bioavailability of free EPB was 100%, the BAR of Bio-CS-PLGA NPs calculated through the equation above by the AUC0–24 h was 197%, much higher than the free drug. According to the previous report, we believed that the longer t1/2 in plasma may be due to the enhanced permeability and retention (EPR) effect of NPs. As prolonged plasma circulation is the driving force for increased tumor targeting prepared long circulating NPs to improve therapeutic efficacy of doxorubicin (Guo et al. Citation2014; Pereverzeva et al. Citation2008). In this study, the slow release of EPB from NPs in the blood suggested that the bioavailability of EPB improved when it was loaded into Bio-CS-PLGA NPs. However, whether it could enhance the uptake of passive permeability in targeting tumor tissues or not needs to be further proved by the experiments such as bio-distribution study and pharmacodynamic test.
Figure 6. Plasma drug concentration of EPB and Bio-CS-PLGA NPs after i.v. injection.
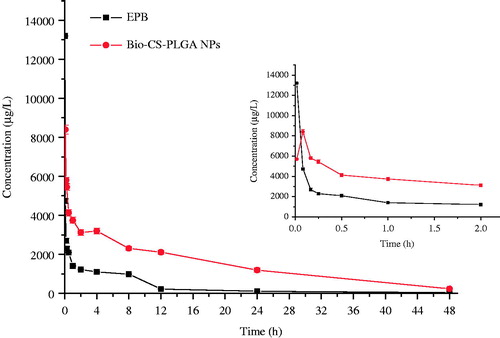
Table 4. Pharmacokinetic parameters of EPB and EPB loaded Bio-CS-PLGA NPs i.v. injected in rats at the equivalent 10 mg/kg dose.
In vivo antitumor activity of NPs
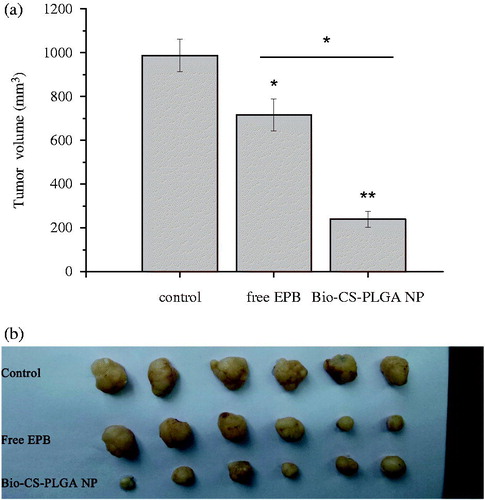
EPB has a wide range of anti-tumor activity and is used to treat various carcinomas. However, EPB therapy may cause some serious side effects such as allergic reactions, cardiotoxicity, and blood problems (Florescu et al. Citation2014). To overcome these limitations, one of the approaches is to use nanotechnology and develop nanoformulation to reduce the inherent toxicity (Ju et al. Citation2014; Ruenraroengsak et al. Citation2010), and the nanoformulation is believed as an effective strategy for improving the drug bioavailability and realizing the tumor-targeted drug delivery (Ju et al. Citation2014; Mohammad et al. 2015). Thus, we established a xenograft model by implanting MCF-7 cells subcutaneously into nude mice and further evaluated the in vivo antitumor activity of EPB-loaded Bio-CS-PLGA NPs using free EPB as the controls. The mice were injected with free EPB and NPs at EPB doses of 3.0 mg/kg every 3 days for consecutive five times. After the first injection, the tumor volume changes were detected for 21 days. Obviously, EPB-loaded Bio-CS-PLGA NPs significantly inhibited the tumor growth even at a low EPB dose of 3.0 mg/kg and the average tumor size was markedly smaller than those of the control group and the treatment groups of free EPB (). In our previous study, the distributions of Cy5.5 Bio-CS-PLGA NPs in the liver, kidney, and tumor were evidently observed in tumor-bearing mouse at 12 h (Chen et al. Citation2016). We believed that the effective activity of EPB-loaded Bio-CS-PLGA NPs on the tumor growth was due to their in vivo tumor-targeted delivery for EPB.
Figure 7. The inhibition effects of EPB-loaded NPs on the in vivo tumor growth. (a: the volume of tumors in nude mice bearing MCF-7 tumors after intravenous injections of free EPB and EPB-loaded NPs; b: the picture of tumors removed from tumor-bearing nude mice at 21 days after the first injections. * and ** represent P < 0.05 and P < 0.01, respectively).
Conclusions
In this study, Bio-CS-PLGA NPs with regularly spherical shapes was prepared. The NPs were stable in normal saline for at least 3 weeks in vitro. Furthermore, Bio-CS-PLGA NPs were safe in mice at 300 mg/kg and showed the potential to improve the bioavailability of the loaded drug in vivo. In MCF-7 tumor-bearing nude mice, Bio-CS-PLGA NPs evidently inhibited the tumor growth at EPB dose of 3 mg/kg. In conclusion, Bio-CS-PLGA NPs displayed a great potential as a novel carrier for anticancer drugs.
Funding information
This research was supported by the grants from the National Natural Sciences Foundation (No.U1304819, No.81401519), the Key Technologies R&D Program of Henan Province (No. 122102210148), and National Training Programs of Innovation and Entrepreneurship for Undergraduates (No. 201410472030).
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Silva AH, Locatelli C, Filippin-Monteiro FB, Martin P, Liptrott NJ, Zanetti-Ramos BG, et al. 2016. Toxicity and inflammatory response in Swiss albino mice after intraperitoneal and oral administration of polyurethane nanoparticles. Toxicol Lett. 246:17–27.
- Alexander-Bryant AA, van den Berg-Foels WS, Wen XJ. 2013. Bioengineering strategies for designing targeted cancer therapies. Adv Cancer Res. 118:1–59.
- Chen HL, Wang YX, Zhou P, Liu R, Nan WB, Wang YS. 2014. Chitosan surface modified PLGA nanoparticle: preparation, characterization, in vitro drug release behavior and cytotoxicity. Curr Nanosci. 10:255–262.
- Chen HL, Xie LQ, Qin JW, Jia YJ, Cai XH, Nan WB. 2016. Surface modification of PLGA nanoparticles with biotinylated chitosan for the sustained in vitro release and the enhanced cytotoxicity of epirubicin. Colloids Surf B Biointerfaces. 138:1–9.
- Chen HL, Yang WZ, Chen H, Liu LR, Gao FP, Yang XD. 2009. Surface modification of mitoxantrone-loaded PLGA nanospheres with chitosan. Colloids Surf B Biointerfaces. 73:212–218.
- Farokhzad OC, Langer R. 2009. Impact of nanotechnology on drug delivery. ACS Nano. 3:16–20.
- Fatemeh S, Abolfazl A, Morteza M, Zarghami N. 2016. Comparison between the cytotoxic effects of pure curcumin and curcumin-loaded PLGA-PEG nanoparticles on the MCF-7 human breast cancer cell line. Artif Cells Nanomed Biotechnol. 44:423–430.
- Florescu M, Magda LS, Enescu OA, Jinga D, Vinereanu D. 2014. Early detection of epirubicin-induced cardiotoxicity in patients with breast cancer. J Am Soc Echocardiogr. 27:83–92.
- Gabizon A, Shmeeda H, Barenholz Y. 2003. Pharmacokinetics of pegylated liposomal doxorubicin: review of animal and human studies. Clin Pharmacokinet. 42:419–436.
- Giarra S, Serri C, Russo L, et al. 2016. Spontaneous arrangement of a tumor targeting hyaluronic acid shell on irinotecan loaded PLGA nanoparticles. Carbohydr Polym. 140:400–407.
- Guo H, Zhang D, Li T, Li CY, Guo YY, Liu GP. 2014. In vitro and in vivo study of Gal-OS self-assembled nanoparticles for liver-targeting delivery of doxorubicin. J Pharm Sci. 103:987–993.
- Ju R, Li X, Shi J, Li XY, Sun MG, Zeng F. 2014. Liposomes, modified with PTD(HIV-1) peptide, containing epirubicin and celecoxib, to target vasculogenic mimicry channels in invasive breast cancer. Biomaterials. 35:7610–7621.
- Kristina BK, Helle N, Pramod K, Permin A, Gjetting T, Andresen TL. 2015. In vivo toxicity of cationic micelles and liposomes. Nanomed Nanotechnol Biol Med 11:467–477.
- Laura C, Mara M, Maria FG, Cametti C, Laura CD, Mariella D. 2013. Chitosan-coated PLGA nanoparticles: a sustained drug release strategy for cell cultures. Colloids Surf B Biointerfaces. 103:310–317.
- Laura GF, Lalit HM, Dennis N, Hargrove D, Manautou J, Liang BT. 2016. Reduced in vivo toxicity of doxorubicin by encapsulation in cholesterol-containing self-assembled nanoparticles. Pharmacol Res. 107:93–101.
- Moghimi SM, Hunter AC, Murray JC. 2001. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 53:283–318.
- Muhammad IS, Haliza K Mohd C, Shiow-Fern N, Mohd HZ, Fhataheya B. 2015. Minimization of local and systemic adverse effects of topical glucocorticoids by nanoencapsulation: in vivo safety of hydrocortisone–hydroxytyrosol loaded chitosan nanoparticles. J Pharm Sci. 104:4276–4286.
- Nel A, Xia T, Madler L, Li N. 2006. Toxic potential of materials at the nanolevel. Science. 311:622–627.
- Paolo B, Aurelie S, Giovanna T, Giovagnoli S, Alessandro DM, Maurizio R. 2013. Lipid nanoparticles for brain targeting III. Long-term stability and in vivo toxicity. Int J Pharm. 454:316–323.
- Pereverzeva E, Treschalin I, Bodyagin D, Maksimenko O, Kreuter J, Gelperina S. 2008. Intravenous tolerance of a nanoparticle-based formulation of doxorubicin in healthy rats. Toxicol Lett. 178:9–19.
- Ruenraroengsak P, Cook J, Florence A. 2010. Nanosystem drug targeting: facing up to complex realities. J Control Release. 141:265–276.
- Sedigheh FA, Abolfazl A, Mohammad RY, Zarghami F, Nejati-Koshki K, Zarghami N. 2016. Gene silencing effect of SiRNA-magnetic modified with biodegradable copolymer nanoparticles on hTERT gene expression in lung cancer cell line. Artif Cells Nanomed Biotechnol. 44:188–193.
- Tang H, Li L, Chen H, Zhou ZM, Chen HL, Li XM. 2010. Stability and in vivo evaluation of pullulan acetate as a drug nanocarrier. Drug Deliv. 17:552–558.
- Vander R, Vehmeijer LJ, Kok RJ, Storm G, Gaal EVBV. 2013. Ligand-targeted particulate nanomedicines undergoing clinical evaluation. Curr Stat Adv Drug Deliv Rev. 65:1284–1298.
- Vega-Villa KR, Takemoto JK, Yanez JA, Remsberg CM, Forrest ML, Davies Neal M. 2008. Clinical toxicities of nanocarrier systems. Adv Drug Deliv Rev. 60:929–938.
- Walter ER, Adriana P, Abuzar A, Lane MA, Aminabhavi TM. 2016. Targeted delivery of small interfering RNA to colon cancer cells using chitosan and PEGylated chitosan nanoparticles. Carbohydr Polym. 147:323–332.
- Xiong XY, Guo L, Gong YC, Li ZL, Li YP, Liu ZY. 2012. In vitro and in vivo targeting behaviors of biotinylated Pluronic F127/poly(lactic acid) nanoparticles through biotin-avidin interaction. Eur J Pharm Sci. 46:537–544.
- Yu H, Tang Z, Zhang D, Song WT, Zhang Y, Yang Y. 2015. Pharmacokinetics, biodistribution and in vivo efficacy of cisplatin loaded poly(l-glutamic acid)-g-methoxy poly(ethylene glycol) complex nanoparticles for tumor therapy. J Control Release 205:89–97.
- Zhu HJ, Chen HB, Zeng XM, Wang ZY, Zhang XD, Xu YP. 2014. Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance. Biomaterials. 35:2391–2400.