
Abstract
The aim was to enhance the oral bioavailability of olmesartan medoxomil (OM) by preparing solid lipid nanoparticles (SLNs) and comparing with nanosuspension (OM-NS). OM-SLNs and OM-NS were prepared by known methods. Prepared SLNs were evaluated for physical characters and in vivo pharmacokinetic (PK) performance in rats. OM-NS showed more than four-fold increase in the solubility. During DSC and XRD studies, drug incorporated in SLNs was found to be in amorphous form. The relative bioavailability of OM-SLN and OM-NS was 7.21- and 3.52-fold when compared with that of coarse suspension. Further, OM-SLNs also increased the oral bioavailability by two-fold over that of OM-NS.
Introduction
Olmesartan medoxomil (OM), an ester prodrug of olmesartan, is an angiotensin II type 1 (AT1) receptor antagonist, and is used in the treatment of hypertension [Citation1]. The major drawback in the therapeutic application and efficacy of OM as an oral dosage form is its very low aqueous solubility. The unabsorbed drug leads to gastrointestinal side effects such as abdominal pain, dyspepsia, gastroenteritis and nausea. Based on its solubility in physiologically relevant pH conditions and absorption characteristics, OM is classified in the Biopharmaceutics Classification System (BCS) as a class II drug. Improvement in oral bioavailability of OM is possible by incorporating into the SLNs [Citation2]. SLNs increase clinical efficacy, reduce the oral dose required to achieve the same effect, and hence reduce the side effects.
Previous reports suggested that the co-administration of lipophilic drug along with a meal rich in fat resulted in improvement of oral bioavailability [Citation3,Citation4]. This observation helped to develop the formulation of colloidal lipid carrier systems as a means to improve the drug solubilization and transport across the GI barrier [Citation5,Citation6]. Among the various conventional and colloidal drug delivery systems, solid lipid nanoparticles (SLNs) confer distinct advantages like biocompatibility and scale up [Citation7]. In addition, due to lymphatic transport the first pass metabolism is reduced with an increase in bioavailability. Such an effect seems to be due to the drain of SLN directly into the systemic circulation via thoracic duct circumventing the portal circulation [Citation8].
Nanosuspension (NS) is a sub-micron colloidal dispersion of pure particles of drug, and stabilized by surfactants in an aqueous vehicle with average particle size below 1 μm [Citation9]. Attractive features, such as increased dissolution velocity, increased saturation solubility, versatility in surface modification, and ease of post-production processing, have widened the applications of NSs for various routes. The NSs were studied for parenteral and oral delivery extensively, and also recently pulmonary and ocular delivery [Citation10,Citation11]. Previously, NSs of OM were reported, being prepared by media milling technique, without any oral bioavailability studies [Citation12]. Till now, OM SLNs were not reported. SLNs were also showed to have enhanced pharmacodynamic effect [Citation13].
In the literature, SLNs and NS formulations having great potential for improving bioavailability of poorly water soluble drugs were reported earlier. Further, in some cases comparisons of SLN and NS were also reported for their relative improvement in oral bioavailabilities. Shamsunder et al. [Citation14] reported the relative bioavailabilities for saquinavir NS and SLN in rats and were found to be 37.39% and 66.53%, respectively, compared with 18.87% bioavailability obtained after administration of suspension. Hanafy et al. [Citation15] reported that NSs and SLN formulation of fenofibrate both exhibited similar two-fold improvement in oral bioavailability when compared with drug suspension. Similar studies with OM SLN and NS were not reported. Thus, this study was aimed at the formulation and comparative evaluation of nanosized formulations, i.e. SLNs and NSs for improvement of bioavailability of OM. Accordingly, OM-SLNs and OM-NSs were prepared by hot homogenization followed by ultrasonication and high-pressure homogenization method, respectively. Further, they were characterized for size, in vitro release and optimal systems of SLN and NS were evaluated for pharmacokinetic (PK) effects in comparison with a coarse suspension of OM in Wistar rats.
Material and methods
Materials
Olmesartan medoxomil was a kind gift sample from M/s. Dr. Reddy’s labs, India. Tristearin (TS, Dynasan-118) were purchased from M/s. Sigma-Aldrich Chemicals, Hyderabad, India. Stearic acid (SA) and glyceryl monostearte (GMS) were purchased from M/s. Hi-Media, Mumbai, India. Egg Lecithin E-80 was a gift sample from M/s. Lipoid, Ludwigshafen, Germany. Losartan potassium (internal standard) and Poloxamer-188 were gift samples from M/s. Aurobindo Labs, Hyderabad, India. PVP k30 and tween 80 were purchased from M/s. Hi-Media, Hyderabad, India. Methanol, acetonitrile, and chloroform were of HPLC grade (Merck, Bombay, India). Centrisart filters (molecular weight cut off 20,000) were purchased from M/s. Sartorius, Goettingen, Germany.
Methods
Preparation of OM-SLNs
OM-loaded SLNs were prepared by hot homogenization followed by the ultrasonication [Citation16,Citation17]. OM, lipid, and egg lecithin were dissolved in 5 mL mixture of chloroform and methanol (1:1). Organic solvents were completely removed using a rota evaporator (Heidolph, Germany). The drug embedded lipid layer was molten by heating at 5 °C above melting point of the lipid. An aqueous phase was prepared by dissolving Poloxamer 188 in double distilled water and heated to same temperature (based on lipid melting point) of oil phase. Hot aqueous phase was added to the oil phase, and homogenization was carried out (at 12,000 rpm) using homogenizer (Diax900, Heidolph, Germany) for 4 min. The coarse hot oil in water emulsion so obtained was ultrasonicated using a 12 T probe sonicator (Vibracell, Sonics, Newtown, CT) for 20 min. OM-loaded solid lipid nanoparticles were obtained by allowing hot nanoemulsion to cool to room temperature. The compositions of various formulations are shown in .
Table 1. Composition of olmesartan medoxomil-loaded SLNs, nanosuspension and coarse suspension.
Preparation of OM-NSs
OM NSs were prepared by high-pressure homogenization method [Citation18]. Initially, 400 mg of OM coarse powder was ground in mortar and then PVPk30 and tween 80 as stabilizers (1.01% and 1.05%) were dispersed in 20 mL of water. The obtained pre-mix was further processed using homogenizer (Diax 900, Heidolph, Germany) at 12,000 rpm for 4 min in a beaker to form a pre-suspension then subjected to high-pressure homogenization at 5000–15,000 psi pressure for about 10–20 cycles using Emusiflex C5 high-pressure homogenizer (Avestin Inc., Ottawa, Canada). Finally, a freeze-dried OM NS powder was obtained after lyophilization with 10% w/v trehalose as cryoprotectant and used for further studies.
Preparation of OM coarse suspension
Coarse suspension of OM was prepared, by taking 50 mg of sodium carboxy methyl cellulose in a mortar and triturating for 3 min, then 10 mg of OM was added to it, and further triturated for 3 min. To it, 10 mL of water was added and again triturated for 5 min to get coarse suspension of drug ().
Characterization of solid lipid nanoparticles and NSs
Measurement of size, PDI and zeta potential (ZP)
The average particle size, PDI and ZP of OM-SLNs and OM-NS were measured by photon correlation spectroscopy (PCS) using Zetasizer (Nano ZS 90, Malvern Instruments, Malvern, UK) at room temperature. From the prepared SLN and NS dispersion, 100 μL was diluted to 5 mL with double distilled water to get optimum kilo counts per second (Kcps) of 50–200 for measurements. The sample was used for size and then ZP measurements and was obtained directly from the instrument. All the samples were analyzed in triplicate and data were represented in mean ± SD.
Entrapment efficiency (EE) and drug content
EE and content OM of OM-SLNs were determined by high-performance liquid chromatography (HPLC). The chromatographic system consisted of a LC-10AT solvent delivery module containing double reciprocating plunger pump (Shimadzu, Tokyo, Japan), a LC-10 A UV–visible variable wavelength detector with deuterium lamp and a RP C18 analytical column, Merck (250 mm × 4.6 mm i.d., 5 μm). The mobile phase consisting of acetonitrile: 0.05 M phosphate buffer (pH 4.5 was adjusted with ortho phosphoric acid) (50:50) ratio at a flow rate of 1 mL/min was employed for the detection of drug at λmax of 257 nm. EE was determined by measuring the concentration of free drug (unentrapped) in aqueous medium as reported previously [Citation19]. To determine EE, centrifugation was performed using Centrisart (Sartorius, USA) tubes at 20,000 rpm (Remi Equipment, Mumbai, India) for 30 min, which consisted of a filter membrane (molecular weight cut off of 20,000 Da) to separate the ultrafiltrate. The EE of the system was determined by measuring the concentration of free drug in the ultrafiltrate using HPLC, along with the following formula:
where Wtotal is the weight of drug added in the system.
For total drug content, about 0.1 mL of formulation was dissolved in chloroform and methanol (1:1) mixture. The final dilution of solution was made with the mobile phase. OM content was determined by HPLC.
Assay of OM-NS
For drug content determination, about 1 mL of OM-NS was taken and diluted appropriately with 0.1NHCl and then drug content was estimated by using UV–visible spectrophotometer at λmax 257 nm [Citation20].
Determination of saturation solubility of OM-NS
The saturation solubility of pure OM and OM-NS was determined by adding excess material in distilled water (10 mL) in screw-capped vials and shaking for 48 h on a mechanical shaker. The dispersion was filtered through a 0.22-μm membrane filter and the filtrate was diluted appropriately with distilled water and the drug content was analyzed by UV–visible spectrophotometer at λmax 257 nm [Citation21].
In vitro drug-release studies
In vitro drug-release studies were performed using dialysis method. Dialysis membrane (Hi-Media, Mumbai, India) having a pore size of 2.4 nm and molecular weight cut-off between 12,000 and 14,000 was used for the drug-release studies. Dialysis membrane was soaked overnight in double-distilled water prior to the release studies. Hydrochloric acid (0.1 N) for 2 h followed by pH 6.8 phosphate buffer for 22 h were used as release media [Citation22]. The experimental unit had donor and receptor compartments. Donor compartment consisted of a boiling tube which was cut open at one end and tied with dialysis membrane at the other end into which 1 mL of SLN dispersion was taken for drug-release study. Receptor compartment consisted of a 250 mL beaker which was filled with 100 mL of drug-release medium and the temperature was maintained at 37 ± 0.5 °C. At 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h time points, 2 mL samples were withdrawn from receiver compartment and replenished with the same volume of release medium. The collected samples were suitably diluted and analyzed by UV–visible spectrophotometer (SL-150, ELICO) at λmax of 257 nm.
Dissolution studies of OM-NS
For determining the drug release from the NS, the dissolution test was carried out in 900 mL of 0.1NHCl by using USP type II dissolution tester (TDT-06 P, Electrolab, Hyderabad, India). For this purpose, OM-NS and plain suspensions equivalent to 20 mg were placed directly into the vessels [Citation16]. The bath temperature and the rotating speed of the apparatus were maintained at 37.0 ± 0.5 °C and 50 rpm, respectively. About 5 mL samples were withdrawn at predetermined time intervals and filtered through 0.45 μm filter. The absorbances of the filtrates were analysed spectrophotometrically at λmax of 257 nm against the respective blank. All experiments were repeated thrice and the average values were recorded [Citation21].
Stability studies
Optimized formulation of OM-loaded solid lipid nanoparticles was stored at room temperature and refrigerated temperature for 2 months. The average size, PDI, ZP, assay, and EE were determined periodically after first day, 15 d, 1 month, and 2 months [Citation23,Citation24].
Lyophilization
OM-SLN and NS formulations were subjected to lyophilization process to get the powder forms, which were used for further characterization. The SLN and NS of OM containing 10% w/v trehalose [Citation25] were prepared and kept in deep freezer at −40 °C (Sanyo, Tokyo, Japan) for overnight. The frozen samples were then transferred into freeze-dryer (Lyodel, Delvac Pumps Pvt. Ltd, Chennai, India). Vacuum was applied and the sample was subjected to various drying phases for about 48 h to get powdered lyophilized product [Citation26,Citation27].
Solid-state characterization
Drug-excipient compatibility studies by differentialscanning calorimetry (DSC)
DSC thermograms of pure OM, TS, SA, GMS, physical mixtures (PM) in 1:1 ratio, and lyophilized OM-SLNs and NS were obtained using Perkin Elmer instrument (DSC-4000, Perkin Elmer Inc., Waltham, MA). Initially, the instrument was calibrated with indium. All the samples (∼10 mg) were heated in aluminium pans using dry nitrogen as the effluent gas. The analysis was performed within a heating range of 20–200 °C and at a rate of 20 °C/min.
Characterization of crystallinity by powder X-ray diffractometry (PXRD)
PXRD (XRD-6000, Shimazdu, Tokyo, Japan) was used for diffraction studies [Citation28]. Powder XRD studies were performed on the samples by exposing them to nickel filtered CuKα radiation (40 kV, 30 mA) and scanned from 2° to 70°, 2θ at a step size of 0.045°, and a step time of 0.5 s. Samples used for PXRD analysis were pure OM, pure TS, SA + GMS, and lyophilized OM-loaded solid lipid nanoparticles [Citation29].
Morphology by scanning electron microscopy (SEM)
The morphology of both SLNs and NS was studied by SEM (Hitachi, Tokyo, Japan). Freeze-dried solid lipid nanoparticles and NS of OM were diluted with double-distilled water (1 in 100) and a drop of formulation was placed on sample holder and air dried. Then the sample was observed at accelerating voltage of 15,000 V at various magnifications [Citation30].
Bioavailability study
Study design and sampling schedule
A single-dose bioavailability study was designed in male Wistar rats under fasting conditions. The oral bioavailabilities of the optimized OM-SLN, OM-NS formulation, and coarse suspension were estimated in male Wistar rats with an oral dose of 10 mg/kg body weight. All experimental procedures were reviewed and approved by the institutional animal ethical committee, University College of Pharmaceutical Sciences, Kakatiya University (Warangal, India). Male Wistar rats weighing 200–250 g were taken for study (06 animals per group). Blood samples were withdrawn by retro-orbital venous plexus puncture at 0, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h post-dose. About 0.5 mL of blood samples were withdrawn in eppendorf tubes and centrifuged at 3000 rpm for 30 min. The serum samples were separated and stored at –20 °C until analysis by HPLC.
HPLC analysis
The HPLC column, Lichrosphere C18, (250 × 4.6 mm), was equilibrated with an eluent mixture of acetonitrile:0.05 M phosphate buffer adjusted to pH 4.5 with ortho phosphoric acid (50:50v/v) at a flow rate of 1 mL/min. The peaks were detected at λmax of 256 nm wavelength without any interferences from serum [Citation31].
Extraction procedure
To 100 μL of serum, 100 μL of internal standard (losartan potassium, 1 μg/mL) and 500 μL of acetonitrile were added. After vortex mixing for 10 min at room temperature, the samples were centrifuged at 5000 rpm for 15 min. The upper organic layer was separated and evaporated by using vacuum oven. The dry residue was reconstituted with 100 μL of mobile phase and vortexed well for 2 min, and 20 μL of this solution was injected into HPLC.
Estimation of pharmacokinetic parameters and statistical significance
The PK parameters such as peak serum concentration (Cmax), time for peak serum concentration (tmax), AUCtotal, biological half-life (t½), and mean residence time (MRT) were calculated by using the Kinetica software (version 5.0, 2000, GraphPad Software Inc., La Jolla, CA). The values were expressed as mean ± SD. The statistical comparison of data of three samples were done with one-way ANOVA using Graph pad prism software (version 5.02.2013, GraphPad Software Inc., La Jolla, CA) and p < 0.01 was considered as statistically significant.
Results and discussion
Solid lipid nanoparticles of OM were prepared involving hot homogenization followed by ultrasonication and NS by high-pressure homogenization (HPH) methods.
In case of SLNs, based on the particle size and uniformity of dispersion, the homogenization time and sonication time were optimized to 4 and 20 min, respectively [Citation23]. The obtained polydispersity indices were within the acceptable limits (<0.3) for all the SLN formulations and suggested that the particles formed were of narrow size distribution [Citation2]. Homogeneous systems with particle size ranging from 50 to 200 nm, with a surface charge of −20 to −30 mV and drug entrapment (80–96%) were obtained with higher concentration of surfactant (1.5% w/v) [Citation32].
In case of NSs, to select a suitable stabilizer, OM (400 mg) was homogenized using different concentrations (0.5%, 1% and 2% w/v) of tween 80, poloxamer 188 and sodium lauryl sulphate. The number of homogenization cycles (10) and homogenization pressure 15,000 psi were kept the same for all the experiments after preliminary trials with variable cycles and pressure (data not shown). From the preliminary trials, it was observed that a single surfactant was not enough for long-term stability. So a combination of surfactant and polymer was used. PVP k30 was selected as polymer and used at a concentration of 1.01%.
Mean particle size (MPS) of nano-suspension stabilized with poloxamer 188 was found to be high, ranging from 667.8 to 1240 nm with high PDI values, which could influence the Ostwald ripening during storage. In this study, poloxamers were not able to reduce the particles to desired range. MPS of NSs stabilized with SLS was found to be in the range of 606.3–722 nm with PDI values ranging from 0.115 to 0.213 and ZP values were in the range of −32.5 to −48.3. Similarly, tween 80 also yielded lower MPS ranging from 605 to 717 nm with PDI ranging from 0.151 to 0.430. Tween 80 at 1% level yielded lower particle size when compared with 0.5% and 2% levels. ZP of formulations was also in the range of −26.2 to −32.8 mV.
Measurement of size, PDI and ZP of SLN formulation
All the prepared SLN formulations were analyzed for their particle size, PDI and ZP values. The particle size of SLNs ranged from 82.8 ± 2.9 nm to 369.9 ± 11.31 nm, PDI from 0.176 ± 0.13 to 0.758 ± 0.07 and ZP from −11.5 ± 1.38 to −28.3 ± 4.68 mV. Initially, OM-SLNs were prepared with 100 mg of single fatty acid or fatty amine as lipid. Both stearyl amine (F2) and stearic acid (SA) (F3) were used in preparation of SLNs and the size, PDI and ZP of F2, F3 formulations were found to be 106.1 ± 7.3 nm and 194.1 ± 14.3 nm; 0.208 ± 0.01 and 0.267 ± 0.11; +6.3 ± 0.6 and −17.6 ± 1.52 mV, respectively. When the content of lipids increased from 100 mg to 200 mg of stearyl amine (F4) and stearic acid (F6), size, PDI and ZP of the SLNs were also increased (). Solid lipid nanoparticles, prepared without any charge modifier, possessed negative ZP [Citation30]. However, in order to possess good stability, the particles should have ZP more than +20 mV. Addition of stearylamine increased the ZP value to +33.7 ± 0.7 mV. It was reported that positively charged solid lipid nanoparticles had better uptake by intestinal lymphatics than neutral or negatively charged particles [Citation16,Citation33].
Table 2. Physical characters – size, PDI, EE and content of OM-SLNs and nanosuspension (mean ± SD, n = 3).
Similarly, formulation of SLN with glyceryl monostearate (GMS) (F7) resulted in 170.9 ± 2.1 nm, 0.469 ± 0.05 and –8.13 ± 0.37 mV of size, PDI and ZP. Further, SLNs (F1 and F5) prepared with triglyceride, such as tristearate resulted in size from 82.8 ± 2.9 to 138.3 ± 4.5, PDI from 0.176 ± 0.13 to 0.270 ± 0.03 and ZP from −14.92 ± 0.9 to –18.3 ± 0.59 mV based on the content of the lipid. In this work, an attempt was made to prepare SLNs with tristearate, glyceryl monostearate and glyceryl monostearate with stearic acid combinations. Combinations of SA and GMS (F8, F9 and F10) resulted in particle size ranging from 122.7 ± 10.8 nm to 369.9 ± 11.31 nm, PDI from 0.369 ± 0.02 to 0.75 ± 0.07 and ZP from −20.6 ± 0.28 mV to –28.3 ± 1.9 mV. The size of the SLNs decreased by the addition of small quantities of stearic acid (F9 and F10 when compared with F7). If more stearic acid was added the size increased (F8).
From the results obtained, formulations containing Dynasan-118 (F1 and F5) and GMS + SA (F10) showed lower particle sizes, and the ZP was higher for GMS + SA (F10) formulation. The optimized NS showed size of 573.53 ± 38.25 nm, PDI of 0.308 ± 0.052 and ZP of -25.7 ± 2.34 mV.
Entrapment efficiency and drug content
A total drug content of OM in the SLN formulations was determined and was ranging from 93.6 ± 0.89 to 99.8 ± 1.54%. EE of the SLN formulations was found to be 77.4 ± 0.61% to 94.2 ± 0.61%. High lipophilicity of OM (Log p 5.6) resulted in high EE of drug in triglyceride nanoparticles (). This might be because due to the long-chain fatty acids of the glyceride resulting in increased accommodation of lipophilic drugs. Similarly, the drug was also entrapped in SLNs prepared with a combination of SA + GMS. The less ordered lipid matrix created imperfections leading to void spaces in which drug molecules could be entrapped [Citation34]. In this method of preparation, drug was dissolved in molten lipid at temperature above the melting point of lipid and there was no drug leakage or precipitation of drug during the preparation. Drug content in the NS formulation was found to be 99.0 ± 0.18%.
Saturation solubility of OM-NS
The saturation solubility of OM NS and the plain drug were found to be 405.3 ± 0.03 μg/mL and 81.2 ± 0.01 μg/mL, respectively. Thus, there was five-fold increase in the saturation solubility of olmesartan when it was formulated as a NS. This great increase in saturation solubility was attributed to the dual role played by the presence of surfactant and smaller particle size and subsequent increase in surface area. As OM belongs to BCS class II drug, i.e. low solubility and high permeability, an increase in the solubility is expected to improve the absorption and thus bioavailability [Citation32].
In vitro drug release studies of OM-SLNs
In vitro release studies of SLNs were conducted in 01 N HCl (2 h) followed by pH 6.8 phosphate buffer (22 h), by using dialysis method. Release of olmesartan ranged from 52.99 ± 3.38% (F4) to 75.10 ± 2.15% (F3), while F10 formulation showed 74.76 ± 1.44% during 24 h. Release profiles of SLNs are shown in . The drug release from F8 was relatively faster for about 6 h when compared with that of F7, F9 and F10. Poor aqueous solubility of drug may be a reason for the slow release of drug from the lipid matrices. Further, drug release was decreased when increased the lipid concentration. This might be due to the drug’s location within the inner core rather than in outer shell. On the contrary, the increase of lipid concentration prolonged the olmesartan release from SLN, and could be due to the solubility and homogeneous distribution of drug within the lipid matrix.
Figure 1. (A) In vitro release of OM-SLNs in 0.1 N HCl followed by pH 6.8 phosphate buffer (mean ± SD, n = 3). (B) Dissolution profile of OM-NS (mean ± SD, n = 3).
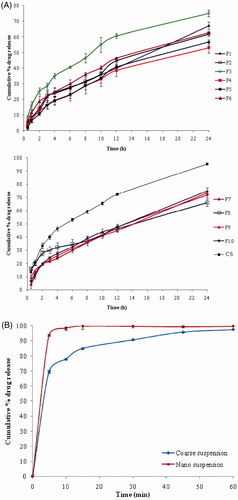
Dissolution studies of OM-NS
OM-NS showed 93.71 ± 0.89% of drug release in first 5 min when compared with that of a coarse suspension, which was 69.49 ± 1.10%. There was a significant difference between the dissolution rate of the plain drug and the NS of OM (). This drastic increase in the dissolution rate was due to the increased surface area of the drug as a follow up of smaller particle size, increased saturation solubility [Citation35] and also due to presence of the stabilizers, according to Noyes–Whitney equation.
Stability studies
Stability studies were conducted for optimized formulation (F10) of olmesartan medoxomil-loaded solid lipid nanoparticles by storing at room and refrigerated temperature for 60 d. The number of samples estimated was in triplicate. Results are shown in and average particle size, ZP, PDI and EE were determined. Formulation showed changes in size and PDI, but not in ZP and EE characteristics at both conditions. This indicated the instability of SLNs in both conditions.
Table 3. Physical stability studies of the optimized formulation (F10) at room temperature (25 °C) and refrigerated temperature (4 °C) (mean ± SD, n = 3).
Solid-state characterization
DSC of OM-SLNs
The compatibility status of the lipids in the SLN formulation was investigated by DSC and was based on the fact that different lipids possessed different melting points and enthalpies. DSC thermograms of pure drug, lipids, physical mixtures and lyophilized SLN formulation are shown in . The DSC thermogram of OM exhibited a sharp endothermic peak at 183.86 °C corresponding to its melting point. The DSC thermograms of stearic acid, stearyl amine, dynasan-118 and glyceryl monostearate showed sharp endothermic peaks at 60.18 °C, 56.55 °C, 80.12 °C and 76.78 °C, corresponding to their respective melting points. Physical mixture of OM and Dynasan118 showed endothermic peaks at 183.06 °C and 71.99 °C and physical mixture of OM with combination of GMS + SA (1:1) showed endothermic peaks at 172.01 °C, 74.26 °C (GMS) and 67.38 °C (SA). The melting points of colloidal systems were distinctly decreased by about 3–8 °C. In accordance with the literature reports, the phase transition temperature and enthalpy of SLN formulation was significantly lower than the corresponding anhydrous physical mixture [Citation36]. Freeze-dried SLN formulations containing Dynasan118 (F1) and GMS + SA (F10) showed the absence of endothermic peak of OM indicating that the drug was encapsulated, molecularly dispersed in the lipid matrix. No incompatibilities among the used lipids and drug were observed.
Figure 2. (A) DSC thermograms of (a) OM, (b) stearic acid, (c) stearyl amine, (d) dynasan118, (e) GMS, (f) lyophilized dynasan118 SLNs, (g) lyophilized GMS-SA SLNs, (h) physical mixture of GMS-SA + OM and (i) physical mixture of dynasan118 + OM. (B) DSC thermograms of (a) pure drug, (b) PVPk30, (c) physical mixture of PVPk30 + tween 80 + drug and (d) lyophilized nanosuspension.

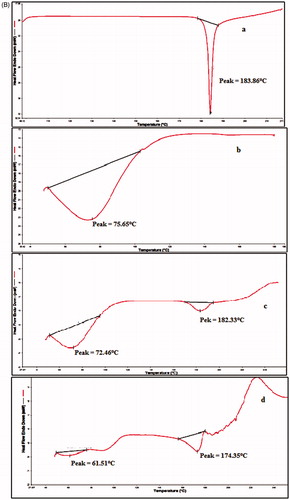
DSC of OM-NS
The crystalline structure of NS was assessed by DSC. The DSC curves for bulk drug, pure PVP k30, physical mixture (drug + PVP k30 + tween80) and lyophilized NS are shown in . The DSC curve of OM exhibited melting endotherm at 183.86 °C, PVP k30 showed decomposed melting point of 75.65 °C and, in physical mixture, the melting point of drug was shifted to 182.33 °C and PVP k30 showed 72.46 °C, while NS showed a melting endotherm at almost the same temperature, i.e. 174.35 °C. However, the peak was much broader compared with peak of bulk drug indicating a reduction in crystallinity of drug, during nanosizing. This could be attributed to increased lattice defects in the drug crystals, which in turn might have reduced the degree of crystallinity [Citation13,Citation37].
X-ray diffraction (XRD) studies
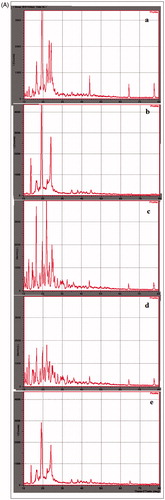
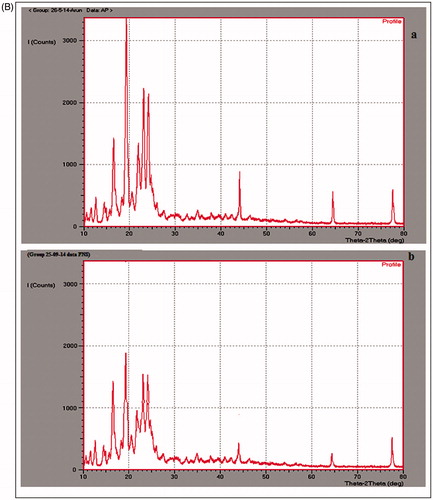
The XRD patterns of plain drug (OM), pure lipids (Dynasan118, GMS + SA), lyophilized SLNs and NS formulation are shown in . Diffractograms were obtained from the initial angle 2θ of 10° to the final angle of 80°. The diffractogram of OM exhibited several sharp peaks and the highest one at 2θ equaling at 19°, 22° and 23° indicating the crystalline nature of OM [Citation38]. Pure lipid of GMS + SA exhibited several major sharp peaks at 2θ values 11°, 16°, 20° and 24°. Dynasan118 exhibited several major sharp peaks at 2θ value of 14°, 19° and 23°. The peak intensities of drug in lyophilized SLNs were reduced indicating that the degree of the crystallinity was lower in the nanoparticles than in the pure lipids. Further, it was clearly shown that no peak of OM was detected in OM-SLNs. These results confirmed that drug was molecularly dissolved in the blend of GMS + SA and Dynasan118. Similarly, in NS formulation, the reduction in intensity of peaks was observed at the corresponding angles which might be due to small particle size (nanometer range), high specific surface area and presence of surfactant in NS (). The results are in accordance with previous report on DSC observations, which indicated the decrease in crystallinity due to nanosizing [Citation36].
Figure 3. (A) XRD spectra of (a) pure OM, (b) pure dynasan118 lipid, (c) pure GMS + SA lipid, (d) lyophilized GMS + SA SLNs and (e) lyophilized dynasan118 SLNs. (B) XRD spectra of (a) pure OM and (b) lyophilized nanosuspension.
SEM studies of SLN
The morphology of lyophilized solid lipid nanoparticles (F1 & F10) was studied by SEM (Hitachi, Tokyo, Japan). These studies (SEM) revealed that solid lipid nanoparticles were not spherical but in different shapes ().
Figure 4. SEM photographs of (a) dynasan118 SLNs (at magnification ×30k) (F1) and, (b) GMS-SA SLNs (at magnification ×15k) (F10).

Bioavailability study
This study was focused to investigate the feasibility of SLN and NS for improved oral delivery of OM. The PK parameters of OM in rats for coarse powder suspension, solid lipid nanoparticles (F10) and NS formulations were calculated by non-compartmental estimations using Kinetica 2000 software (GraphPad Software Inc., La Jolla, CA). The serum concentration versus time profiles following single-dose administration of OM-SLN, OM-NS and suspension formulation are shown in . The PK parameters, AUCtot, tmax. Cmax, MRT and t1/2 were calculated and shown in . The statistical comparison of data was done by one-way ANOVA at a significance level of p values <.01 using Graph pad prism (version 5.0, 2007, GraphPad Software Inc., La Jolla, CA) software. From the results, it was found that optimized SLN formulation showed high Cmax, AUCtot, MRT and t1/2 values when compared with NS and coarse powder suspension, which was statistically significant. The tmax of SLN formulation was higher (4 h) when compared with coarse suspension (1 h) and NS (1.3 h) formulations. No significant difference in Cmax between SLNs and NS formulation was observed. AUCtot of SLN, NS and coarse suspension was 77.19 ± 7.25, 33.6 ± 2.59 and 10.68±.86 μg h/mL, respectively. The bioavailability of optimized NS and optimized SLNs was found to be 3.52 and 7.21 times fold when compared with a control coarse suspension. Further, when AUC was compared for SLN and NS, it was more than twice. In case of SLNs, the MRT and half-life of the drug was found to be more than thrice as that of the NS and coarse suspension, which was an indicator for the prolonged release of the drug from the solid lipid nanoparticle formulation [Citation39]. The improved oral bioavailability of SLNs and NS of OM could be due to the contribution of individual and/or combined effects of nanosized particles of the SLNs and NS. The effective surface would influence the adhesion to GI tract. Consequently, there is increased contact time of the SLN particles. In addition, the phosphatidylcholine and poloxamer could alter the permeability characters of the GI membrane. The fatty acid chains present in the lipids of SLNs improve the uptake by lymphatic transport. This lymphatic transport minimizes the first-pass effect of the drug [Citation40]. Increased saturation solubility of NS could be due to the reduced particle size.
Figure 5. Mean serum concentration versus time profiles of OM coarse suspension, nanosuspension, solid lipid nanoparticles upon oral administration to male albino Wistar rats (mean ± SD, n = 6).
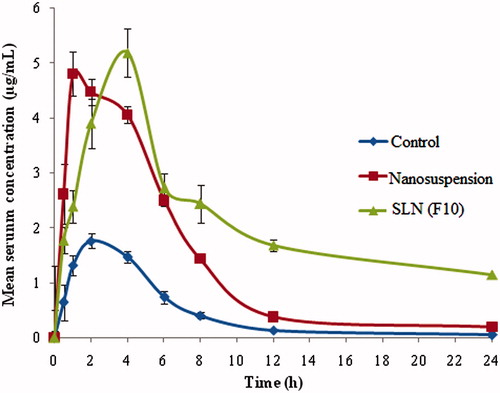
Table 4. PK Parameters of optimized OM nanosuspension, SLNs formulation (F10) and coarse suspension (mean ± SD., n = 6).
Conclusion
In this study, OM-loaded SLN and NS were developed and characterized. A comparison was made by including different quantities of GMS and stearic acid in SLN preparation. Formulation with very low SA level yielded better SLNs over all, with less size, ZP and EE. The solid-state characterization revealed the transformation of crystalline state of the drug to amorphous form in SLNs. Controlled release and enhanced dissolution were accomplished by incorporating OM into the SLN and NS. The SLN (F10) was rather not stable at RT and 4 °C. The PK studies in rats of optimized SLN and NS showed 7.21- and 3.52-folds improvement in oral bioavailability when compared with coarse suspension, respectively. Hence, these results confirmed the potential of SLN and NS as suitable delivery system for oral delivery of olmesartan in improving the oral bioavailability. In this study, the OM-SLNs were found to be superior in performance than OM-NS.
Disclosure statement
Authors declare that no conflict of interest is there in this study.
Additional information
Funding
References
- Warne GT, Jarvis B. Olmesartan medoxomil. Drugs. 2002;62:1345–1353.
- Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196.
- Charman WN, Porter CJH, Mithani S, et al. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J Pharm Sci. 1997;86:269–282.
- Sunesen VH, Vedesdal R, Kristensen HG, et al. Effect of liquid volume and food intake on the absolute bioavailability of danazol, a poorly soluble drug. Eur J Pharm Sci. 2005;24:297–303.
- Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287.
- Porter CJH, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–248.
- Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177.
- Porter CJH, Charman WN. Intestinal lymphatic drug transport: an update. Adv Drug Deliv Rev. 2001;50:61–80.
- Müller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47:3–19.
- Yuan Z, Jianhua Z. Preparation of budesonide nanosuspensions for pulmonary delivery: characterization, in vitro release and in vivo lung distribution studies. Artif Cells Nanomed Biotechnol. 2016;44:285–289.
- Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56:827–840.
- Thakkar HP, Patel BV, Thakkar SP. Development and characterization of nanosuspensions of olmesartan medoxomil for bioavailability enhancement. J Pharm Bioallied Sci. 2011;3:426–434.
- Narendar D, Kishan V. Improved anti-hyperlipidemic activity of Rosuvastatin Calcium via lipid nanoparticles: pharmacokinetic and pharmacodynamic evaluation. Eur J Pharm Biopharm. 2017;110:47–57.
- Shamsunder SD, Sandip SC, Krutika KS, et al. Solid lipid nanoparticles and nanosuspension formulation of saquinavir: preparation, characterization, pharmacokinetics and biodistribution studies. J Microencapsul. 2011;28:515–527.
- Hanafy A, Spahn-Langguth H, Vergnault G, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59:419–426.
- Manjunath K, Venkateswarlu V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J Control Release. 2005;107:215–228.
- Bina G, Amber V. Preparation, characterization, and optimization of altretamine-loaded solid lipid nanoparticles using Box–Behnken design and response surface methodology. Artif Cells Nanomed Biotechnol. 2016;44:571–580.
- Li W, Yang Y, Tian Y, et al. Preparation and in vitro/in vivo evaluation of revaprazan hydrochloride nanosuspension. Int J Pharm. 2011;408:157–162.
- Venkateswarlu V, Manjunath K. Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release. 2004;95:627–638.
- Jaydeep P, Garala K, Anjali P, et al. Development of the UV spectrophotometric method of Olmesartan medoxomil in bulk drug and pharmaceutical formulation and stress degradation studies. Pharm Met. 2011;2:36–41.
- Pardeike J, Daniela MS, Nina S, et al. Nanosuspensions as advanced printing ink for accurate dosing of poorly soluble drugs in personalized medicines. Int J Pharm. 2011;420:93–100.
- Aji Alex MR, Chacko AJ, Jose S, et al. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42:11–18.
- Narendar D, Kishan V. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016;23:395–404.
- Vinay Kumar V, Raghavendra C, Rojarani K, et al. Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: a novel strategy to avoid intraduodenal administration. Colloids Surf B. 2012;95:1–9.
- Doktorovova S, Shegokar R, Fernandes L, et al. Trehalose is not a universal solution for solid lipid nanoparticles freeze-drying. Pharm Dev Technol. 2014;19:922–929.
- Schwarz C, Mehnert W. Freeze-drying of drug-free and drug-loaded solid lipid nanoparticles (SLN). Int J Pharm. 1997;157:171–179.
- Cavalli R, Caputo O, Carlotti ME, et al. Sterilization and freeze-drying of drug-free and drug-loaded solid lipid nanoparticles. Int J Pharm. 1997;148:47–54.
- Vinay Kumar V, Chandrasekhar D, Ramakrishna S, et al. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335:167–175.
- Madhulika P, Deependra S, Manju S. Influence of selected variables on fabrication of Triamcinolone acetonide loaded solid lipid nanoparticles for topical treatment of dermal disorders. Artif Cells Nanomed Biotechnol. 2016;44:392–400.
- Vijaykumar N, Raviraj P, Venkateshwarlu V, et al. Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm Dev Technol. 2009;14:290–298.
- Shah SK, Asnani AJ, Kawade DP, et al. Simultaneous quantitative analysis of olmesartan medoxomil and amlodipine besylate in plasma by high performance liquid chromatography technique. J Young Pharmacists. 2012;4:88–94.
- Müller RH, Becker R, Kruss B, et al. Pharmaceutical nanosuspensions for medicament administration as system of increased saturation solubility and rate of solution. 1998; US Patent, No. 5858410.
- Sumeet S, Natarajan J, Kunal J, et al. Olanzapine loaded cationic solid lipid nanoparticles for improved oral bioavailability. Curr Nanosci. 2013;9:26–34.
- Rawat MK, Jain A, Singh S. Studies on binary lipid matrix based solid lipid nanoparticles of repaglinide: in vitro and in vivo evaluation. J Pharm Sci. 2011;100:2366–2378.
- Grant DJW, Brittian HG. Solubility of pharmaceutical solids. In: Brittian HG, editor. Physical characterization of pharmaceutical solids. New York: Marcel Dekker; 1995. p. 322–380.
- Narendar D, Kishan V. Pharmacokinetic and pharmacodynamic studies of nisoldipine loaded solid lipid nanoparticles by central composite design. Drug Dev Ind Pharm. 2015;41:1968–1977.
- Otsuka M, Kaneniwa N. Effect of seed crystals on solid-state transformation of polymorphs of chloramphenicol palmitate during grinding. J Pharm Sci. 1986;75:506–511.
- Venkata SKB, Bansilal PG, Sharad CPN, et al. Polymorphic form of olmesartan and process for its preparation. 2006; US Patent No. 20060281800 A1.
- Rabea PFJ, Ahmad Z, Iqbal MS, et al. Solid lipid nanoparticles of anticancer drug andrographolide: formulation, in vitro and in vivo studies. Drug Dev Ind Pharm. 2014;40:1206–1212.
- Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129:1–10.