
Abstract
We constructed a dual ligands-modified nanostructured lipid carrier (NLC) called PAR-NLC, in which the epidermal growth factor receptor (EGFR)-targeted small peptide AEYLR was attached to the distal end of PEG2000 anchored on the NLC surface naming PEG-AEYLR, and poly-arginine (R8) as a classic cell-penetrating peptide was attached directly to the NLC surface. PAR-NLC was near-spherical particle with average size ∼50 nm and zeta potential at +14.09 mV; the cellular uptake of PAR-NLC showed synergistic effect of the two peptides, presented as significant superior cellular uptake in EGFR-positive cells NCI-H1299 and S180 over EGFR-negative cell K562. In the animal optical imaging study, 2 h after the administration of the Dir-loaded PAR-NLC, maximum Dir signal appeared in tumor tissue, indicating prompt tumor targeting effect, as time prolonged to 48 h, the Dir signal attenuated in the organs except tumor, suggesting constant clearance from the body. In the in vivo antitumor study, in premise of the same dose, paclitaxel-loaded PAR-NLC exhibited better antitumor and safety effect than Taxol, the body weight of the mice was more stable and tumor size was smaller. In summary, PAR-NLC was a potential drug carrier to deliver anticancer drugs safely and effectively.
Introduction
Although equipped with angiogenesis, tumor tissue still experiences far less blood than the reticuloendothelial system (RES) [Citation1], in this case to prepare nanoparticles (NPs) that are less captured by the RES could prolong their systemic circulation time and improve the chances to enter the tumor tissue [Citation2]. Since PEG could form a layer of neutral generic hydrophilic polymer outside of the NPs, PEGylated NPs could exhibit stealth effect towards RES thus extending the circulation time and adding the chances to enter the tumor tissue [Citation3–5]. Nevertheless, the same reason could lead to lower cellular uptake of the PEGylated NPs, for example, the stealth liposomes (Doxil®) shows little cellular uptake when incubated with tumor cells for 24 h [Citation1]. To solve this problem, researchers conjugate ligands that could be recognized by the receptors on the cell membrane on the distal end of the PEG aiming to elevate the cellular uptake of the NPs via the receptor-ligand recognition effect while maintaining the stealth effect of PEG [Citation6–9]; however, the internalized particles are sometimes trapped inside the vesicles that would be further trafficked through the endo-lysosomal pathway to lysosomes for degradation, which would compromise the efforts and shorten the anticancer effect [Citation10].
As a member of the cell-penetrating peptides, polyarginine possesses a distinguished feature that it could slower the acidification of the endosome and strive for suitable condition and time for itself and for polyarginine-modified NPs to escape from the endosome [Citation10]. Besides, due to the positive electric potential, polyarginine are liable to contact with the negative cell membrane, so polyarginine-modified NPs could be internalized to the cells in a faster speed and greater amount [Citation11,Citation12]. For this reason, polyarginine-decorated NPs often show superior cellular uptake and concomitant outstanding in vitro antitumor effect [Citation13,Citation14]. However, deficiency in the tumor cell specificity makes it run risks when used in vivo because NPs decorated with polyarginine could probably cause unwanted toxicity to the organs abundant in blood like spleen, heart and liver [Citation15].
According to the basis above, we constructed a nanostructured lipid carrier (NLC) called PAR-NLC with two peptides, one is the epidermal growth factor receptor (EGFR)-targeted small peptide AEYLR (developed by our group) [Citation16,Citation17], it was attached to the distal end of PEG2000, the other is polyarginine (RRRRRRRR, R8) anchored directly to the NLC surface. As the drug carrier used in this study, NLC exhibits advantages like good biocompatibility and easy modification and so forth [Citation18]. Another advantage makes it a better cargo over liposome is its greater surface decoration potential. Since the phospholipid bilayer could be disrupted by the detergent-like motifs like DSPE-PEG, the surface decoration amount on liposomes are often <10 mol% [Citation1]. By contrast, we find that the stability of NLC anchored with >10 mol% was even better than the ones with <10%, and the size was smaller, too. This was possibly because of the beneficial surfactant-like effect of stearyl-PEG in the NLC process; in addition, as for NLC, the less the structure uniformity, the more stable the NLC would be, and the less probability of drug precipitation [Citation19]. So we chose NLC instead of liposome to be the drug delivery carrier. To guarantee the longer blood circulation time and less RES capture, we decorated PEG2000 on the NLC surface and conjugated the EGFR-targeting small peptide AEYLR to its the distal end. EGFR is a member of the receptor tyrosine kinase, it overexpresses on the membrane of many malignant tumor cells and correlates with their proliferation, differentiation and migration [Citation20]. EGFR is a transmembrane protein, the extracellular ligand-binding domains could bind with the epidermal growth factor (EGF) or other ligands, after which the intracellular autophosphorylation domains and the catalytic domains are activated initiating the cell proliferation [Citation21]. The small peptide AEYLR developed by our group could combine specifically with EGFR, the in vitro and in vivo EGFR-targeting ability of AEYLR-modified NLC has been conformed [Citation16,Citation17]. In the present study, we attached it to the distal end of PEG2000 excepting it to mediate the positive targeting effect and improve the cellular uptake. The combination of the stealth effect of PEG2000 and the active EGFR-targeting effect of AEYLR could ensure better accumulation in tumor tissue by EPR effect; however, nanosized drug carrier still have the opportunity to leak back to the blood through the over 400 nm interspace of the blood vessel [Citation1]. In this case, to build a nano drug delivery system which internalizes into the cells faster could lessen the inverse flow to the blood circulation thus guarantee better antitumor effect. For this purpose, we employed R8 for its outstanding cell penetrating ability. Unlike AEYLR which was attached to the distal end of PEG2000, R8 was conjugated directly to the NLC surface thus hidden in the PEG2000 cluster, hence, the in vivo adverse effect of R8 could be relieved. By the cooperation of the peptides and PEG2000, PAR-NLC could escape from the RES and leak to the tumor tissue by EPR effect, then the AEYLR in the structure could mediate a positive-targeting effect to EGFR-positive cancer cells, as PAR-NLC get closer to the cells, R8 could be exposed profit from the swinging of the PEG2000 chain and perform a synergistic effect with AEYLR for better cellular uptake.
Materials and methods
Materials
Stearyl-polyarginine (STR-R8) and stearyl-PEG2000-AEYLR (STR-PEG2000-AEYLR) with greater than 98% purity were synthesized by TeraBio Technology Co., Ltd., Guangzhou, China. Glycerin monostearate (GMS), Kolliphar ELP and Kolliphor HS15 were generously presented by BASF Co., Ltd., Ludwigshafen, Germany. Medium chain triglyceride (MCT) was purchased from Yuhao Chemical Co., Ltd., Hangzhou, China. Comarin 6 (Cou6) was purchased from J&K Scientific Ltd., Beijing, China. Paclitaxel (PTX) was purchased from Jiangsu Hengrui Pharmaceutical Co., Ltd. Jiangsu, China. DiR iodide (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide) was purchased from Fanbo Co., Ltd. Beijing, China.
NCI-H1299, S180 and K562 cells were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI-1640 culture medium (Hyclone Co., Ltd., Thermo Fisher Scientific, Perth, UK), supplemented with 10% foetal bovine serum (Sijiqing Co., Ltd., Hangzhou, China). For NCI-H1299 cell, when the cells grew to 80–90% confluence, the cells were trypsined and passaged in the ratio of 1:3. S180 and K562 cells were passaged every 2 days in the ratio of 1:3.
All other chemicals and reagents were of analytical or cell culture grade.
Methods
We have prepared PEG2000-AEYLR modified NLC (PA-NLC), R8-modified NLC (R-NLC) and PEG2000-AEYLR and R8-comodified NLC (PAR-NLC) in the present study, first to verdict the distinguished EGFR-targeting capacity and the faster cellular uptake of PAR-NLC by in vitro cellular uptake and in vivo optical imaging study; then as a drug carrier, PAR-NLC was loaded with antitumor drug PTX, of which the morphology and in vitro drug release profile were tested; after that, the in vivo antitumor effect of PTX-loaded PAR-NLC was assessed in comparison with Taxol to learn whether PAR-NLC was a safe and effective antitumor drug delivery system.
The demands of the Animal Ethics committee have been carefully followed in the experiments related with mice.
Preparation of PA-NLC, R-NLC and PAR-NLC
Melt emulsification method was employed in this study. Briefly, Compritol® 888 ATO, GMS, MCT and Kolliphor ELP were mixed and melted at 80 °C to get the transparent oil phase, Kolliphor HS 15 was dispersed in distilled water at the same temperature and added into the oil phase dropwisely, after which the primary emulsion was cooled immediately at 4 °C to obtain the unmodified NLC with transparent light blue opalescence.
To prepare PA-NLC, STR-PEG2000-AEYLR was added into the oil phase at the ratio of 10 mol% to the total lipid; to prepare R-NLC, STR-R8 was added to the oil phase at the ratio of 2 mol% to the total lipid; and to prepare PAR-NLC, STR-PEG2000-AEYLR and STR-R8 were added into the oil phase at the same ratio as described above. The process was the same as unmodified NLC.
The constructed preparations were diluted 50 times by distilled water and measured by Malvern Zetasizer ZS (Malvern, Worcestershire, UK) for the size and zeta potential, each sample was tested for three times.
Cou6 or Dir was added in the oil phase to be loaded in PA-NLC, R-NLC and PAR-NLC to learn their in vitro cellular uptake and in vivo and ex vivo tissue distribution, respectively; PTX was employed to assess the potential of PAR-NLC to be an antitumor drug carrier, it was added to the oil phase and the process was the same as mentioned above.
Cellular uptake of PA-NLC, R-NLC and PAR-NLC
EGFR-positive cell NCI-H1299 and S180 together with EGFR-negative cell K562 were selected to evaluate the cellular uptake of the preparations.
NCI-H1299 cells were seeded in 24-well plate at the density of 1 × 105 cells/well and incubated overnight to allow the cell attachment, then the culture medium was replaced by fetal bovine serum (FBS)-free PRIM 1640 culture medium-containing Cou6-loaded PA-NLC, R-NLC and PAR-NLC with the Cou6 concentration at 2 μg/ml and incubated for 1 and 4 h, respectively; then the culture medium was abandoned and the cells were rinsed for three times with PBS, after that, the cells were trypsined and resuspended in PBS and assayed by flow cytometry (FACSCalibur, Becton, Dickinson and Company, NY, USA.).
Moreover, NCI-H1299 cells were seeded at the density of 1 × 105 cells/well in the 24-well plate preplaced with sterile cover glass and incubated overnight, at the second day, the cells were treated with the preparations as described above. After the 1 or 4 h incubation, the cells were washed with PBS for three times, fixed with paraformaldehyde, stained with DAPI and observed with the laser confocal microscopy (Carl Zeiss, Oberkochen, Germany).
S180 or K562 cells were placed in 24-well plate at the density of 1 × 105 cells/well and incubated overnight, at the second day, the cells were centrifuged and the culture medium was changed to FBS-free fresh medium containing various Cou6-loaded preparations with Cou6 concentration at 2 μg/ml and incubated for 1 and 4 h, then the cells were centrifuged at 1 × 103 rpm, the supernatant was discarded and the cells were washed for three times with PBS, resuspended in PBS and measured by flow cytometry.
In vivo and ex vivo tissue distribution of Dir-loaded PA-NLC, R-NLC and PAR-NLC in S180 tumor-bearing mice
S180 cells were planted in the peritoneal cavity of Kunming Mice, and the ascites was used for the cell passage. The ascites was diluted one time and injected to the left armpit of Kunming mice at the volume of 0.2 ml, the experiment was conducted 7 days after the tumor cell injection. The mice were randomly divided into four groups with six mice in each group.
Dir-loaded PA-NLC, R-NLC and PAR-NLC were intravenously injected to the mice with Dir concentration at 2.5 mg/kg, the pictures of the mice were taken at predicted time points (2, 6 and 48 h), three mice in each group were sacrificed at 6 and 48 h to collect the tumor, heart, liver, spleen, lung and kidney, and the pictures of which were taken to see the fluorescent signal in each organ.
Morphology and in vitro drug release of PAR-NLC
After conformation of the high cellular uptake and effective targeting ability of PAR-NLC, we observed its morphology and in vitro drug release profile using PTX as the model drug in this section.
PTX-loaded PAR-NLC was diluted 100 times and added on the carbon-coated copper screen, negatively stained with 20 g/L phosphotungstic acid, and observed by transmission electron microscopy (TEM, Hitachi TEM system, Tokyo, Japan).
To explore the in vitro drug release profile, PTX-loaded PAR-NLC was placed in the dialysis bag [molecular weight (MW) 8–14 KDa] with both end sealed, immersed in 50 ml drug release medium (the release medium was pH 7.4 PBS with 0.5% Tween-80) and shaken at 37 °C at 100 rpm. An aliquot of 1 ml release medium was withdrawn at predicted time point (2, 4, 6, 8, 10, 24, 36 and 48 h), and a supplement was added immediately. The withdrawn release medium was analyzed by high-performance liquid chromatography (HPLC) to know the PTX concentration, and the accumulative drug release profile was calculated.
Study of the in vivo antitumor efficacy
S180 cells were planted to the left armpit of Kunming mice with the ascites in “In vivo and ex vivo tissue distribution of Dir-loaded PA-NLC, R-NLC and PAR-NLC in S180 tumor-bearing mice” section, when the tumor volume reached about 100 mm3 (tumor volume = length × width2/2), the mice were randomly divided into three groups with five mice in each group (saline, Taxol and PTX-loaded PAR-NLC groups). Corresponding preparation was intravenously injected to the mice at predicted time points (0, 3, 6, 9 and 12 day). The administration dose of PTX was 10 mg/kg in Taxol and PTX-loaded PAR-NLC groups.
The body weight and tumor volume were recorded immediately after each administration. After the course of 15 days, the mice were sacrificed and the tumors were peeled off, weighted and pictured.
Results and discussion
Size and zeta potential of PA-NLC, R-NLC and PAR-NLC
The size and zeta potential of PA-NLC, R-NLC and PAR-NLC were listed in , as the emulsifier like effect of STR-PEG2000-AEYLR or STR-R8 exerted, the average size of the preparations were all relatively small at ∼50 nm with acceptable polydispersity index (PDI) < 0.3, such size distribution could ensure the successful leakage through the over 400 nm interspace on the tumor blood vessel to the tumor tissue using EPR effect [Citation1]. The zeta potential of PA-NLC was +2.36 mV, and R-NLC and PAR-NLC showed strong positive zeta potential at ∼+14 mV due to the guanidine group in the R8 structure. Although the zeta potential absolute value of the preparations were all beyond the value that was widely believed for the stability of the NPs (higher than 20–25 mV) [Citation19], they still exhibited good stability as the average size did not show obvious change in 1 week, this was possibly because of the layer of neutral generic hydrophilic polymer formed by PEG that coated outside of the NLC and the ununiformity structure of the NLC.
Table 1. Size, PDI and zeta potential of PA-NLC, R-NLC and PAR-NLC.
In vitro cellular uptake study
According to , for EGFR-positive cells NCI-H1299 and S180, the Cou6 signal was stronger in PAR-NLC than the other groups in as short as 1 h, exhibiting a faster cellular uptake profile; and as time prolonged to 4 h, the Cou6 signal was much stronger than the other groups suggesting constant effective internalization to the cells. As for EGFR-negative cell K562, the Cou6 signal was nearly identical in PAR-NLC and R-NLC, indicating little effect of AEYLR in PAR-NLC due to its splendid EGFR selective effect.
Figure 1. Cellular uptake of PA-NLC, R-NLC and PAR-NLC in different cell lines at different incubation time. (A) EGFR-positive cell NCI-H1299, left for 1 h and right for 4 h; (B) EGFR-positive cell S180, left for 1 h and right for 4 h; (C) EGFR-negative cell K562, left for 1 h and right for 4 h.
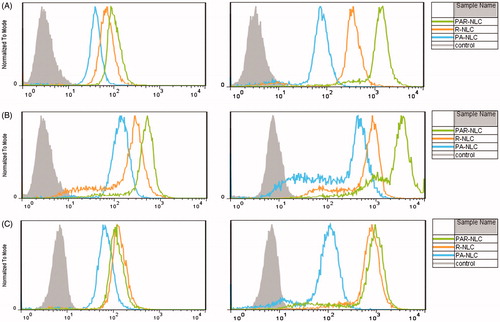
The results also implied that the PEG2000 chain did not interfere the cell-penetrating property of R8 because the cellular uptake of PAR-NLC was higher than R-NLC, this outcome seemed to contradict with our recognition. As the length of PEG2000 was much longer than R8, it should sterically hinder R8 which was the main reason why researchers developed the cleavable PEG to eliminate the hindrance effect [Citation13,Citation22]. Our results, on the other hand, suggested that 10 mol% PEG2000 would not bring problems like this. According to references, for 100 nm liposomes, 10 mol% PEG could guarantee the brush conformation; however, for particles smaller than 100 nm, the PEG concentration should be higher [Citation23]. In this case, 10 mol% PEG2000 that we added to the formula could not guarantee the brush conformation since the size of our preparations were much smaller than 100 nm, the PEG2000 was more likely to be the mushroom–brush transition conformation which gave R8 great chances to be exposed; besides, although buried inside the PEG2000 chain, R8 sill possessed the ability to attract positive molecules in the cell culture medium to obtain a positive electric potential (presented as the similar zeta potential as R-NLC) which could assist the PAR-NLC to be absorbed to the negative cell membrane and initiate the cellular uptake process. Meanwhile, the stretched brush like PEG2000 could guarantee the prior combination between the attached AEYLR and the EGFR on the cell membrane and exert the effect of AEYLR.
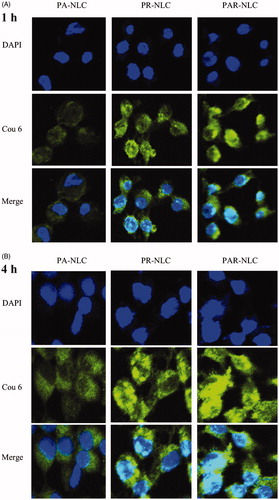
also revealed the superior cellular uptake of PAR-NLC over the other ones, presented as more Cou6 signals in the cells in 1 and 4 h, the nucleic delivery of PAR-NLC was also significant which correlated closely with the outstanding membrane penetrating capacity of R8 [Citation24]; meanwhile, the more cell internalization of PAR-NLC than R-NLC also indicated the unnegligible effect of AEYLR which targeted to the EGFR on the cell membrane.
Figure 2. Confocal laser scanning microscopy (CLSM) images of NCI-H1299 cells incubated with PA-NLC, R-NLC and PAR-NLC at different incubation time. (A) 1 h and (B) 4 h. Green signal is Cou6, blue signal is the cell nuclei stained by DAPI.
In vivo and ex vivo tumor-targeting study of PA-NLC, R-NLC and PAR-NLC using the optical imaging system
We employed the optical imaging system to learn the in vivo and ex vivo tumor-targeting ability and tissue distribution of PA-NLC, R-NLC and PAR-NLC. According to , distinct tumor accumulation was observed in as short as 2 h in all the three preparations suggesting excellent tumor-targeting capacity which we believe was contributed by the relative small size of the particles; nevertheless, same reason could cause the back flow of the particles to the blood circulation leading to the decrease of the accumulation presented as the attenuating of the Dir signal.
Figure 3. In vivo and ex vivo images of S180 tumor-bearing mice injected with Dir-loaded PA-NLC, R-NLC and PAR-NLC. The Dir concentration was 2.5 mg/kg. The maximum and minimum Dir signal was 900 and 4000, respectively.
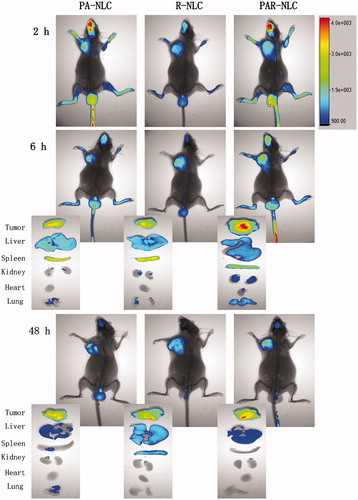
The most significant Dir fading appeared in PA-NLC group, which could be speculated from the in vitro cellular uptake study, as the cellular uptake of PA-NLC was the slowest, the extra ones that were not internalized to the tumor cells could flow back to the blood circulation due to the small size. However in R-NLC group, the in vivo pictures showed relatively consistent Dir signal from 2 to 48 h, the ex vivo pictures even revealed a uplifting accumulation as the time prolonged suggesting more effective tumor accumulation since R-NLC could enter the cells in a faster speed and more amount. Although performed a better tumor accumulation, R-NLC was not a satisfactory antitumor drug delivery system, since signals in other tissues were still significant especially in liver and spleen at 48 h, implying the longer time that R-NLC exposed to them which may arouse severe side effect. In this case, PAR-NLC which possessed both R8 and PEGylated AEYLR was constructed. The in vivo and ex vivo images of PAR-NLC group have confirmed that the tumor accumulation was significantly higher than PA-NLC and R-NLC groups indicating the synergistic effect of the two peptides, moreover, the Dir signal in other organs were weaker than either peptide-modified NLC, such distribution kinetics was reasonable and welcomed because someone has declared that the introduction of the tumor creates a new compartment for the distribution [Citation1], then more tumor accumulation means less amount in other organs thus leading to less toxicity.
Morphology and in vitro drug release profile of PAR-NLC
showed the near-spherical morphology of PAR-NLC with the size slightly smaller than 50 nm. Someone has claimed that the NP size was often smaller under the TEM than measured by DLS, which was because the hydrodynamic radius tested by DLS were often consisted with the unavoidable absorbance of molecules to the NP surface while the TEM images related only with the particle [Citation25].
Figure 4. Morphology and in vitro drug release of PAR-NLC. (A) TEM image of PAR-NLC, the scale bar is 100 nm. (B) In vitro drug release of PAR-NLC, the result was presented as mean ± standard deviation (n = 3).
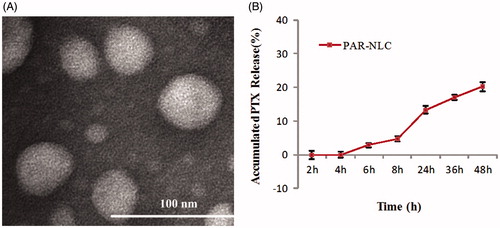
The in vitro drug release curve of PAR-NLC was showed in , a sustained drug release profile was observed with little drug release before 4 h and below 30% at 48 h which we believe was contributed by the non-uniformity of the NLC structure and the PEG2000 on the surface. As widely accepted, the more complex the NLC structure, the more stable it was [Citation26]; the PAR-NLC that we developed was consisted with glyceryl palmitate, GMS, stearyl group and MCT, such complicated constituents have guaranteed the mass structure which could exhibit a sustained drug release profile, besides, the spatial barrier formed of the PEG2000 could also slow the drug release down [Citation27]. The sustained drug released profile was welcomed since it could ensure less drugs exposed to the healthy organs.
In vivo antitumor efficacy
In this section, S180 cells were selected to build the tumor model. The body weight and tumor volume of the mice were recorded during the 15 days course, and saline group was known as the normal speed of the tumor growth without drug treatment. From the results in , we could see that at the Day 6 after the first administration, Taxol and PTX-loaded PAR-NLC exhibited 55 and 83% tumor inhibition ratio [TIR, calculated by TIR (%) = (1 − V/Vsaline) × 100%, where V was the average tumor volume of Taxol or PAR-NLC group and Vsaline was the average tumor volume of the saline group), respectively; after the 15 days treatment, the TIR of PAR-NLC has reached 90% and was higher than the Taxol group (p < .001).
Figure 5. Antitumor effect of PAR-NLC and Taxol, the drugs were injected to S180 tumor-bearing mice through the tail vein at 10 mg/kg. The drug administration was conducted at 0, 3, 6, 9 and 12 days, and the mice were sacrificed at the Day 15. (A) Body weight. (B) Tumor volume. (C) Tumor weight after the sacrifice of the mice. (D) Image of the tumors in Saline, Taxol and PAR-NLC groups (n = 5).
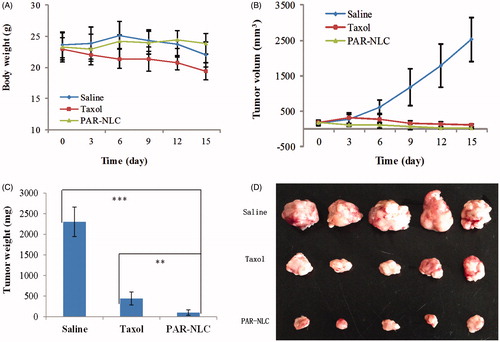
Along with the good TIR of Taxol was the reduction of the mice body weight which was possibly caused by the Cremophor EL in the solvent and the injury to the healthy organs by PTX due to its absence of tumor selectivity [Citation28]; in contrast to Taxol, PAR-NLC group showed more stable body weight and higher TIR, which we believe was contributed by the synergistic effect of AEYLR, PEG2000 and R8 as with their modification PAR-NLC exerted excellent tumor accumulation and tumor cell uptake, in addition, the sustained drug release also guaranteed the less PTX exposure to healthy tissues thus reduced the adverse effect to the body.
Conclusions
In the present study, we successfully constructed PAR-NLC which was comodified with R8 and PEGylated AEYLR; the in vitro cellular uptake study has confirmed a faster and better cell internalization than either peptide-modified NLC in EGFR-positive cells indicating the synergistic effect of the two peptides which maintained both the membrane penetrating ability of R8 and the EGFR targeting capacity of AEYLR; the in vivo and ex vivo tissue distribution study has revealed that PAR-NLC could accumulate in tumor tissue in a short time and remained there for at least 48 h, in addition, the fast clearance in other organs implied its safety as the exposure to healthy tissues was decreased; the results in the in vivo antitumor efficacy has verified that PTX-loaded PAR-NLC could exert excellent antitumor effect while decrease the adverse effect to the body indicating PAR-NLC was an outstanding candidate as the antitumor drug delivery system.
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–181.
- Lo CL, Chou MH, Lu PL, et al. The effect of PEG-5K grafting level and particle size on tumor accumulation and cellular uptake. Int J Pharm. 2013;456:424–431.
- Stepniewski M, Pasenkiewicz-Gierula M, Rog T, et al. Study of PEGylated lipid layers as a model for PEGylated liposome surfaces: molecular dynamics simulation and Langmuir monolayer studies. Langmuir. 2011;27:7788–7798.
- Koren E, Apte A, Jani A, et al. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264–273.
- Tabatabaei Mirakabad FS, Akbarzadeh A, Milani M, et al. A Comparison between the cytotoxic effects of pure curcumin and curcumin-loaded PLGA-PEG nanoparticles on the MCF-7 human breast cancer cell line. Artif Cells, Nanomed Biotechnol. 2016;44:423–430.
- Kim SK, Huang L. Nanoparticle delivery of a peptide targeting EGFR signaling. J Control Release. 2012;157:279–286.
- Arabi L, Badiee A, Mosaffa F, et al. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J Control Release. 2015;220:275–286.
- Dubey PK, Singodia D, Vyas SP. Polymeric nanospheres modified with YIGSR peptide for tumor targeting. Drug Deliv. 2010;17:541–551.
- Kaur S, Mehra NK, Jain K, et al. Development and evaluation of targeting ligand-anchored CNTs as prospective targeted drug delivery system. Artif Cells Nanomed Biotechnol. 2017;45:242–250.
- Raagel H, Saalik P, Hansen M, et al. CPP-protein constructs induce a population of non-acidic vesicles during trafficking through endo-lysosomal pathway. J Control Release. 2009;139:108–117.
- Torchilin VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev. 2008;60:548–558.
- Trehin R, Merkle HP. Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur J Pharm Biopharm. 2004;58:209–223.
- Ruoslahti E. Tumor penetrating peptides for improved drug delivery. Adv Drug Deliv Rev. 2016;pii:S0169-409X(16)30094-1.
- Sun M, Gao Y, Zhu Z, et al. A systematic in vitro investigation on poly-arginine modified nanostructured lipid carrier: pharmaceutical characteristics, cellular uptake, mechanisms and cytotoxicity. Asian J Pharm Sci. 2016;12:51–58.
- Park K. Arginine-rich CPPs for improved drug delivery to tumors. J Control Release. 2012;159:153.
- Han C, Li Y, Sun M, et al. Small peptide-modified nanostructured lipid carriers distribution and targeting to EGFR-overexpressing tumor in vivo. Artif Cells Nanomed Biotechnol. 2014;42:161–166.
- Han CY, Yue LL, Tai LY, et al. A novel small peptide as an epidermal growth factor receptor targeting ligand for nanodelivery in vitro. Int J Nanomedicine. 2013;8:1541–1549.
- Yang X, Zhao L, Almasy L, et al. Preparation and characterization of 4-dedimethylamino sancycline (CMT-3) loaded nanostructured lipid carrier (CMT-3/NLC) formulations. Int J Pharm. 2013;450:225–234.
- Zhang W, Li X, Ye T, et al. Design, characterization, and in vitro cellular inhibition and uptake of optimized genistein-loaded NLC for the prevention of posterior capsular opacification using response surface methodology. Int J Pharm. 2013;454:354–366.
- Wang XB, Zhou HY. Molecularly targeted gemcitabine-loaded nanoparticulate system towards the treatment of EGFR overexpressing lung cancer. Biomed Pharmacother. 2015;70:123–128.
- Abe M, Kuroda Y, Hirose M, et al. Inhibition of autophosphorylation of epidermal growth factor receptor by small peptides in vitro. Br J Pharmacol. 2006;147:402–411.
- Liu J, Zhang B, Luo Z, et al. Enzyme responsive mesoporous silica nanoparticles for targeted tumor therapy in vitro and in vivo. Nanoscale. 2015;7:3614–3626.
- Lee H. Molecular dynamics studies of PEGylated single-walled carbon nanotubes: the effect of PEG size and grafting density. J Phys Chem C. 2013;117:26334–26341.
- Kang MH, Park MJ, Yoo HJ, et al. RIPL peptide (IPLVVPLRRRRRRRRC)-conjugated liposomes for enhanced intracellular drug delivery to hepsin-expressing cancer cells. Eur J Pharm Biopharm. 2014;87:489–499.
- Bhattacharjee S. DLS and zeta potential - what they are and what they are not? J Control Release. 2016;235:337–351.
- Aditya NP, Macedo AS, Doktorovova S, et al. Development and evaluation of lipid nanocarriers for quercetin delivery: a comparative study of solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and lipid nanoemulsions (LNE). Food Sci Technol. 2014;59:115–121.
- Guo P, Song S, Li Z, et al. In vitro and in vivo evaluation of APRPG-modified angiogenic vessel targeting micelles for anticancer therapy. Int J Pharm. 2015;486:356–366.
- Yoshizawa Y, Kono Y, Ogawara K, et al. PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int J Pharm. 2011;412:132–141.