
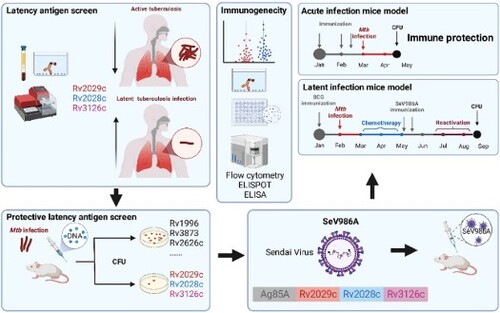
ABSTRACT
One-quarter of the world’s population is infected with Mycobacterium tuberculosis (Mtb). After initial exposure, more immune-competent persons develop asymptomatic latent tuberculosis infection (LTBI) but not active diseases, creates an extensive reservoir at risk of developing active tuberculosis. Previously, we constructed a novel recombinant Sendai virus (SeV)-vectored vaccine encoding two dominant antigens of Mtb, which elicited immune protection against acute Mtb infection. In this study, nine Mtb latency-associated antigens were screened as potential supplementary vaccine candidate antigens, and three antigens (Rv2029c, Rv2028c, and Rv3126c) were selected based on their immune-therapeutic effect in mice, and their elevated immune responses in LTBI human populations. Then, a recombinant SeV-vectored vaccine, termed SeV986A, that expresses three latency-associated antigens and Ag85A was constructed. In murine models, the doses, titers, and inoculation sites of SeV986A were optimized, and its immunogenicity in BCG-primed and BCG-naive mice were determined. Enhanced immune protection against the Mtb challenge was shown in both acute-infection and latent-infection murine models. The expression levels of several T-cell exhaustion markers were significantly lower in the SeV986A-vaccinated group, suggesting that the expression of latency-associated antigens inhibited the T-cell exhaustion process in LTBI infection. Hence, the multistage quarter-antigenic SeV986A vaccine holds considerable promise as a novel post-exposure prophylaxis vaccine against tuberculosis.
GRAPHICAL ABSTRACT
Introduction
Tuberculosis (TB) remains one of the major causes of infectious disease mortality, and with 10.6 million new cases and 1.6 million deaths in 2021, it was the world’s most lethal infectious disease, only being surpassed by COVID-19 during the 2019–2021 pandemic [Citation1]. One-quarter of the world’s population is estimated to have been infected with the causative agent Mycobacterium tuberculosis (Mtb) [Citation2]. After initial exposure, 30% will develop primary infection, among which, 85% to 90% of immune-competent people develop asymptomatic latent TB infection (LTBI), which cumulatively could amount to 2 billion people living with LTBI [Citation3]. In LTBI, there is a state of persistent immune response to Mtb antigenic stimulation in which bacillary replication is controlled and there is no clinical evidence of active TB disease [Citation4]. Once asymptomatic LTBI is established after initial exposure, there is a 5% to 10% lifetime risk of developing into active TB disease, among the disease progressors, nearly one-half of LTBI progress within two years after exposure, and the remaining risk of progression is distributed over the rest of the life span [Citation5,Citation6]. Thus, the current LTBI burden includes an extensive reservoir of individuals at risk of developing active TB.
Mycobacterium bovis Bacille Calmette-Guérin (BCG) is the only licensed TB vaccine. Vaccination in infancy offers protection against the aggressive childhood forms of the disease: meningeal and miliary TB [Citation7]. However, its protective efficacy against TB diseases ranges from 0% to 80% in adults [Citation8]. BCG vaccination fails to block the establishment of latent infection during the acute Mtb infection phase or to eliminate established latent Mtb and prevent its subsequent reactivation [Citation9,Citation10]. Most novel TB vaccines that have entered clinical trials have been based on Mtb antigens that are expressed during stages of bacterial replication [Citation11] and therefore might favor the prevention of active Mtb infection rather than the reactivation of latent infection. Accordingly, vaccines that target LTBI by incorporating latency-associated antigens might overcome this deficiency [Citation12,Citation13]. To this end, several multistage protein-based TB vaccines that included latency-associated antigens have been constructed and shown to give immune protection in animal models [Citation12]. However, considering that Mtb is mainly resident in the lung tissues after latent infection establishment [Citation14,Citation15], systemic immunization with protein-based vaccines might fail to induce sufficient immune memory in the lung mucosa [Citation16]. Thus, other vaccine platforms might overcome this limitation.
Among these, recombinant virus-vectored vaccines are capable of inducing robust immune responses by mimicking the processes of pathogenic invasion that result in the establishment of long-lasting immune memory in the lung [Citation17]. Sendai virus (SeV) is a negative sense, single-stranded, and non-integrating RNA virus of the family paramyxoviridae respiratory virus. It has low pathogenicity, a powerful capacity for foreign gene expression, a broad host range, and constitutes a vector capable of inducing mucosal immunity in the lung. We previously constructed a replication-deficient SeV [Citation18]-based TB vaccine that expressed Mtb immunodominant antigens Ag85A and Ag85B (SeV85AB) and showed that it induced high levels of lung tissue-resident memory T cell-mediated immune protection, which provided a new research direction for TB vaccines [Citation19]. Notably, SeV85AB could be used to optimize the systemic immune protection against Mtb infection induced by combined BCG/DNA vaccination [Citation20,Citation21].
In this study, nine Mtb latency-associated antigens [Citation22–28] were screened as potential vaccine candidate antigens, and three (Rv2029c, Rv2028c, and Rv3126c) were selected based on their therapeutic effect as recombinant DNA vaccines in an Mtb-infected murine model and efficacy against LTBI. Then, a recombinant SeV-vectored vaccine SeV986A was constructed to express these three antigens plus Ag85A (a dominant antigen of Mtb). We optimized the dose and inoculation sites for this new vaccine and determined the immunogenicity in BCG-primed and BCG-naive murine models. Enhanced immune protection against the Mtb challenge was observed in both acute-infection and latent-infection models in BCG-vaccinated mice. Interestingly, the expression levels of several T-cell exhaustion markers were significantly lower in the SeV986A vaccinated group in the LTBI infection model, suggesting that the expression of latency-associated antigens halted the T-cell exhaustion process. Thus, the multi-antigenic recombinant SeV-vectored vaccine holds considerable promise as a novel post-exposure prophylaxis vaccine to combat the TB pandemic.
Methods
Human study subjects
This study was approved by the Ethical Committee of Shanghai Public Health Clinical Center, and written informed consent was obtained from the recruited participants. The cohort comprised 98 patients with active TB diseases (ATB), 20 patients with LTBI, 30 healthy volunteers, and 32 patients with lung diseases but not TB (Non-TB) [Citation29]. All subjects had received the BCG vaccine after birth, were negative for human immunodeficiency virus (HIV)/hepatitis C virus infection, and were not taking immunosuppressive drugs. ATB was identified based on culture/GeneXpert MTB/RIF (Xpert; Cepheid, USA) positivity of sputum samples, with polymerase chain reaction (PCR) amplification of the Mtb complex-specific gene IS6110 used to exclude non-TB mycobacterial infection (MeltPro, Zeesan Biotech, China). LTBI was defined as QuantiFERON-TB Gold (Qiagen, Germany) or T-SPOT.TB (Oxford, UK) positive, culture/Xpert negative, and without clinical manifestations of Mtb infection. Healthy volunteers were recruited based on the negativity of QuantiFERON-TB Gold/T-SPOT.TB and absence of clinical signs of disease. Patients without TB had other lung diseases, such as lung cancer, pneumonia, chronic bronchitis, and bronchiectasis, and were negative in Mtb culture/Xpert/non-tuberculous mycobacteria (NTM) and QuantiFERON-TB Gold/T-SPOT.TB tests.
Construction of recombinant DNA vaccines and recombinant proteins
The genes coding Rv3126c, Rv2626, Rv1813, Rv2031, Rv3426, Rv1996, Rv3873, Rv2028c, Rv2029c and Ag85A (Rv3804c) were amplified by PCR using Mtb H37Rv ATCC genomic DNA as a template. After digestion with EcoR I and BamH I, the PCR products were inserted into the pVAX1 plasmid. The recombinant DNA vaccines were purified with Qiagen columns (EndoFree Plasmid Giga kit, Qiagen, Germany), quantified by Nano-Drop 2000 (Thermo Fisher Scientific, USA), and then eluted in pyrogen-free deionized water to the concentration of 1 mg/mL. To obtain the encoded antigens, the recombinant plasmids were transformed into E. coli BL21 (DE3) cells, which were then induced with 1 mM isopropyl thio-β-D-galactoside (IPTG; Sigma, Germany) and disrupted via sonication. The expressed proteins were purified using Ni-nitrilotriacetic acid resin.
Construction of SeV986A
The chimeric DNA vaccine sequence encoding antigens Ag85A, Rv2029c, Rv2028c, and Rv3126c was ligated by HL5 linker and constructed by standard cloning procedures (Figure 2A), and was inserted into pSeV(+)18/dF cDNA containing the F-gene-defective SeV genome, the ligated DNA construct was mixed with plasmids pGEM-L, pGEM-N, and pGEM-P that had the SeV L, NP and P/C genes cloned downstream of the T7 promoter [Citation18]. The mixture of plasmids was transfected into LLC-MK2 cells infected with vaccinia virus vTF7-3 containing the phage T7 RNA polymerase gene. Cytoplasmic extracts derived from the transfected LLC-MK2 cells were used to detect expression of the chimeric antigen, termed antigen 986A (incorporating Rv2029c, Rv2028c, Rv3126c and Ag85A) and 986AB (incorporating Rv2029c, Rv2028c, Rv3126c, Ag85A, and Ag85B), by western blotting assay 2 h post-transfection. The virus was recovered, cloned, and amplified to provide a recombinant virus for immunization.
Animals and immunization
Specific pathogen-free (SPF) female Balb/c mice aged six weeks were purchased from Shanghai SLAC Laboratory Animal Co., Ltd and maintained in the animal centre of Shanghai Public Health Clinical Center under SPF conditions. To screen latency-associated antigen-expressing DNA vaccines, mice were infected by aerosol with the Mtb H37Rv strain, as described below. After five weeks post-infection, the Mtb-infected mice were immunized intramuscularly (i.m.) with three doses of 100 μg recombinant DNA vaccines in each hind leg in a volume of 100 μL at 2-week intervals. The mice were sacrificed four weeks after the final inoculation, and the lungs were aseptically removed to determine the bacterial burden. For the optimization of inoculation times and inoculation methods, the mice were immunized two or three times intranasally (i.n.) or i.m., with high (1 × 107 cell infectious units [CIU]) or low (2 × 106 CIU) virus titers and then were sacrificed two weeks after the final inoculation. The lungs were aseptically removed to determine T-cell immune responses. The SeV vector expressing an Mtb-irrelevant antigen SARS-CoV-2 S1 protein [Citation30] (SeVS1) was used as a negative control. For the BCG prime-SeV986A boost immunization, the mice were primed by BCG vaccine subcutaneously (s.c.) with a single dose of 1 × 106 colony forming units (CFU) and immunized i.m. with two doses of 1 × 107 CIU SeV986A at 2-week intervals. The immune responses in the lung were determined two weeks after the final inoculation. For the Mtb acute infection and LTBI infection models, the mice were challenged with Mtb as described below, treated with anti-TB chemotherapy in the LTBI model, then immunized with the different vaccines and sacrificed at indicated time points. All the animal experiments were approved by the Institutional Animal Care and Use Committee and were performed under the supervision of the Laboratory Animal Ethical Board of Shanghai Public Health Clinical Center.
Human IFN-γ enzyme-linked immunosorbent assay (ELISA)
Peripheral venous blood from different population groups was collected in vacutainers containing sodium heparin. The whole blood was diluted 1:2 (v/v) in RPMI-1640 medium containing 10% fetal bovine serum (FBS) and 1% penicillin & streptomycin, plated in 96-well plates, and stimulated with recombinant ESAT-6/CFP10 protein, Rv2029c protein, Rv2028c protein and Rv3126c protein (all 5 μg/mL), respectively, with a final volume of 200 μL. Incubation with the medium was used as a negative control. After stimulation at 37°C in incubators with 5% CO2 for 48 h, the suspension was centrifuged, and the supernatant was harvested, cryopreserved in a −80°C refrigerator, and analyzed by a commercial human IFN-γ ELISA kit for scientific research (eBioscience, USA). Briefly, anti-human IFN-γ monoclonal antibody was coated in a 96-well plate overnight at 4°C, the wells were washed with PBST-20 (PBS buffer containing 0.05% Tween-20) and then blocked using PBS buffer containing 1% bovine serum albumin for 2 h. The supernatant was added and incubated at 37°C for 2 h. After washing, a biotinylated anti-rabbit detection antibody was added and incubated for 45 min at room temperature, and a final colorogenic step was performed by adding avidin-peroxidase. The reaction was stopped using 2 M H2SO4, and the plates were read at OD490 on a microplate reader. Readings from negative controls (double values) were subtracted from test values.
Single-cell suspensions from spleen and lung tissues
After sacrifice, the spleens of mice were aseptically removed, mechanically disrupted, and splenocytes were filtered through mesh gauze. The red blood cells (RBCs) were lysed with lysis buffer and splenocytes were counted. The lungs were minced with surgical scissors and incubated at 37°C with 1 mg/mL of Collagenase IV (Invitrogen, USA) and 10 U of DNase I (R&D Systems, USA) in 10 mL of RPMI-1640 medium containing 10% FBS and 1% penicillin & streptomycin for 30 min. The digested lung pieces were gently squashed and filtered through a 70-μm cell strainer (Thermo Fisher Scientific, USA) and the cell suspension was washed and subjected to RBCs lysis.
IFN-γ enzyme-linked immunospot assay (ELISPOT)
The commercial mouse IFN-γ ELISPOT kit for scientific research was performed according to the manufacturer’s protocol (BD Biosciences, USA). Briefly, the 96-well plates were coated with a monoclonal antibody against mouse IFN-γ at 4°C overnight. The plates were blocked with RPMI 1640 medium containing 10% FBS for 2 h at room temperature, rinsed with medium, then 2 × 105 isolated lung cells were added with 5 μg/mL proteins per well in a total volume of 200 μL and stimulated in a 37°C incubator with 5% CO2 for 16 h. After stimulation, the cells were removed, the plates were washed with PBST-20, and then incubated with anti-mouse IFN-γ biotinylated antibody for 2 h at room temperature. After washing with PBST-20, the cells were incubated with the streptavidin-horseradish peroxidase (HRP)-conjugated anti-biotin antibody for 1 h at room temperature. After washing with PBST-20 and PBS, the 3-amino-9-ethylcarbazole (AEC) substrate was added and incubated for 20 min until the colour was developed. The plates were washed with water, air-dried, and the spots were counted with an Immunospot Reader (Champspot III, Beijing Sage Creation Science).
Intracellular cytokine staining (ICS) and flow cytometry
The isolated lung cells were added to 96-well plates with 1 × 106 cells, 5 μg/mL proteins per well in a total volume of 200 μL and stimulated in a 37°C incubator with 5% CO2 for 16 h in the presence of 1 μL/mL monensin/Golgistop and BFA/Golgiplug (BD Biosciences, USA). The stimulated cells were washed with wash buffer (PBS containing 2% FBS) and then stained with antibodies against surface markers for 30 min at 4°C in the dark. After surface staining, the cells were washed and subjected to fixation and permeation using fix/perm solution (BD Biosciences, USA) for 20 min at 4°C in the dark. After washing, the cells were stained with antibodies against intracellular cytokines for another 30 min at 4°C. After washing, the cells were resuspended and filtered for flow cytometry analysis (BD Forterssa, BD Biosciences, USA). At least 300,000 events were collected for each sample, and responses were analyzed using FlowJo software version 10 (Tree Star, Ashland, USA).
The antibodies used were: CD3-eFluor450 (clone 17A2), CD4-APC-eFluor780 (clone RM4-5), CD44-FITC (clone IM7), CD62L-PerCP-Cyanine5.5 (clone MEL-14), IFN-γ-APC (clone XMG1.2), and IL-2-PE (clone JES6-5H4), and TNF-α-PE-Cyanine7 (clone MP6-XT22) from eBioscience (USA), CD38-BV711 (clone Ab90), LAG3- BV510 (clone C9B7W), PD1-BV605 (clone J43), and TIM3-PE-CF594 (clone 5D12) from BD Biosciences (USA).
The 3D structure prediction of fusion antigen
The FastAF2 server was used to predict the 3D Structure of the 986A protein by uploading the amino acid sequences encoded by the chimeric 986A gene. The accuracy of the predicted protein structure was measured by average accuracy (pLDDT), in which the structures with pLDDT > 70 values were considered to be well-folded regions [Citation31]. The prediction structure was displayed using PyMol programme.
Antigen-specific IgG antibody titers
Recombinant Ag85A/Rv2029c/Rv2028c/Rv3126c proteins at a concentration of 0.5 μg/mL were coated into 96-well plates at 4°C overnight. After washing, the plates were blocked using PBS buffer containing 1% bovine serum albumin for 2 h. Whole blood collected from immunized mice was incubated at 37°C for 30 min and then was transferred to 4°C for 1 h. The separated serum was serially diluted 2-fold from a starting ratio of 1:100 in PBS and incubated in the plates at 37°C for 1 h. The plates were washed and incubated with HRP-labeled anti-mouse IgG at 37°C for 1 h. The plates were washed and incubated with TMB substrate solution for an additional 10 min. The reaction was stopped by 2M H2SO4. The OD450 was measured on a microplate reader. Readings from negative controls (double values) were subtracted from test values.
Mycobacterial challenge and bacterial culture
The murine model of the Mtb challenge was previously described [Citation32]. Briefly, the mice were aerosol exposed to Mtb H37Rv to deposit a dose of ∼100 CFU per lung through an inhalation exposure system (Glas-Col, Terre Haute, USA) in the animal facility of Animal Biosafety Level-3 laboratory in Shanghai Public Health Clinical Center. The Mtb burden was quantified by plating homogenates of lung and spleen onto Middlebrook 7H11 (BD Biosciences, USA) agar plates supplemented with 10% OADC nutrient (BD Biosciences, USA) and a mixture of four antibiotics to prevent the growth of contaminants (a final concentration of 4 mg/mL amphotericin, 40 U/mL polymyxin B, 50 mg/mL carbenicillin, and 2 mg/mL trimethoprim).
The latent infection murine model
The murine model of latent Mtb infection was established as described previously with modifications [Citation33,Citation34]. Briefly, mice were immunized with BCG four weeks before aerosol challenge with the virulent Mtb H37Rv (∼100 CFU). Four weeks post-infection, the mice were treated with isoniazid (INH; Sigma, Germany) and pyrazinamide (PZA; Sigma, Germany) at 100 μg/mL and 8 mg/mL respectively per day through drinking water. After eight weeks of chemotherapy, CFUs were rarely detectable in the lungs and the mice were immunized i.m. with two doses, two weeks apart, of 1 × 107 CIU recombinant SeV-based virus vaccine, either SeV85AB or SeV986A. Four weeks after the final immunization, mice received three s.c. injections of dexamethasone (Sigma, Germany; 6 mg per kg body weight) with 2-day intervals to induce Mtb reactivation. The mice were sacrificed to determine Mtb burdens in the lungs and spleen eight weeks after reactivation.
Histopathological analysis
The right superior lobes of the Mtb-infected lungs were fixed with 4% neutral formaldehyde, embedded in paraffin, serially sectioned in thickness of 4 μm, stained with hematoxylin and eosin (H&E), and photographed using an Olympus CKX41 microscope fitted with a DP20 camera (Japan) connected to a computer. The Image Pro Plus programme was utilized to assess the levels of inflammation present in each image objectively.
Statistical analysis
The statistical analysis was performed using GraphPad Prism 9.0 software. One-way ANOVA test was used to analyze the statistical significance for the comparisons of multiple groups, and two-way ANOVA was used for the sub-grouped analysis. The value of p < 0.05 was considered significant.
Results
Selection of latency-associated antigens
The candidate antigens screened in this study were chosen based on published reports showing or predicting association with Mtb latency () [Citation22–28]. The DNA sequences encoding the nine candidate antigens and one dominant antigen, Ag85A, were amplified from Mtb and incorporated into DNA vectors to form ten recombinant DNA vaccines. In the acute Mtb infection murine model, five weeks after aerosol infection the mice were immunized with three doses of a recombinant DNA vaccine ((A)). Each of the five vaccines encoding a latency-associated antigen gave some benefit with a log10 CFU decrease of more than 0.5, namely, antigens Rv3126c, Rv1813, Rv2031, Rv2028c, and Rv2029c ((B)). Vaccines expressing several combinations of these five antigens and Ag85A/Ag85B were made and similarly tested. Among these, a fusion construct encoding Rv2029c-Rv2028c-Rv3126c-Ag85A antigens (986A) showed enhanced immune protection against Mtb infection in a preliminary experiment (Figure S1).
Figure 1. The screen of latency-associated antigens. (A) Schema of latency-associated antigens screen in an acute infection murine model. (B) Numbers of live bacteria in homogenates of lungs were counted as CFU after 3-week incubation on 7H11 agar and transformed as log10. The mock group receiving PBS immunization, and the rifampicin (RFP) chemotherapy group through drinking water at a concentration of 20 μg/mL were used as controls, when compared with the mock control group. The data are derived from one experiment with five mice per group. A one-way ANOVA followed by a Tukey test was performed to compare the values of different groups. * p < 0.05, *** p < 0.01, and **** p < 0.001 (C-F) The antigen-specific IFN-γ responses in peripheral blood from different human population groups after ex vivo stimulation. Samples from ATB patients, LTBI populations, healthy controls, and other lung diseases were stimulated with Rv2029c (C), Rv2028c (D), and Rv3126c (E), and ESAT-6/CFP10 (F) proteins in parallel, and the release of IFN-γ was quantified by ELISA assay. A one-way ANOVA followed by a Tukey test was performed to compare the values of different groups. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001, when compared with PBS control or compared with each other as indicated.

Table 1. Nine Mtb latency-associated antigens selected as vaccine candidates.
We confirmed that elevated responses against these three latency-associated antigens were observed in our previous study of human cohorts [Citation29]. As expected, after the ex vivo stimulation with Rv2029c, Rv2028c, and Rv3126c proteins, the secretion of IFN-γ was consistently significantly higher in the LTBI population than in ATB patients, healthy volunteers, and non-TB patients with other lung diseases (Figure 1(C–E)). In contrast, the antigen-specific responses against Mtb acute infection antigen ESAT-6 were comparable between LTBI and ATB and were significantly higher than those from healthy volunteers and non-TB patients ((F)).
Taken together, the Rv2029c, Rv2028c, and Rv3126c antigens were chosen based on their protective efficacy against Mtb infection and elevated immune responses in LTBI populations. They were ligated with Mtb dominant antigen Ag85A to give a chimeric DNA construct to express 986A.
Construction and verification of novel recombinant SeV986A vaccine
The DNA sequence of chimeric antigen 986A ((A)) was inserted into the SeV vector to construct the SeV986A vaccine. The expression of 986A chimeric protein was confirmed in the cell lysate from LLC-MK2 cells infected with SeV986A. The expression of chimeric protein was confirmed in the cell lysate from SeV986A-infected LLC-MK2 cells by western blotting with mouse antiserum to 986A ((B), Figure S2). Structural prediction from amino acids encoded by the chimeric gene 986A was done by using FastAF2. The predicted 3D structure with a pLDDT value of 70.240 was shown in (C). The overall structure showed that the four antigen proteins formed a relatively tight architecture, which were connected by flexible linkers. By comparing the structure of each antigen protein with its corresponding monomer structure, it was found that the structures of Ag85A and Rv2029c were similar to the corresponding monomer structure, with RMSD values less than 0.2, while the structures of Rv2028c and Rv3126c were significantly different from the corresponding monomer structures, which might be due to the obstruction of spatial location.
Figure 2. Construction and verification of recombinant SeV986A vaccine. (A) Transgene cassette diagram for 986A antigen, assembled with HL5 linkers for a final construct containing 3273 bp. (B) Western blotting of cell lysate from SeV986A/mock-infected cells or recombinant proteins using antiserum to Ag85A (5 s exposure). M, maker; lane 1, LLC-MK2 cells without infection; lanes 2 & 3, SeV986AB-infected LLC-MK2 cells; lane 4, SeV986A-infected LLC-MK2 cells; lane 5, recombinant Ag85A protein; Lane 6, recombinant 986A protein. (C) The 3D structure of chimeric A986 protein based on the homology modelling the amino acids.

The optimization of doses, titers, and routes of SeV986A noculation
We previously reported that the SeV-based TB vaccine showed superior immunogenicity at a dose of 1 × 107 CIU compared with a dose of 2 × 106 CIU; here we immunized mice i.m. with single, double, or triple doses of recombinant SeV986A at a larger titer of 1 × 107 CIU ((A)). As shown in (B), detectable antigen-specific IFN-γ responses were observed in all immunization groups. However, two doses showed the highest responses, suggesting that two doses would be enough for antigen-specific immune responses with this recombinant vaccine. When delivery of two doses of a low titer (2 × 106 CIU) by the i.m. route was compared with the delivery of two standard doses of 1 × 107 CIU by i.n. route and i.m. route ((C)) the low titer SeV986A failed to elicit detectable IFN-γ immune responses ((D)). Although i.n. immunization showed slightly higher immunogenicity, the i.m. route was selected for subsequent studies based on its convenience for potential human use.
Figure 3. The optimization of doses, titers, and routes of SeV986A inoculation. PBS and a recombinant SeV vaccine expressing an Mtb-irrelevant antigen (SeVS1) were used as negative controls and BCG was used as a positive control. (A) Schema of dose optimization (B) The antigen-specific IFN-γ responses in lung cells at two weeks after the final vaccination with repeated i.m. doses were determined by ELISPOT assay. (C) Schema of titer and route optimizations. (D) The antigen-specific IFN-γ responses in the lung cells at two weeks after final vaccination by different routes were determined by ELISPOT assay. The data are representative of two independent experiments with at least four mice per group. A two-way ANOVA test followed by Tukey’s multiple comparisons test was performed for multiple comparisons between groups. * p < 0.05 and ** p < 0.01 when compared with the PBS control group or compared with each other as indicated. ns indicates no significant difference.

Sev986a boost increased antigen-specific T-cell responses primed by BCG vaccination
Considering that the majority of the world’s population is immunized with BCG in infancy, a booster vaccine that improves the efficacy of BCG could significantly impact the TB pandemic. We previously showed that SeV85AB boosted BCG-primed immunity and protection against TB [Citation19]. Herein, we tested the effectiveness of this novel SeV-based vaccine, SeV986A, expressing latency-associated antigens as a booster vaccine administered i.m. to mice 4 and 6 weeks after BCG immunization ((A)). A recombinant SeV vaccine expressing an Mtb-irrelevant antigen (SeVS1) was used as a negative control. Two weeks after the last dose, the mice were sacrificed to test the antigen-specific immune responses in the lung cells using ELISPOT and ICS assays. Lung cell suspensions were obtained and stimulated with the Ag85A, Rv2029c, Rv2028c, and Rv3126c proteins, respectively, or incubated with the medium as control. As expected, SeV986A vaccine induced modest specific responses to these individual specific antigens (< 25 IFN-γ spot-forming units [SFUs]/106 cells), whereas SeV986A, when given after BCG, boosted the BCG-induced responses to Ag85A about 10-fold (∼ 250 IFN-γ SFUs/106 cells). The responses to Rv2029c, Rv2028c, and Rv3126c were smaller and only Rv2029c was significantly enhanced ((B)). In the ICS assay, the levels of IFN-γ, IL-2, and TNF-α-positive T cells were compared between vaccine groups (the gating strategy and representative plots are shown in Figure S3 in Supplementary Material). Consistent with the ELISPOT assay, the SeV986A boost to BCG significantly enhanced the responses to in vitro stimulation with Ag85A, as seen in the production of IFN-γ and TNF-α cytokines in both CD4+ and CD8+ T cells ((C, D)). Notably, the boost with SeV986A also significantly enhanced the responses of CD4+ cells to stimulation with antigen 986, with increased production of IFN-γ and IL-2 cytokines, whereas in CD8+ T cells only IFN-γ was increased ((E, F)). Moreover, the boost with SeV986A modestly increased the secretion of IL-17 after Ag85A stimulation, and significantly higher IL-17 responses were observed after stimulation with antigen 986 in CD4+ T cells (Figure S4(A–B)). The antigen-specific T-cell immune responses in the splenocytes were also determined by ELISPOT (Figure S4(C)) and ICS (Figure S4(D–E)) assays, higher levels of Ag85A-specific CD4+ T cells were induced in both BCG and SeV986A boost groups (Figure S4(D)), and enhanced 986-specific CD4+ T cell responses were induced in SeV986A boost group (Figure S4(E)). Consistent with other reports, the BCG vaccine failed to elicit detectable antigen-specific IgG responses; SeV986A only elicited low levels of Ag85A-specific IgG response (Figure S5). Together, these data demonstrated that the SeV986 boost markedly enhanced the levels of antigen-specific CD4+ and CD8+ T cell responses primed by the BCG vaccine.
Figure 4. SeV986A boost increased antigen-specific T cell responses primed by BCG vaccination. (A) Schema of the BCG prime-SeV986A boost immunization murine model. PBS and SeV encoding a non-relevant antigen were used as negative controls. (B) The antigen-specific IFN-γ response in the lung cells at two weeks after final vaccination was determined by ELISPOT assay. (C-F) Flow cytometric analysis of intracellular staining in the lung cells of vaccinated mice. The lung cells were collected two weeks after the last inoculation, incubated with Ag85A protein (C, D) or 986A protein (E, F) in the presence of monensin and brefeldin A, and analyzed for cytokine production by ICS assay. The single- (C, E) and multi-functional (D, F) antigen-specific T cell responses in the lung were shown, respectively. CD3 + CD4+ cells (CD4+ T cells) and CD3 + CD4− cells (CD8+ T cells) producing IFN-γ, IL-2, and TNF-α were analyzed. A one-way ANOVA followed by a Tukey test was performed to compare the groups, and two-way ANOVA was used for the sub-grouped analysis. * p < 0.05, ** p < 0.01, and *** p < 0.001, when compared with the PBS control group or compared with each other as indicated. The data are representative of two independent experiments with at least four mice per group.

Enhanced protection against acute Mtb infection after boosting BCG-primed mice with SeV986A
We boosted BCG-primed mice with two doses of SeV986A at weeks 4 and 6 and challenged them with low-dose aerosol infection with Mtb after two weeks. The protective effect was assessed by counting bacteria in lungs and spleens five weeks later in comparison against counts from controls that were given PBS, BCG, or SeV986A alone, or a recombinant SeV vaccine expressing an Mtb-irrelevant antigen (SeVS1), as shown in (A). Compared to SeVS1, both BCG and SeV986A immunization significantly reduced bacterial loads in the lung ((B)) and spleen ((C)). Notably, compared to vaccination with either BCG or SeV986A alone, a boost with SeV986A resulted in markedly enhanced protection against Mtb challenge, evaluated by the further reduction in bacterial loads in the lung ((B)) although dissemination to spleens was not reduced ((C)). In addition, decreased inflammation scores were observed in H&E-stained lung sections derived from BCG-, SeV986A-, and BCG-SeV986A- vaccinated mice ((D, E)). This result indicated that boosting BCG-primed mice with a recombinant SeV-vectored multi-antigenic vaccine expressing latency-associated antigens can enhance immune protection against Mtb aerosol challenge.
Figure 5. Boosting BCG-primed mice with SeV986A enhanced protection against acute Mtb infection. (A) Schema of protection evaluation in an acute infection murine model. (B-C) Bacterial burden in infected organs. Five weeks post-infection, the numbers of live bacteria in homogenates of lungs (B) and spleen (C) were counted as CFU after incubation for three weeks on 7H11 agar and transformed as log10. A one-way ANOVA followed by a Tukey test was performed to compare the values of different groups. (D-E) The tissue sections from the right superior lobes of the infected lung were stained for histopathology with HE. The representative histological appearances are shown in (D), and the average values are shown in (E). Show ×200, scale bar, 100 μm. The data are representative of two independent experiments with four to five mice per group. * p < 0.05, ** p < 0.01, *** p < 0.001, and **** p < 0.0001, when compared with the PBS control group or compared with each other as indicated.

Enhanced protection against latent Mtb infection in BCG-primed mice by treatment with SeV986A
We further asked whether the latency-associated antigen-expressing SeV986A could be used as an immunotherapeutic vaccine against latent Mtb infection. As shown in (A), in the latent infection model, mice were inoculated with BCG, aerosol challenged with Mtb four weeks later then rested for four weeks to establish infection. The mice were given isoniazid plus pyrazinamide chemotherapy in the diet from week 8 to week 16 to clear chemotherapy-sensitive bacteria and establish latent infection. Two doses of SeV986A immunization were given immediately after chemotherapy to elicit antigen-specific immune responses to inhibit the reactivation of persistent bacteria. Treatments with PBS and SeV85AB, a SeV-vectored vaccine expressing two dominant antigens (Ag85A and Ag85B) but not latency-associated antigens, were used as controls. Dexamethasone was administrated at week 22 to re-activate the Mtb infection. The bacterial loads and antigen-specific immune responses were evaluated eight weeks later. Compared to PBS and SeV85AB, SeV986A significantly lowered CFU in the lung ((B)) and spleen ((C)). The expression profiles of four T-cell exhaustion markers (CD38, LAG3, PD1, and TIM3) were assessed by flow cytometry with the gating strategy shown in Figure S6. The Mtb-specific CD4+ T cells in the lung were defined as CD4+ T cells that secreted at least one of the cytokines IFN-γ, IL-2 or TNF-α after Mtb purified protein derivative (PPD) stimulation. The expression levels of these four T-cell exhaustion markers were all significantly lower in the SeV986A immunized group compared with the PBS group. In particular, the expression levels of CD38 and TIM3 were significantly lower in the SeV986A group than in SeV85AB ((D–G)), suggesting that vaccination with SeV986A tunes the T cells in the chronic latent infection into a less exhausted profile. Moreover, in H&E-stained lung sections, the SeV986A group had significantly fewer inflammatory infiltrates with lower inflammation scores ((H, I)).
Figure 6. Treatment with SeV986A reduced latent Mtb infection in BCG-primed mice. (A) Schema of treatment evaluation in a latent infection murine model. (B-C) Bacterial burden in infected organs. Eight weeks after reactivation with dexamethasone, the numbers of live bacteria in homogenates of lung (B) and spleen (C) were counted as CFU after 3-week incubation on 7H11 agar. (D-G) The expression profiles of four T-cell exhaustion markers in the latent infection model. The lung cells from latent-infected mice were stimulated with PPD and analyzed for the expression levels of TIM3 (D), PD1 (E), CD38 (F), and LAG3 (G). A one-way ANOVA followed by a Tukey test was performed to compare the values of different groups. (H-I) The tissue sections from the right superior lobes of the infected lung were stained for histopathology with HE. The representative histological appearances are shown in (H), and the average values are shown in (I). Show ×200, scale bar, 100 μm. * p < 0.05, ** p < 0.01, and *** p < 0.001, in comparisons as indicated. The data are derived from one experiment with five mice per group.

Discussion
Latent TB infection is medically defined as a state of a persistent immune response to stimulation by Mtb-specific antigens in the absence of any clinically manifest TB disease [Citation35,Citation36]. During latent infection, Mtb can survive in an inactive/dormant state for a long time and retains its ability to resuscitate under appropriate circumstances, such as weakened immunological status, as in HIV infection, immunosuppressive therapies, or immunosenescence caused by aging [Citation37–39]. For example, in a cohort study of people who were known or likely to have been exposed to active TB in recent years, it was found that the annual incidence of active TB diseases in participants who tested positive for T-SPOT.TB was 8.8 times higher than those who tested negative [Citation40]. Thus, populations with LTBI are major reservoirs of TB infection and to achieve the WHO goals for globally limiting the spread of TB by 2035, it is necessary to develop methods for LTBI treatment to prevent progression to active TB disease. Recently, it was shown that prophylactic anti-TB chemotherapy with four months of rifampicin or nine months of isoniazid might prevent the development of active TB disease from latent infection [Citation41]. However, poor adherence, toxic effects, drug resistance, and low cost-effectiveness ratio hinder the application of LTBI-targeted therapies in high-burden, low-income countries [Citation42].
Prophylactic stimulation of immunity to eliminate latent infection is an alternative approach. Immune responses to latency-associated antigens were found to be elevated during LTBI compared with active TB patients and these antigen-specific immune responses were considered to contribute to preventing populations with LTBI from progressing to active TB disease [Citation43]. Accordingly, the antigen-specific immune responses elicited by vaccines containing latency-associated antigens might help to prevent the reactivation of latent infection. Some multistage subunit vaccines containing latency-associated antigens have already been shown to elicit appropriate and protective immune responses against Mtb infection in animal models and have already entered clinical trials. A multistage subunit protein plus adjuvant vaccine H56:IC31 containing Ag85B, ESAT-6 proteins, and the nutrient stress-induced antigen Rv2660c in the adjuvant IC31 was constructed, and this prevented the reactivation of latent infection as a boost of BCG in a cynomolgus macaque model [Citation44]. The safety and immunogenicity of H56:IC31 were shown in a randomized open-label phase I/II clinical trial [Citation45]. Its effect on preventing TB recurrence is now being evaluated in a phase II clinical trial (NCT03512249). A different multistage subunit vaccine, ID93/GLA-SE, was also formulated containing virulence proteins Rv2608/Rv3620/Rv1813 and hypoxia-associated antigen Rv3619, with GLA-SE (a synthetic TLR-4 agonist formulated in the oil-in-water emulsion) as an adjuvant [Citation46]. The safety and immunogenicity of ID93/GLA-SE in healthy adults and in patients with previously treated TB were shown in randomized, double-blind, placebo-controlled phase I and IIa clinical trials [Citation47,Citation48]. Thus, these protein-based vaccines have demonstrated the feasibility of TB vaccines targeting latency-associated antigens. However, adjuvanted protein vaccines may not be ideal and viral vectors offer some advantages [Citation17].
As one of the most widely used vaccine vectors, virus-vectored vaccines are able to induce robust antigen-specific immune responses by mimicking the process of pathogenic invasion, with the ability to form long-lasting immune memory. Basically, as vaccine vectors, most candidate vectors have the following advantages: (1) the ability to accommodate relatively sizeable foreign antigen genes; (2) the stable exogenous gene expression efficiency; (3) the ability to induce high levels of cellular immune responses, especially intracellular-replicated virus; (4) the adjuvant effects induced by the vector itself that might augment the antigen-specific immune responses; (5) low-cost in operate and culture; (6) safety use in humans when choosing the attenuated or replication-deficient viruses [Citation17]. Specially, since Mtb can survive intracellularly for a long time after primary infection, the specific ability of viral vectors to induce cellular immune responses might be crucial to the clearance of intracellular Mtb during latent infection. Recently, a chimpanzee adenovirus-vectored multi-stage vaccine containing Ag85A, TB10.4, and a dormancy-associated antigen, RpfB, showed immune protection against Mtb infection in murine models [Citation49], indicating the effectiveness of virus-vectored TB vaccines containing latency-associated antigens.
SeV is an alternative attractive vector. It was well-tolerated and immunogenic when used as a vector of vaccines against human parainfluenza virus due to their similarity in sequence, structure, and antigenicity [Citation50]. Moreover, recombinant vaccines based on replication-deficient SeV vectors have been developed against HIV [Citation51], respiratory syncytial virus [Citation52], and influenza [Citation53]. We previously showed the potential utility of the SeV vector as TB vaccines; its immunogenicity and protective efficacy against Mtb infection in murine models were obtained by expressing immunodominant Ag85A and Ag85B [Citation19]. Although the Sendai virus encoding the fusion antigens of Ag85A and Ag85B showed strong immunogenicity and immune protection in the acute infection murine models in our previous studies [Citation19–21], the combination of 986A fusion showed no inferior to 986AB, the same trend was also observed in another preliminary experiment when comparing SeV986A and SeV986AB. The reason why the Ag85B addition did not enhance the protective efficacy of 986A might be due to the foreign gene expression efficacy of the SeV vector or the immune competition between antigens. Thus, we choose SeV986A in subsequent studies.
Although genes of the latency antigens are absent from BCG, some background of responses in the control PBS, SeVS1, and BCG groups suggested the presence of some inherent cross-reactivity to unrelated antigens that may facilitate responses to these antigens. When given as a booster after BCG vaccination, SeV986A markedly enhanced the IFN-γ and TNF-α responses to Ag85A, responses which are complicit in protective immunity. The IFN-γ responses to the latency antigens were also greater than those after SeV986A alone, suggesting a costimulatory effect of the strong reaction to Ag85A and this was seen in both CD4+ and CD8+ cells (). The boosted generation of strong responses in both CD4+ and CD8+ cells is consistent with both the protection seen against subsequent aerosol challenge infection () and the reduction of reactivation in the latent infection model (). This reduction in reactivation was greater when SeV986A was used as the boost than when SeV85AB was used, confirming the important role of the latency-associated antigens in this effect.
To be noted, considering it was well proved that Th1 cytokines are associated with tuberculosis control, only one representative Th1 cytokine, IFN-γ, was used in the screening stage ((C–F)), considering the expense of effort and cost in this cohort study. Thus, although the three antigens screened from the nine latency-associated antigens showed protective efficacy in subsequent experiments, we cannot rule out the protective potential of the other six antigens.
In this study, only Ag85A showed modest antibody responses in SeV986A-vaccinated mice. The reason might be explained by the limited ability of these three latency-associated antigens to induce antibody responses, or the antigen production in this novel replication-deficient recombinant virus-vectored vaccine did not reach the threshold value of inducing antibody responses. Considering the role of antibodies in anti-TB immune protection has gradually been disclosed recently, further studies incorporating antibody-inducing antigens or increasing the antigen production of the current vaccine might further enhance the protective efficacy of the current vaccine.
The bases of establishment and breakdown of latent infection are not fully understood. However, during LTBI, the chronic exposure to mycobacterial antigens and the established cytokines milieu leads the T cells of the host to undergo a process of immune exhaustion [Citation54,Citation55]. The secretion of cytokines such as IFN-γ by intact CD4+ and CD8+ T cells is an essential mediator of macrophage function in the local sites of infection, the T helper cells also contribute to the antibody secretion ability of B cells [Citation56]. Exhausted T cells are inferior at supporting the function of macrophages and B cells and inhibiting the proliferation of invading Mtb [Citation57,Citation58]. Consequently, T-cell exhaustion is regarded as one of the reasons for the progression from LTBI to active TB diseases. We found here that the expression levels of T-cell exhaustion markers CD38, PD-1, LAG3, and TIM3 were significantly lower in the SeV986A groups compared with the PBS group, especially the expression of TIM3 and CD38 were lower in the SeV986A group than SeV85AB, suggesting that the expression of latency-associated antigens halted the T-cell exhaustion process in the LTBI infection model, which led to the enhanced immune protection, although the decreased T-cell exhaustion state might also be a result of downregulating Mtb burden in the lung. Together, the multi-antigenic recombinant SeV vectored vaccine SeV986A holds considerable promise as a novel post-exposure prophylaxis vaccine against TB infection.
The current study has several limitations. Firstly, although the intranasal delivery showed slightly higher immunogenicity than the intramuscular route, the latter was selected for subsequent studies based on its convenience for potential human use. However, during the past several years, we also recognized that the highly efficacious COVID-19 vaccines have accelerated the vaccine development process in human use and increased the public acceptance of an inhaled vaccine, with an example of Ad5-nCoV inhaled vaccine. Thus, further studies evaluating the protection afforded by the intranasal delivery of SeV986A are warranted. Secondly, although the antigen combination of Rv2029c, Rv2028c, Rv3126c, and Ag85A showed significant protection in both acute and latent infection murine models, which antigen(s) are less protective are undetectable, further research replacing the less protective antigens with other antigens is also warranted. Thirdly, as mentioned above, only Ag85A showed modest antibody responses in this study, further studies incorporating antibody-inducing antigens might further enhance the protective efficacy of the current vaccine.
In conclusion, a recombinant SeV-vectored vaccine SeV986A that expresses three latency-associated antigens Rv2029c, Rv2028c, Rv3126c and immune dominant antigen Ag85A was constructed. Enhanced immune protection against Mtb challenge was shown in both acute-infection and latent-infection murine models. Thus, the multistage quarter-antigenic SeV986A vaccine holds considerable promise as a novel vaccine against TB.
Author contributions
X.-Y.F, Z.H., and T.S. conceived the project and designed the experiments. Z.H., J.X., J.W., H.Z., P.J., L.G., W.G., Z.C., J.X., X.H., and J.M. performed experiments. Z.H. and X.-Y.F. analyzed the data and wrote the manuscript. A.C. and J.X. assisted with the 3D structure prediction.
20231212_supplement_material
Download MS Word (1.3 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- WHO. World Health Organization Global Tuberculosis Report. 2023.
- Cohen A, Mathiasen VD, Schon T, et al. The global prevalence of latent tuberculosis: a systematic review and meta-analysis. Eur Respir J. 2019;54(3):1900655.
- Flores-Valdez MA, Kupz A, Subbian S. Recent developments in mycobacteria-based live attenuated vaccine candidates for tuberculosis. Biomedicines. 2022;10(11):2749.
- Shah M, Dorman SE. Latent tuberculosis infection. N Engl J Med. 2021;385(24):2271–2280. doi:10.1056/NEJMcp2108501
- Lillebaek T, Dirksen A, Baess I, et al. Molecular evidence of endogenous reactivation of Mycobacterium tuberculosis after 33 years of latent infection. J Infect Dis. 2002;185(3):401–404. doi:10.1086/338342
- Targeted tuberculin testing and treatment of latent tuberculosis infection. This official statement of the American Thoracic Society was adopted by the ATS Board of Directors, July 1999. This is a joint statement of the American Thoracic Society (ATS) and the Centers for Disease Control and Prevention (CDC). This statement was endorsed by the Council of the Infectious Diseases Society of America. (IDSA), September 1999, and the sections of this statement. Am J Respir Crit Care Med. 2000;161(4 Pt 2):S221–S247.
- Trunz BB, Fine P, Dye C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet. 2006;367(9517):1173–1180. doi:10.1016/S0140-6736(06)68507-3
- Mangtani P, Abubakar I, Ariti C, et al. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin Infect Dis. 2014;58(4):470–480. doi:10.1093/cid/cit790
- Singh S, Saavedra-Avila NA, Tiwari S, et al. A century of BCG vaccination: immune mechanisms, animal models, non-traditional routes and implications for COVID-19. Front Immunol. 2022;13:959656. doi:10.3389/fimmu.2022.1011209
- Moliva JI, Turner J, Torrelles JB. Prospects in Mycobacterium bovis Bacille Calmette et Guérin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis? Vaccine. 2015;33(39):5035–5041. doi:10.1016/j.vaccine.2015.08.033
- Schrager LK, Vekemens J, Drager N, et al. The status of tuberculosis vaccine development. Lancet Infect Dis. 2020;20(3):e28–e37. doi:10.1016/S1473-3099(19)30625-5
- Khademi F, Derakhshan M, Yousefi-Avarvand A, et al. Multi-stage subunit vaccines against Mycobacterium tuberculosis: an alternative to the BCG vaccine or a BCG-prime boost? Expert Rev Vaccines. 2018;17(1):31–44. doi:10.1080/14760584.2018.1406309
- Mascart F, Locht C. Integrating knowledge of Mycobacterium tuberculosis pathogenesis for the design of better vaccines. Expert Rev Vaccines. 2015;14(12):1573–1585. doi:10.1586/14760584.2015.1102638
- Hernandez-Pando R, Jeyanathan M, Mengistu G, et al. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet. 2000;356(9248):2133–2138. doi:10.1016/S0140-6736(00)03493-0
- Ahmad S. Pathogenesis, immunology, and diagnosis of latent Mycobacterium tuberculosis infection. Clin Dev Immunol. 2011;2011:814943.
- Zhang Y, Xu JC, Hu ZD, et al. Advances in protein subunit vaccines against tuberculosis. Front Immunol. 2023;14:1238586. doi:10.3389/fimmu.2023.1238586
- Hu Z, Lu SH, Lowrie DB, et al. Research advances for virus-vectored tuberculosis vaccines and latest findings on tuberculosis vaccine development. Front Immunol. 2022;13:895020, doi:10.3389/fimmu.2022.895020
- Li HO, Zhu YF, Asakawa M, et al. A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J Virol. 2000;74(14):6564–6569. doi:10.1128/JVI.74.14.6564-6569.2000
- Hu Z, Wong KW, Zhao HM, et al. Sendai virus mucosal vaccination establishes lung-resident memory CD8 T cell immunity and boosts BCG-primed protection against TB in mice. Mol Ther. 2017;25(5):1222–1233. doi:10.1016/j.ymthe.2017.02.018
- Hu Z, Gu L, Li CL, et al. The profile of T cell responses in bacille calmette–guérin-primed mice boosted by a novel sendai virus vectored anti-tuberculosis vaccine. Front Immunol. 2018;9:1796. doi:10.3389/fimmu.2018.01796
- Hu Z, Jiang W, Gu L, et al. Heterologous prime-boost vaccination against tuberculosis with recombinant Sendai virus and DNA vaccines. J Mol Med (Berl). 2019;97(12):1685–1694. doi:10.1007/s00109-019-01844-3
- Shi SD, Hsueh PR, Yang PC, et al. Use of DosR Dormancy Antigens from Mycobacterium tuberculosis for Serodiagnosis of Active and Latent Tuberculosis. ACS Infect Dis. 2020;6(2):272–280. doi:10.1021/acsinfecdis.9b00329
- Rosenkrands I, Slayden RA, Crawford J, et al. Hypoxic response of Mycobacterium tuberculosis studied by metabolic labeling and proteome analysis of cellular and extracellular proteins. J Bacteriol. 2002;184(13):3485–3491. doi:10.1128/JB.184.13.3485-3491.2002
- Bretl DJ, He H, Demetriadou C, et al. MprA and DosR coregulate a Mycobacterium tuberculosis virulence operon encoding Rv1813c and Rv1812c. Infect Immun. 2012;80(9):3018–3033. doi:10.1128/IAI.00520-12
- Fleischmann RD, Alland D, Eisen JA, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184(19):5479–5490. doi:10.1128/JB.184.19.5479-5490.2002
- Hingley-Wilson SM, Lougheed KE, Ferguson K, et al. Individual Mycobacterium tuberculosis universal stress protein homologues are dispensable in vitro. Tuberculosis (Edinb). 2010;90(4):236–244. doi:10.1016/j.tube.2010.03.013
- Hinks TS, Dosanjh DP, Innes JA, et al. Frequencies of region of difference 1 antigen-specific but not purified protein derivative-specific gamma interferon-secreting T cells correlate with the presence of tuberculosis disease but do not distinguish recent from remote latent infections. Infect Immun. 2009;77(12):5486–5495. doi:10.1128/IAI.01436-08
- Leyten EM, Lin MY, Franken KL, et al. Human T-cell responses to 25 novel antigens encoded by genes of the dormancy regulon of Mycobacterium tuberculosis. Microbes Infect. 2006;8(8):2052–2060. doi:10.1016/j.micinf.2006.03.018
- Zhao HM, Du R, Li CL, et al. Differential T cell responses against DosR-associated antigen Rv2028c in BCG-vaccinated populations with tuberculosis infection. J Infect. 2019;78(4):275–280. doi:10.1016/j.jinf.2018.10.016
- Hu Z, Chen JP, Xu JC, et al. A two-dose optimum for recombinant S1 protein-based COVID-19 vaccination. Virology. 2022;566:56–59. doi:10.1016/j.virol.2021.11.011
- Krapp LF, Abriata LA, Cortes Rodriguez F, et al. PeSTo: parameter-free geometric deep learning for accurate prediction of protein binding interfaces. Nat Commun. 2023;14(1):2175. doi:10.1038/s41467-023-37701-8
- Ji P, Hu ZD, Kang H, et al. Boosting BCG-primed mice with chimeric DNA vaccine HG856A induces potent multifunctional T cell responses and enhanced protection against Mycobacterium tuberculosis. Immunol Res. 2016;64(1):64–72. doi:10.1007/s12026-015-8674-9
- Lowrie DB, Tascon RE, Bonato VL, et al. Therapy of tuberculosis in mice by DNA vaccination. Nature. 1999;400(6741):269–271. doi:10.1038/22326
- Aagaard C, Hoang T, Dietrich J, et al. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat Med. 2011;17(2):189–194. doi:10.1038/nm.2285
- Padmapriyadarsini C, Sachdeva KS, Nair D, et al. The paradigm shift in the approach to management of latent tuberculosis infection in high tuberculosis burden countries. Expert Rev Respir Med. 2021;15(7):899–910. doi:10.1080/17476348.2021.1862652
- Behr MA, Edelstein PH, Ramakrishnan L. Revisiting the timetable of tuberculosis. Br Med J. 2018;362:k2738.
- Bergeron A, Mikulska M, De Greef J, et al. Mycobacterial infections in adults with haematological malignancies and haematopoietic stem cell transplants: guidelines from the 8th European Conference on Infections in Leukaemia. Lancet Infect Dis. 2022;22(12):e359–ee69.
- Olmo-Fontanez AM, Turner J. Tuberculosis in an aging world. Pathogens. 2022;11(10):1101.
- Pawlowski A, Jansson M, Skold M, et al. Tuberculosis and HIV co-infection. PLoS Pathog. 2012;8(2):e1002464. doi:10.1371/journal.ppat.1002464
- Abubakar I, Drobniewski F, Southern J, et al. Prognostic value of interferon-gamma release assays and tuberculin skin test in predicting the development of active tuberculosis (UK PREDICT TB): a prospective cohort study. Lancet Infect Dis. 2018;18(10):1077–1087. doi:10.1016/S1473-3099(18)30355-4
- Menzies D, Adjobimey M, Ruslami R, et al. Four months of rifampin or nine months of isoniazid for latent tuberculosis in adults. N Engl J Med. 2018;379(5):440–453. doi:10.1056/NEJMoa1714283
- Hu Z, Lu SH, Lowrie DB, et al. The predictive values of the tuberculin skin test and interferon-gamma release assays for active tuberculosis development. Lancet Infect Dis. 2019;19(1):19–20. doi:10.1016/S1473-3099(18)30712-6
- Gong W, Wu X. Differential diagnosis of latent tuberculosis infection and active tuberculosis: a key to a successful tuberculosis control strategy. Front Microbiol. 2021;12:745592. doi:10.3389/fmicb.2021.745592
- Lin PL, Dietrich J, Tan E, et al. The multistage vaccine H56 boosts the effects of BCG to protect cynomolgus macaques against active tuberculosis and reactivation of latent Mycobacterium tuberculosis infection. J Clin Invest. 2012;122(1):303–314. doi:10.1172/JCI46252
- Jenum S, Tonby K, Rueegg CS, et al. A Phase I/II randomized trial of H56:IC31 vaccination and adjunctive cyclooxygenase-2-inhibitor treatment in tuberculosis patients. Nat Commun. 2021;12(1):6774. doi:10.1038/s41467-021-27029-6
- Bertholet S, Ireton GC, Ordway DJ, et al. A defined tuberculosis vaccine candidate boosts BCG and protects against multidrug-resistant Mycobacterium tuberculosis. Sci Transl Med. 2010;2(53):53ra74.
- Day TA, Penn-Nicholson A, Luabeya AKK, et al. Safety and immunogenicity of the adjunct therapeutic vaccine ID93 + GLA-SE in adults who have completed treatment for tuberculosis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet Respir Med. 2021;9(4):373–386. doi:10.1016/S2213-2600(20)30319-2
- Sagawa ZK, Goman C, Frevol A, et al. Safety and immunogenicity of a thermostable ID93 + GLA-SE tuberculosis vaccine candidate in healthy adults. Nat Commun. 2023;14(1):1138. doi:10.1038/s41467-023-36789-2
- Afkhami S, D'Agostino MR, Vaseghi-Shanjani M, et al. Intranasal multivalent adenoviral-vectored vaccine protects against replicating and dormant M.tb in conventional and humanized mice. NPJ Vaccines. 2023;8(1):25. doi:10.1038/s41541-023-00623-z
- Slobod KS, Shenep JL, Lujan-Zilbermann J, et al. Safety and immunogenicity of intranasal murine parainfluenza virus type 1 (Sendai virus) in healthy human adults. Vaccine. 2004;22(23–24):3182–3186. doi:10.1016/j.vaccine.2004.01.053
- Takeda A, Igarashi H, Nakamura H, et al. Protective efficacy of an AIDS vaccine, a single DNA priming followed by a single booster with a recombinant replication-defective Sendai virus vector, in a macaque AIDS model. J Virol. 2003;77(17):9710–9715. doi:10.1128/JVI.77.17.9710-9715.2003
- Scaggs Huang F, Bernstein DI, Slobod KS, et al. Safety and immunogenicity of an intranasal sendai virus-based vaccine for human parainfluenza virus type I and respiratory syncytial virus (SeVRSV) in adults. Hum Vaccin Immunother. 2021;17(2):554–559. doi:10.1080/21645515.2020.1779517
- Imamura T, Oshitani H. Mucosal immunity against influenza induced by attenuated recombinant Sendai virus. Expert Rev Vaccines. 2011;10(10):1393–1395. doi:10.1586/erv.11.123
- Lombardi A, Villa S, Castelli V, et al. T-Cell exhaustion in mycobacterium tuberculosis and nontuberculous mycobacteria infection: pathophysiology and therapeutic perspectives. Microorganisms. 2021;9(12):2460.
- Hu Z, Zhao HM, Li CL, et al. The role of KLRG1 in human CD4+ T-cell immunity against tuberculosis. J Infect Dis. 2018;217(9):1491–1503. doi:10.1093/infdis/jiy046
- Khan N, Vidyarthi A, Amir M, et al. T-cell exhaustion in tuberculosis: pitfalls and prospects. Crit Rev Microbiol. 2017;43(2):133–141. doi:10.1080/1040841X.2016.1185603
- Ankley L, Thomas S, Olive AJ. Fighting Persistence: how Chronic Infections with Mycobacterium tuberculosis Evade T Cell-Mediated Clearance and New Strategies To Defeat Them. Infect Immun. 2020;88(7). doi:10.1128/IAI.00931-19
- Barry C3, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7(12):845–855. doi:10.1038/nrmicro2236