Abstract
Baculoviruses infecting Lepidoptera (butterflies and moths) encodes an enzyme known as ecdysosteroid UDP-glycosyltransferase (EGT), which inactivates insect host ecdysosteroid hormones, thereby preventing molt and pupation and permitting a build-up of the viral population within the host. Baculovirus EGT shows evidence of homology to insect UDP-glycosyltransferases (UGTs), and a phylogenetic analysis supported that the closest relatives of baculovirus EGT are the UGT33 and UGT34 families of lepidopteran UGTs. The phylogenetic analysis thus supported that baculovirus EGT arose by horizontal gene transfer of a UGT from a lepidopteran host, an event that occurred 70 million years ago at the earliest but possibly much more recently. Three amino acid replacements unique to baculovirus EGTs and conserved in all available baculovirus sequences were identified in the N-terminal region of the molecule. Because of their conservation, these amino acids are candidates for playing an important functional role in baculovirus EGT function.
The genomes of large double-stranded DNA (dsDNA) viruses include a number of genes that show homology to genes of cellular organisms (Hughes & Friedman Citation2003, Citation2005; Shackleton & Holmes Citation2004; Hughes et al. Citation2010). Although some of these genes may have formed part of the viral genome since its origin, certain other viral genes show close sequence similarity to host genes, suggesting that such genes represent host genes that have been “captured” by the virus; that is, horizontally transferred from the host to the virus (Barry & McFadden Citation1997; Wall et al. Citation1998; Lalani & McFadden Citation1999; Hughes Citation2002a). In ds-DNA viruses of vertebrates, many of the putative “captured” genes play a role in disrupting host immune system signaling, thus conferring an advantage on the virus (Barry & McFadden Citation1997; Wall et al. Citation1998; Lalani & McFadden Citation1999). Thus, the capture and incorporation of host genes plays an important role in coevolution of these viruses with their hosts.
The baculoviruses (Baculoviridae) are dsDNA viruses infecting arthropods (Miller Citation1997). Currently, four genera are recognized in the family (Jehle et al. Citation2006). The genera Alphabaculovirus and Betabaculovirus infect members of the insect order Lepidoptera (butterflies and moths); the genus Gammabaculovirus infects members of the insect order Hymenoptera; and the genus Deltabaculovirus infects members of the insect order Diptera (Jehle et al. Citation2006). Like those of other dsDNA viruses, baculovirus genomes include several genes with homology to those of cellular organisms (Hughes Citation2002b; Hughes & Friedman Citation2003). One of these, the gene encoding the inhibitor of apoptosis (IAP), is known to play a role in disrupting host immune defense. Because apoptosis is a host defense mechanism against viruses, interference with apoptosis is advantageous to viral fitness (Birnbaum et al. Citation1994; Clem Citation1997; Teodoro & Branton Citation1997; Carpes et al. Citation2005). IAP homologs are found throughout the animal kingdom (Hughes Citation2002b; Bergman et al. Citation2003). A phylogenetic analysis showed that certain IAP genes of baculoviruses infecting Lepidoptera are closely related to those of their hosts, supporting the hypothesis that these baculovirus genes have recently been transferred from their insect hosts (Hughes Citation2002b).
The egt gene, which encodes an enzyme known as ecdysosteroid UDP-glycosyltransferase (EGT), is another well-studied baculovirus gene that has homologs in cellular organisms (O’Reilly Citation1995, Citation1997). This gene is found in many viruses belonging to the two baculovirus genera that infect Lepidoptera, Alphabaculovirus and Betabaculovirus (O’Reilly Citation1995, Citation1997; Ahn et al. Citation2012). UDP-glycosyltransferases (UGTs) catalyze the conjugation of a sugar donated by a UDP-glycoside to a lipophilic molecule; baculovirus EGT inactivates host ecdysosteroid hormones by glycosulating them (O’Reilly Citation1995). One result is to prevent molting and thus pupation in infected lepidopteran larvae, thereby permitting a build-up of the viral population within the larva and increasing the likelihood of further infection (O’Reilly & Miller 1991; O’Reilly Citation1995). Baculovirus EGT has numerous additional effects on the host (Cory et al. Citation2004), including behavioral effects on the European gypsy moth Lymantria dispar, leading infected larvae to climb to the top of host trees before dying, thereby maximizing dispersal of virus particles (Hoover et al. Citation2011).
Phylogenetic analyses can provide evidence of horizontal gene transfer (HGT) from host to virus, but the ability to rule out alternative hypotheses depends on the availability of sequences from both host species and other cellular organisms in order to document the relationships of viral sequences. In their survey of putative cases of HGT from hosts to baculoviruses, Hughes and Friedman (Citation2003) were unable to determine whether baculovirus EGT arose by HGT from their insect hosts because of the lack of sequences from insects of the order Lepidoptera, the hosts of all baculoviruses known to encode EGTs. More recently, numerous sequences of UGTs from Lepidoptera have become available, including those from complete genomes of the silkworm moth Bombyx mori and the monarch butterfly Danaus plexippus (Ahn et al. Citation2012). Using this larger database, Ahn et al. (Citation2012) noted that baculovirus EGT sequences showed greatest similarity to a Lepidoptera-specific family of UGTs, which they designated UGT33. However, those authors did not comment on the evolutionary origin of baculovirus EGT. Here, reconstructing a rooted phylogenetic tree, I conduct a formal phylogenetic test of the hypothesis that baculovirus EGT has arisen from horizontal transfer of a host gene. In addition, I use phylogenetic methods to reconstruct ancestral amino acid sequences in order to identify amino acid replacements unique to baculovirus EGT.
Methods
Phylogenetic analyses were based on 141 selected UGT sequences of insects; 47 EGT sequences of baculovirus EGT; and six sequences from nematodes (phylum Nematoda) used as an out-group to root the tree (for accession numbers see Supplementary Figure S1). The insect species chosen (Table ) represented two orders of exopterygotes (insects with incomplete metamorphosis) from the infraclass Paraneoptera and four orders of the infraclass Endopterygota (endopterygotes or insects with complete metamorphosis). The latter included six species of Lepidoptera represent six different families (Table ). Baculovirus homologs of UGT were identified by BLASTP homology search. Multiple BLAST searches were conducted using UGT amino acid sequences of a variety of insect and baculovirus species. Homologs (Table ) were identified from the genera Alphabaculovirus (corresponding in large part to the older generic name Nucleopolyhedrovirus) and Betabaculovirus (a synonym of the older generic name Granulovirus), both of which are specific to Lepidoptera (Jehle et al. Citation2006). Homology search uncovered no UGTs in the baculovirus genera Gammabaculovirus and Deltabaculovirus.
Table 1. Insect species from which UDP-glucosyltransferase sequences are analyzed.
Table 2. Baculovirus species from which UDP-glucosyltransferase sequences are analyzed.
Amino acid sequences were aligned by the CLUSTAL algorithm in MEGA 5.10 (Tamura et al. Citation2011), and any site at which the alignment postulated a gap in any of a set of aligned sequences was excluded from analyses involving that set of sequences (Supplementary Figure S1). Phylogenetic trees were reconstructed by two methods: (1) the minimum evolution (ME) method (Rzhetsky & Nei 1992) based on the JTT + G distance and (2) the maximum likelihood (ML) method based on the WAG + G + F model; the model for the ML analysis was chosen in MEGA 5.10 using the Bayes Information Criterion (Tamura et al. Citation2011). The hypothesis that a given internal branch within the ME tree was equal to zero was tested by the interior branch test, with standard errors of branch lengths estimated by the bootstrap method (Nei & Kumar Citation2000). A branch with 95% or greater confidence level was considered significant. The reliability of branching patterns in the ML tree was tested by bootstrapping; 1000 bootstrap pseudo-samples were used. The ML method was used to reconstruct ancestral sequences (most probable ancestors) at major nodes within the phylogenetic tree in MEGA 5.10.
Results
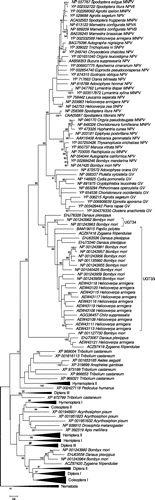
The ME phylogenetic tree of insect and baculovirus UGTs, based on 169 aligned amino acid positions, is shown in partially condensed form in Figure and in full detail in Supplementary Figure S2. For the ease of presentation, certain clades are shown in condensed form in Figure . The baculovirus EGTs formed a distinct cluster, which was supported by a highly significant internal branch (Figure ). The baculovirus EGT cluster in turn fell within a large group of sequences from Lepidoptera, including sequences from all six of the lepidopteran species included in the analysis; and this cluster was supported by a significant branch (Figure ). This latter group included a large cluster corresponding to the Lepidoptera-specific family UGT33 (Ahn et al. Citation2012). In addition, two sequences from the UGT34 family, known only from B. mori, clustered as a sister group to all of the baculovirus sequences (Figure ). The latter pattern was supported by a significant internal branch (Figure ).
Besides UGT33 and UGT34 families, there were additional sequences from Lepidoptera in the phylogenetic analysis. The latter sequences included several clusters that were not close to the baculovirus sequences; rather, certain lepidopteran sequences clustered among sequences from insects of different orders (Figure and Supplementary Figure S2).
The overall topology of the ML tree (Figure and Supplementary Figure S3) was similar to that of the ME tree, but there were significant differences in detail. In the ML tree, the UGT33 and UGT34 sequences clustered together, along with EHJ78328 of the monarch butterfly D. plexippus, in a group that received 70% bootstrap support (Figure ). The latter group of lepidopteran sequences formed a sister group to baculoviris EGTs, a pattern that received 95% bootstrap support. Thus, unlike the ME tree, in the ML tree the sister group to baculovirus EGT was a large cluster of lepidopteran sequences including both UGT33 and UGT34 families (Figure ). However, relationships within this cluster were not well resolved; thus, the ML tree did not provide a basis for decisively rejecting the hypothesis that UGT34 is the sister group of baculovirus EGT, as supported by the ME tree.
Figure 1. ME tree of UGTs of insects based on JTT + G distance at 169 aligned amino acid sites; certain groups are condensed for ease of presentation (for full tree see Supplementary Figure S2). Numbers on the branches are confidence levels of interior branch test; only values ≥95% are shown.
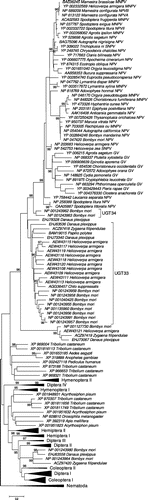
Figure 2. ML tree of UGTs of insects based on WAG + G + F model at 169 aligned amino acid sites; certain groups are condensed for ease of presentation (for full tree see Supplementary Figure S3). Numbers on the branches are percentages of 1000 bootstraps supporting the branch; only values ≥50% are shown.
In order to identify amino acid residues that might be responsible for unique functional properties of baculovirus EGTs, amino acid changes on the branch leading to the baculovirus EGT cluster were reconstructed, assuming the ME tree, that is, changes that occurred on the branch leading from the common ancestor of UGT34 and the baculovirus EGTs to the common ancestor of the baculovirus EGTs (Figure ). Of 31 amino acid changes on that branch, three gave rise to amino acid residues that were conserved in all baculovirus EGTs analyzed. These conserved positions (numbered as in NP_258389, Spodoptera litura NPV EGT) were G179, N185, and Q280 (Figure ). In addition to these conserved baculovirus-specific amino acid replacements, baculovirus EGTs differed from lepidopteran UGT33 and UGT34 family members through an overall reduction in amino acid sequence conservation. Of 46 aligned amino acid residues conserved in all lepidopteran UGT33 and UGT34 family members analyzed, only 13 (28.3%) were conserved in all baculovirus EGTs analyzed.
Figure 3. Segment of alignment of UGT34 family members from Lepidoptera with selected baculovirus EGTs (corresponding to sites 171–283 of NP_258389 from S. litura NPV). Arrows indicate site with amino acid replacements that occurred in the common ancestor of baculovirus EGTs and are conserved in all baculovirus EGTs.

Discussion
When viral genomes contain genes homologous to those of cellular organisms, two alternative hypotheses might be proposed to explain this observation: (1) the gene has been part of the viral genome since its origin; or (2) the gene has been transferred by HGT from a cellular organism (Hughes & Friedman Citation2003). In the latter case, the HGT might have occurred very anciently or more recently. A rooted phylogenetic analysis can be used to decide among these alternatives. In the ME analysis, the EGT genes of baculoviruses were found to have a sister relationship to the UGT34 family of lepidopteran UGTs, so far known only from B. mori. The ML analysis, while providing less clear resolution of the relationship between UGT33 and UGT34, placed both of these lepidopteran gene families as sister groups of baculovirus EGTs. Thus, both ME and ML analysis supported the hypothesis that lepidopteran UDP-glucosyltransferases are the closest relatives of baculovirus EGTs.
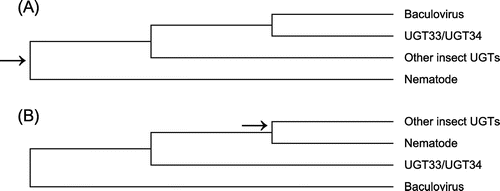
Given the rooting of the phylogenetic tree with sequences from nematodes, a presumed out-group to insects, the fact that baculovirus EGTs clustered with lepidopteran sequences supported the hypothesis of HGT from Lepitoptera to baculoviruses. It might be argued that same tree topology could be rooted on the baculoviruses rather than the nematodes (Figure (B)). This rooting would be consistent with the hypothesis that EGT formed part of the ancestral baculovirus genome, rather than originating from HGT. However, such a rooting would imply that the UGT33 and UGT34 families, found only in Lepidoptera, originated prior to the most recent common ancestor (MRCA) of insects and nematodes (Figure (B)). Therefore, the rooting with nematodes as the out-group (Figure (A)) seems more parsimonious.
Figure 4. Schematic representation of UGT phylogeny rooted (A) with nematodes and (B) with baculoviruses. In each case, arrow indicated putative MRCA of insects and nematodes.
Since the EGT is found in both Alphabaculovirus and Betabaculovirus, it is parsimonious to infer that the HGT event occurred in the common ancestor of these two genera. The widespread occurrence of EGT in these two genera (O’Reilly Citation1995) is most parsimoniously explained by a single HGT event. Although a Betabaculovirus genome without an EGT gene has been reported (Hayakawa et al. Citation1999), this is most easily explained as a secondary loss. The hypothesis that the HGT event involved transfer of a gene from a lepidopteran host is consistent with the apparent absence of the EGT gene in the baculovirus genera Deltabaculovirus and Gammabaculovirus, whose hosts belong to other insect orders (Jehle et al. Citation2006).
Because the baculovirus genes formed a cluster with homologs from Lepidoptera, HGT must have occurred after the origin of the order Lepidoptera, which is believed to have occurred in the late Triassic Period before 250 Mya (Grimaldi & Engel Citation2005). Assuming the topology of the ME tree, in which baculovirus EGT sequences clustered closer to lepidopteran UGT34 than to lepidopteran UGT33, the horizontal transfer event would have occurred after the gene duplication event that gave rise to those two gene families. Members of the UGT34 family are known from at least six families of Lepidoptera (Table ), the MRCA of which occurred in the late Cretaceous about 70 Mya (Grimaldi & Engel Citation2005). Therefore, the gene duplication event giving rise to separate UGT33 and UGT34 families must have occurred prior to that date. The earliest possible data for the HGT event would thus be sometime after 70 Mya, but it may have occurred considerably more recently.
The interruption of host development by baculovirus EGT is advantageous to the virus because it permits the build-up of a large viral within-host population (O’Reilly Citation1995). In an experimental comparison of EGT + and EGT-strains of the AcMNPV, insect larvae infected by EGT + virus persisted in an actively feeding state longer than those infected with EGT-virus, resulting in an estimated 30% greater virus particle production in the former compared to the latter (O’Reilly & Miller 1991). Many lepidopteran larvae migrate away from the host plant in order to pupate; by preventing molt to the pupal stage, baculovirus EGT prevents this migration, thereby increasing the likelihood of virus transmission to the next generation (O’Reilly & Miller 1991). Likewise, the behavioral effects of baculovirus EGT on the behavior of European gypsy moth larva provide a further example of EGT effects aiding virus dispersal, because the death of the larva in an elevated position aids dispersal of virus particles by wind and rain (Hoover et al. Citation2011).
Thus, the HGT event conferred a major adaptive advantage to baculoviruses infecting Lepidoptera, an advantage that may have been responsible for the radiation of these viruses throughout the order Lepidoptera. Nonetheless, there are trade-offs associated with viral adaptations that prevent maturation of the host larva, since the virus thereby becomes unable to take advantage of dispersal by the winged adult insect or of adult to offspring infection (Cory & Myers Citation2003). The drawbacks of this mode of viral transmission are illustrated by the reduced baculovirus infection rates in low-density gypsy moth populations, where the encounter of larvae with virus particles from previous infections is infrequent (Woods & Elkinton Citation1987).
Of the 31 reconstructed amino acid changes on the branch leading to baculovirus EGT, three gave rise to an amino acid residues conserved in all baculovirus EGTs analyzed: G179, N185, and Q280 (numbered as in NP_258389). Based on the alignment of Ahn et al. (Citation2012), the G179 and N185 are located in the N-terminal domain of the protein, not far from the catalytic residues. Q280 is located in the interdomain linker. These residues thus represent unique synapomorphies of the baculovirus EGT clade. Because these residues arose in baculovirus EGTs after the HGT event and have subsequently been conserved, they represent plausible candidates for residues contributing to unique functional properties of baculovirus EGTs.
The origin of viruses remains mysterious. Whereas some have argued that viruses are ancient, representing a pre-cellular stage of the evolution of life on earth (Claverie Citation2006; Koonin Citation2009), others have proposed a more recent origin, with most or even all virus genes originating from horizontal transfer of cellular genes (Moreira & López-García Citation2009). It may not be possible to decide among hypothetical scenarios for virus origin, but there is abundant evidence that acquisition of host genes has been a feature of the evolution of large DNA viruses (Hughes & Friedman Citation2003, Citation2005; Moreira & López-García Citation2009). The evidence that baculoviruses infecting Lepidoptera obtained a host gene that plays a key role in the infection process supports the hypothesis that gene acquisition is an ongoing process in the adaptive evolution of large DNA viruses.
Supplementary material
Download MS Word (595.5 KB)Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health.
References
- AhnS-J,VogelH,HeckelDG.2012.Comparative analysis of the UDP-glycosyltransferase multigene family in insects.Insect Biochem Mol Biol.42:133–147.
- BarryM,McFaddenG.1997.Virus encoded cytokines and cytokine receptors.Parasitology.115:S89–S110.
- BergmanA,YangAY,SrivastaM.2003.Regulators of IAP function: coming to grips with the grim reaper.Curr Opin Cell Biol.15:717–724.
- BirnbaumMJ,ClemRJ,MillerLK.1994.An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs.J Virol.68:2521–2528.
- CarpesMP,deCastroME,SoaresEF,VillelaAG,PinedoFJ,RibeiroBM.2005.The inhibitor of apoptosis gene (iap-3) of Anticarsia gemmatalis multicapsid nucleopolyhedrovirus (AgMNPV) encodes a functional IAP.Arch Virol.150:1549–1562.
- ClaverieJ-M.2006.Viruses take center stage in cellular evolution.Genome Biol.7:10. doi:10.1186/gb-2006-7-6-110
- ClemRJ.1997.Regulation of programmed cell death by baculovirus expression. In:MillerLK, editor.The baculoviruses.New YorkNY:Plenum Press; p.237–266.
- CoryJS,ClarkeEE,BrownML,HailsRS,O’ReillyDR.2004.Microparasite manipulation of an insect: the influence of the egt gene on the interaction between a baculovirus and its lepidopteran host.Functional Ecol.18:443–450.
- CoryJS,MyersJH.2003.The ecology and evolution of baculoviruses.Annu Rev Ecol Evol Syst.34:239–272.
- GrimaldiD,EngelMS.2005.Evolution of the insects.Cambridge:Cambridge University Press.
- HayakawaT,KoR,OkanoK,SeongS-I,GotoC,MaedaS.1999.Sequence analysis of the Xestia c-nigrum granulovirus genome.Virology.262:277–297.
- HooverK,GroveM,GardnerM,HughesDP,McNeilJ,SlavicekJ.2011.A gene for an extended phenotype.Science.333:1401.
- HughesAL.2002a.Origin and evolution of viral interleukin-10 and other DNA virus genes with vertebrate homologues.J Mol Evol.54:90–101.
- HughesAL.2002b.Evolution of inhibitors of apoptosis in baculoviruses and their insect hosts.Infect Genet Evol.2:3–10.
- HughesAL,FriedmanR.2003.Genome-wide survey for genes horizontally transferred from cellular organisms to baculoviruses.Mol Biol Evol.20:979–987.
- HughesAL,FriedmanR.2005.Poxvirus genome evolution by gene gain and loss.Mol Phyl Evol.35:186–195.
- HughesAL,IrausquinSJ,FriedmanR.2010.Evolutionary biology of poxviruses.Infect Genet Evol.10:50–59.
- JehleJA,BlissardGW,BonningBC,CoryJS,HerniouEA,RohrmannGF,TheilmannDA,ThiemSM,VlakJM.2006.On the classification and nomenclature of baculoviruses: a proposal for revision.Arch Virol.151:1257–1266.
- KooninEV.2009.On the origin of cells and viruses: primordial virus world scenario.Ann NY Acad Sci.1178:47–64.
- LalaniAS,McFaddenG.1999.Evasion and exploitation of chemokines by viruses.Cytokine Growth Factor Rev.10:219–233.
- MillerLK.1997.Introduction to the baculoviruses. In:MillerLK, editor.The baculoviruses.New YorkNY:Plenum; p.1–6.
- MoreiraD,López-GarcíaP.2009.Ten reasons to exclude viruses from the tree of life.Nat Rev Microbiol.7:306–311.
- NeiM,KumarS.2000.Molecular evolution and phylogenetics.New YorkNY:Oxford University Press.
- O’ReillyDR.1995.Baculovirus-encoded ecdysosteroid UDP-glycosyltransferases.Insect Biochem Mol Biol.25:541–550.
- O’ReillyDR.1997.Auxiliary genes of baculoviruses. In:MillerLK, editor.The baculoviruses.New YorkNY:Plenum; p.267–300.
- O’Reilly DR, Miller L. 1991. Improvement of a baculovirus pesticide by deletion of the EGT gene. Biotechnology. 9: 1086–1089.
- Rzhetsky A, Nei M. 1992. A simple method for estimating and testing minimum-evolution trees. Mol Biol Evol. 9:945–967.
- ShackletonLA,HolmesEC.2004.The evolution of large DNA viruses: combining genomic information of viruses and their hosts.Trends Microbiol.12:458–465.
- TamuraK,PetersonD,PetersonN,StecherG,NeiM,KumarS.2011.MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.Mol Biol Evol.28:2731–2739.
- TeodoroJG,BrantonPE.1997.Regulation of apoptosis by viral gene products.J Virol.71:1739–1746.
- WallEM,CaoJX,UptonC.1998.Subversion of cytokine networks by viruses.Int Rev Immunol.17:121–155.
- WoodsSA,ElkintonJS.1987.Bimodal patterns of mortality from nuclear polyhedrosis virus in gypsy moth (Lymantria dispar) populations.J Invert Pathol.50:151–157.