
Abstract
The cophylogeny reconstruction problem is central in the inference of ancient relationships among ecologically linked groups of organisms. Although it is known to be NP-Complete, there have been several heuristics proposed that often find good solutions. One such approach is to fix the order of the internal nodes of the host phylogeny and use dynamic programming to recover an optimal reconstruction in polynomial time. A meta-heuristic framework is then leveraged to iterate over the factorial set of fixed node orderings. The original solution used a technique known as node mapping, taking and was designed to handle phylogenetic networks. There has since been a number of
implementations for this problem which use specific properties of phylogenetic trees to reduce their running time. In this paper, we introduce a set of new optimisation techniques to achieve a running time improvement matching these cubic time algorithms, while using node mapping, and discuss how these newly proposed optimisation techniques may lead to an improvement in the generalised case of phylogenetic networks and provide valuable insights into how to construct an optimized algorithm for handling evolutionary systems with widespread parasites.
Cophylogenetics considers the macro-scale coevolutionary associations between two or more phylogenetic histories. One aim of cophylogenetics is the development of algorithms for reconstructing the coevolutionary dependencies between ecologically linked groups of organisms, known as cophylogenetic reconstructions. The aim of the algorithms discussed in this paper is to recover the minimum cost cophylogenetic reconstructions.
Although able to consider any evolutionary systems in which more than two phylogenies coexist, this problem is often discussed in terms of a host phylogeny and a single corresponding parasite phylogeny
. This problem is not constrained to simply analysing host and parasite systems, however, having been applied to a number of other interesting biological phenomena, including herbivore and plant interactions (Percy et al. Citation2004), genes and the species that house them (Libeskind-Hadas & Charleston Citation2009), historical biogeography (Ronquist Citation1997), mutualism between multiple organisms (CitationJackson 1999), and mimicry between species (Hoyal Cuthill & Charleston Citation2012).
This work considers the case where both the host and parasite
phylogenies are rooted trees rather than the more generalised problem of handling phylogenetic networks. We consider the trees as a set of vertices (nodes) and edges where the host tree is defined as
and the parasite tree is defined as
. To simplify the complexity analysis, we consider the number of leaf nodes in
and
to both be
, with the total number of internal nodes and leaf nodes equal to
. This is not a methodological requirement but is used to allow for a consistent comparison with the existing algorithms. This matches the order of the number of edges in
and
as a tree with
nodes has
edges. This is in contrast to a phylogenetic network which can have up to a quadratic number of edges.
Associations between each phylogenetic history are marked only between the leaf nodes of
and
. These associations represent the known interactions between
and
. The problem considered here assumes each parasite inhabits exactly one host, while each host may have zero, one or more than one parasite. Although there is an exponential number of combinations possible for associations between
and
, the requirement that each parasite node is only permitted a single association results in a tight bound for the number of associations
, for a specific instance of
matching the number of leaves in
. Extending the proposed algorithm to the case where many associations are permitted for each leaf node
is discussed in the Further Work section.
The aim of the cophylogeny reconstruction problem is to recover the minimum cost mapping, , of nodes from
into
such the initial associations,
are conserved. The mapping,
, is constructed using the four recoverable biological events, Codivergence, Duplication, Host Switch and Loss.
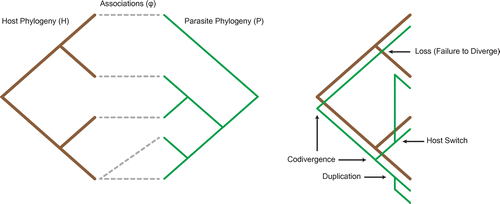
A Codivergence event is a concurrent divergence event of both the parasite and host lineages. A signal of coevolution in an evolutionary system is a high number of codivergence events in the cophylogeny reconstruction (Page Citation2002). A Duplication event is an independent divergence of the parasite lineage with respect to the host, where both new lineages continue to trace the existing host phylogeny (Charleston Citation2003). A Loss event includes three separate phenomena which map to a common reconstruction event, including failure to diverge by the parasite lineage, an extinction event in the parasite lineage, or a sampling error occurring when reconstructing the associations () between the host and parasite (CitationPaterson et al. 2003). A Host Switch event is an independent divergence event in the parasite lineage with respect to the host, where one of the new parasite lineages shifts to a new lineage in the host, with the second lineage continuing to trace the original host lineage (Charleston & Perkins Citation2006). We define the initial host lineage as the take-off edge and the new lineage as the landing edge. A cophylogenetic reconstruction that contains a map
including all four events can be seen in Figure .
Figure 1. A coevolutionary relationship represented using host and parasite phylogenies and a corresponding cophylogenetic reconstruction, which includes all four biological events.
Reconstructing the minimum cost map requires that each event be assigned a penalty cost. This set of costs for each biological event, which we refer to as the cost scheme, is hereby treated as a vector in the form where
,
,
and
represent the penalty costs associated with codivergence duplication, host switch and loss, respectively. The total cost for a cophylogeny mapping can be constructed as follows:
(1)
where ,
,
,
represent the number of events for codivergence duplication, host switch and loss, respectively.
Traditionally, cost schemes aim to promote codivergence events by assigning a reduced penalty score for codivergence. This has been achieved in a number of ways, such as the maximising codivergence cost scheme, where all other event costs are assigned 0 while codivergence is assigned a cost of −1 such that the minimum cost solution is the case with the highest number of codviergence events. This approach has been noted to overcount loss and host switch events (Ronquist Citation2002). The ‘Jungle’ approach was later proposed. In this approach, codivergence was assigned a cost less than the remaining events. This cost scheme has been shown to guarantee that all recovered solutions are from the Pareto front (Libeskind-Hadas & Charleston Citation2009). An alternate approach to minimising the cost of codivergence is to prevent host switch events, which is known as reconciliation (Ronquist Citation2002). Methods employing reconciliation such as Page’s Reconciled Tree Analysis can solve this problem in for all problem instances (Page Citation1994a, Citation1994b). Considering all four biological events, however, significantly increases the complexity of the problem. In fact, this problem has been shown to be NP-Complete when permitting all four biological events (Libeskind-Hadas & Charleston Citation2009; Ovadia et al. Citation2011).
This increase in complexity is due to the handling of host switch events. Host switch events give rise to a factorial number of possible permutations of the internal nodes in the host phylogeny. This increases the complexity of the relative ordering of the nodes and varies the possible placement of the take-off and landing edges for a host switch and the resultant cost.
A number of methods have been proposed to handle this complexity. The most common are two methods that solve modified versions of the cophylogeny reconstruction problem using dynamic programming. The first of these approaches is implemented in the tools Tarzan (Merkle & Middendorf Citation2005) and Core-Pa (Merkle et al. Citation2010). This approach ignores the orderings of the host nodes so as to allow the best host switch to be recovered in polynomial time. This relaxation, however, has been shown to produce solutions which are biologically infeasible (Conow et al. Citation2010).
The second approach is to fix the order of the nodes in the host tree. This approach will be the focus of this paper. This method was proposed by Libeskind-Hadas and Charleston (Citation2009). They proposed a novel solution, where the node ordering is fixed, such that the cophylogeny reconstruction problem can be solved in polynomial time. A meta-heuristic framework is then applied to traverse the factorial combinations of node orderings. A genetic algorithm has since been shown to be particularly successful method for recovering an optimal node ordering. The initial dynamic programming algorithm proposed ran in and was later implemented in the Jane 1 software tool (Conow et al. Citation2010). This algorithm would store the resultant sub-solution for each subtree at the node (see Implementation). This technique has since been called node mapping to differentiate with alternate algorithms which can solve this problem in
time, such as the edge mapping approach proposed by Yodpinyanee et al. (Citation2011) and tree slicing proposed by Doyon, Scornavacca, et al. (Citation2011). The edge mapping and slicing algorithms, unlike the initial node mapping algorithm, which was designed to handle phylogenetic networks, are designed to run where both
and
are phylogenetic trees. Edge mapping, however, is the most efficient, as although both have a running time of
, slicing requires
space complexity as opposed to the space complexity of edge mapping, which is
.
Problem statement
A question that follows from this research is whether the original node mapping algorithm, with an running time, is the best implementation that can be achieved for node mapping, and if not, is there an implementation that can uncover a cophylogeny mapping as efficiently in terms of both time and space complexity, compared to edge mapping, for the special case where
and
are phylogenetic trees. Further, can these improvements translate to a more efficient algorithm for recovering cophylogenetic reconstructions for phylogenetic networks?
Such an algorithmic improvement would be of value as to date only the original node mapping algorithm has offered any extensibility towards handling phylogenetic networks. Algorithms such as edge mapping, when translated to phylogenetic networks, suffer from complexity increases in the size of the dynamic programming table, as the number of mapping locations increases from
for a tree to
for a network, which is not the case for node mapping.
In this paper, we present a number of new optimisation techniques which can be applied to the original node mapping algorithm first presented by Libeskind-Hadas and Charleston, where the worst case running time for recovering a cophylogeny mapping for phylogenetic trees is .
Implementation
The newly proposed algorithm is designed to solve the fixed node ordering permutation of the cophylogeny reconstruction problem using dynamic programming. It is designed to run in three stages. The first stage is the preprocessing step, which allows for optimal coevolutionary event locations to be recovered using an time query. The second stage is the construction of the dynamic programming table of coevolutionary events, where the optimal solutions stored are those which minimise the total event cost for the cophylogenetic reconstruction. Finally, the third stage of the algorithm uses the constructed dynamic programming table to recover an optimal cophylogeny reconstruction.
The node mapping algorithm is designed to store cophylogeny reconstruction events at the nodes in . This approach results in codivergence events mapped to the host node at which they occur, while events that occur on host edges, such as duplication and host switch events, are mapped to the child node of the edge on which they occur.
Reducing to an  set of sub-solutions
set of sub-solutions
The node mapping algorithm runs by solving the sub-solution for each internal node in and then using these sub-solutions to build up to the complete solution. A sub-solution is defined below:
Definition 1 A sub-solution is a cophylogeny mapping of subtree
of
mapped into
.
The first optimisation proposed to reduce the running and space complexity of node mapping, is to reduce the set of sub-solutions stored at each step of the dynamic programming algorithm. Previous node mapping algorithms have stored sub-solutions for each
, requiring
comparisons when processing the parent of sibling sub trees
and
. Rather than considering all
possible sub-solutions, this algorithm restricts the set of sub-solutions considered to
. This reduction is possible by selecting only a single sub-solution for each host node rather than considering the set of all possible permutations which is of size
. That is, for a set of
sub-solutions, only
minimal cost sub-solutions are required to guarantee the optimal solution is recovered. The remaining
sub-solutions are discarded. This can still guarantee the optimal solution is recovered, as all minimum cost sub-solutions are represented in this subset and only sub-solutions with duplicate costs or solutions with worse scores, are discarded.
By reducing the sub-solution stored to at each pass, we ensure that the parent only has
comparisons to process for each node
. As the
comparisons are run for each of the
nodes this algorithm requires
comparisons in total. The set of solutions retained at each step are stored in a dynamic programming table of size
. In this table, the rows and the columns represent parasite and host nodes, respectively. Each column of this table is constructed using the best sub-solutions of
. If
does not have a valid mapping to a node
then this column entry is marked invalid and is not considered when processing the parent of
. The algorithm for the construction of this table is described in Figure .
Figure 2. This is the algorithm that constructs a dynamic programming table for node mapping in . It calls a series of methods including three ‘Opt’ event functions (OptCodivergence, OptDuplication, OptHostSwitch) which query the preprocessed events. The fourth function is the AddMinCost function which ensures that only the minimum cost reconstruction for each host node is retained. As this function only compares two values at a time it runs in
.

To recover the minimum cost mapping using the dynamic programming table requires two algorithms. First, the minimum cost entry
must be recovered, where
is the root of
. Starting with
, the second algorithm makes
recursive calls back to the leaves of
,
. This reconstruction runs after the
algorithm to construct the dynamic programming table, and therefore does not impact on the worst case number of comparisons.
This optimisation of the dynamic programming table relies on the assertion that the number of events considered is a constant factor. As is mapped into
, only
, i.e. a constant number, of optimal events may be recovered for codivergence, duplication and host switch events. This is achieved due to preprocessing and a constant bound of the number of events considered, which are both discussed later.
Reducing the host switch set size
The second optimisation that is applied to the node mapping algorithm is to reduce the number of possible host switch events that are considered. This is to overcome the exponential, , number of locations possible even in the fixed node ordering case if no reduction is enforced.
Of this set of permutations, the original node mapping algorithm considers possible locations for host switches for every set of sub-solutions processed. Using this reduced set of sub-solution results in
comparisons to reconstruct the dynamic programming table, which, although an improvement over existing node mapping, is still significantly worse than the existing algorithms running in
.
To reduce the host switch location’s solution space for node mapping, a reduction of the search space for optimal switch locations is required. We give a definition for the optimal host switch location which is used to reduce the host switch solution set as:
Definition 2 Suppose has children
and
, with
and
. Then an optimal host switch location for
is a pair of edges
and
, where the total number of loss events between
and between
is minimal, and where
and
are edges which share a common interval when considering their distances from the root of
.
This definition does not allow for the recovery of all optimal host switches but does always uncover an optimal host switch instance of the possible optimal solutions. The recovered host switch for
minimises the number of loss events between its children which is always optimal. To ensure all optimal sub-solutions are considered, both host switches where
and
are the take-off and landing edges must be computed as potential sub-solutions. This multiplies the solutions space by a constant factor and therefore does not increase the overall complexity.
Using this method, it is possible to reduce the set of host switch locations considered using the previous implementation down to
. As a result of this optimisation technique, only four items need to be recovered for each node
: the optimal codivergence, duplication, host switch from
to
, and that from
to
. Therefore, the assignment of events for each node
is constant, assuming each event is found in
. This result maintains the reduced running time for construction of the dynamic programming table in
, assuming the optimal codivergence, duplication and host switch locations can be found in
, which is described in the following section.
Applying preprocessing to cache recoverable events
As previously noted, comparisons are required for the construction of the dynamic programming table, but the actual running time cost of each comparison has not yet been discussed. In this section, we show that all comparisons can be reduced to
by preprocessing
. The aim, therefore, is for each comparison to recover the minimum cost codivergence, duplication and host switch event for a pair of children
mapped to
using an
query.
The minimal cost codivergence and duplication locations are the node and parent edge, respectively, of their children’s lowest common ancestor (Doyon, Ranwez, et al. Citation2011). These locations can be uncovered using an query provided preprocessing has been performed. The lowest common ancestor preprocessing for an
query requires an
time pass over the tree with a space requirement of
(Harel & Tarjan Citation1984).
The number of loss events between a pair of parasite nodes mapped into is the number of host nodes separating the pair. This can be queried in
by storing the number of nodes between each node
and the root. This is then subtracted when executing the
query.
The optimal host switch location is the pair of edges that overlap, such that the distance to their mapped children is minimised. To allow this edge pair to be recovered for each edge pair using a query time of
, requires
time and space preprocessing. This result is possible by leveraging Bender and Farach-Colton’s (Citation2004) solution to the level ancestor problem. This allows for a
query for recovering the take-off and landing edges for the host switch as the level ancestor problem allows for a node to be recovered at a set distance from the root in any subtree in
time. Consider a pair of edges
and
where
and
are the ancestral nodes and
is the node of this pair which is closest to the root. Let the distance from the root to node
be
, and the query node
to be
. This construction provides the query information in the form
as required by the level ancestor problem. The node returned from the query is the parent of the edge required to complete the (take-off, landing) edge pair. Therefore, the host switch edge pair can be recovered in
using preprocessing of
.
By applying preprocessing, the running time of this algorithm is , and has a space requirement of
, which matches the running time and space requirement for the best known algorithm for this problem (Yodpinyanee et al. Citation2011). Further to the authors’ knowledge, the solution described in this paper provides the first
time and space complexity for preprocessing algorithm for the fixed node ordering permutation of the cophylogeny reconstruction problem.
Results
Comparison of results using real data
To ensure the consistency of the cophylogeny reconstructions reported by the node mapping algorithm once the proposed optimisations have been applied, we applied our approach on a number of well-studied coevolutionary cases. The validation data-set contains a number of problems which have previously been used to verify algorithms, and the complete set of instances was used by Conow et al. (Citation2010) to validate Jane’s performance against Tarzan. The validation set also offers a variety of coevolutionary scenarios including herbivore and plant interactions (Percy et al. Citation2004), mutualism and symbiosis (CitationJackson 1999) and host–parasite systems (Hafner & Nadler Citation1988) to ensure this algorithm is sufficiently general.
We utilised three independent event cost schemes, based on previous research, to ensure that this algorithm is able to recover Pareto optimal solutions for more than just a single event cost scheme. Event cost vectors were constructed for each event cost scheme selected. These vectors are described in the form where
,
,
and
represent the penalty costs associated with codivergence duplication, host switch and loss, respectively. The event cost schemes included in this analysis are those previously introduced including:
Maximising Codivergence (−1, 0, 0, 0): Under this cost scheme, reconstructions match the solution set reported by cost maximisation algorithms (Ronquist Citation1998, Citation2002).
Jungle (0, 1, 1, 1): Under this cost scheme, all events are assigned the same cost, except for codivergences, which are assigned a cost of 0. This is to promote the selection of codivergences events similar to the maximising codivergence cost scheme, though in contrast, also assigns a penalty to other coevolutionary events to prevent overcounting of such events (Ronquist Citation2002).
Reconciliation (1, 1,
It is important to note that all three cost schemes, including reconcilsation which does not permit host switch events as indicated by the cost for host switch events is able to recover biologically feasible solutions for all problem instances.
For consistency, in our analysis, we applied host switch costs as the cost of a host switch plus the additional cost of a duplication event, handling both the duplication and switch events that arise as part of a host switch, as a single event. This was enforced to match the default behaviour of the Jane software series, along with other existing packages in this field (Merkle & Middendorf Citation2005; Conow et al. Citation2010; Merkle et al. Citation2010).
The algorithms selected for this comparison included first the existing node mapping algorithm implemented in Jane 1 (Libeskind-Hadas Citation2010), which is expected to produce the same solutions set as the newly proposed algorithm, although taking more time to do so and secondly, the most recent implementation of edge mapping implemented in Jane 4 (Libeskind-Hadas Citation2013), which should take approximately the same amount of time as the new node mapping algorithm.
The comparison of the six real data-sets using all three event cost vectors can be seen in Table . This table shows the results achieved for all six test cases and for all three cost schemes. In all cases, the result recovered by both ‘flavours’ of Jane matched the new node mapping algorithm, validating its performance as a method for recovering cophylogenetic reconstructions.
Table 1. Comparing Jane 1 (node mapping), Jane 4 (edge mapping) and the newly proposed node mapping algorithm over six real data-sets using three different event cost vectors. The reported solutions are condensed into one column as all three algorithms reported the same map cost for all event cost vectors in all six test cases.
Comparing actual running time
The new node mapping algorithm was implemented in Java as was Jane 1 and Jane 4. To evaluate the actual execution speed of each algorithm, the largest real data-set, host plants (Leguminosae) and phytophagous insects (Psylloidae) (Percy et al. Citation2004), was selected as the baseline.
Two running time evaluations were carried out on a standard PC with an i5-2400 3.1 GHz CPU and 8 GB of RAM. First, the original node mapping algorithm was compared against the new node mapping algorithm where both were run for all numbers of iterations from 1 to 100, with 100 replicates each, giving 10,000 samples. The second evaluation was to run the existing edge mapping algorithm against the newly proposed node mapping algorithm for 10,000 iterations, repeated 1000 times.
The results of these two simulations are captured in Figure and Table . The original node mapping algorithm was excluded from the second experiment as we estimated that it would have taken approximately half a year of computation to execute the simulation, and because of the imposed constraints of Jane 1, which only allows up to 200 iterations for each data-set. Figure demonstrates the significant running time improvement of the newly proposed approach for cophylogeny reconstruction.
Figure 3. The running time of the original node mapping algorithm (Jane 1) and the new node mapping algorithm plotted on a logarithmic scale.
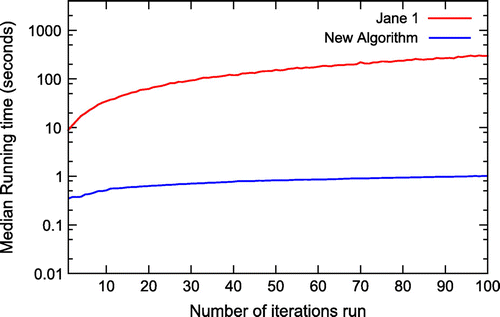
Table 2. The minimum, maximum and median running time for Jane 4 and the newly proposed node mapping algorithm over real data-set 6; host plants (Leguminosae) and phytophagous insects (Psylloidae); for 10,000 iterations repeated 1000 times.
There is, however, a slightly slower running time for the newly proposed node mapping algorithm, compared with the implementation in Jane 4. This margin is a factor of 1.35 (when comparing the median). With further optimisation within the code base, we believe that it is possible to reduce this further to achieve parity with the running time achieved by Jane 4, which has gone through a number of revisions to achieve its current running time.
Discussion and analysis
These results demonstrate that the performance of the newly proposed node mapping algorithm is comparable to the existing edge mapping algorithm’s implementation. It further demonstrates a significant performance improvement over the existing node mapping algorithm, demonstrated by the proposed algorithm taking less than 1 s to perform the same operation that took 300 s by the previous implementation. It is important to note that this result is the case where both algorithms were run for 100 iterations. In fact, both algorithms use 100 populations for 100 iterations each, to ensure with a high probability that they converge on an optimal solution using a genetic algorithm. Therefore, each algorithm requires solving 10,000 instances to recover an optimal solution, which is a difference between solving this problem in approximately 100 s using the newly proposed algorithm or approximately 8.33 h using Jane 1. Further, it is expected that data-sets will continue to increase in size compared to this data-set. One recently constructed system, fig trees and their pollinating wasps (Cruaud et al. Citation2011), contains 5.5 times the number of parasite leaf nodes as the case presented here. Based on the previous performance data Jane 1 would be expected to require two days to complete, compared to approximately 10 min for the proposed algorithm. Data-sets in this field will only continue to increase in size. Therefore, it is essential that algorithms, such as node mapping, are optimised to reduce running time performance while maintaining the same level of accuracy, as has been achieved here.
These results also indicate the potential of node mapping algorithms to be applied further to the research of cophylogenetic reconstruction of phylogenetic networks. Based on the analysis of the best edge mapping and node mapping algorithms, edge mapping is marginally faster when processing phylogenetic trees, but if this newly proposed technique was expanded to phylogenetic networks, a significant running time improvement would be achieved. This is due to edge mapping requiring up to entries in a dynamic programming table because of the increase in the number of edges in the worst case for phylogenetic networks of
. This is in contrast to the newly proposed node mapping algorithm, where there is no need to increase the required size of the dynamic programming table, as there are
nodes in both phylogenetic trees and networks, and therefore storage required for mapping locations does not increase. The additional cost for algorithms handling phylogenetic networks would be incurred in the selection of the best codivergence, duplication and host switch event locations, which still remains an open question for this problem, when considering phylogenetic networks.
Further work
Along with extending this algorithm to handle phylogenetic networks, there are a number of areas for further research involving this newly proposed algorithm. One area is expanding this algorithm to consider cases where a parasite leaf node can have more than one association. The proposed algorithm can be used to handle this problem without an expansion of the dynamic programming table. Currently, all parasite leaf nodes have only a single entry in their respective rows; the mapping to a single host leaf node. For the case where parasites are permitted to have multiple associations, this would be replaced with up to elements which need to be stored in the dynamic programming table for each parasite leaf node depending on the recoverable events used to interpret this permutation of the problem. Therefore, the worst case space complexity would not change as a result of handling widespread (multi-host) parasites using this newly proposed algorithm, and therefore an algorithm for solving this problem where widespread (multi-host) parasites are permitted must exist with a
space complexity.
Also, there is potential to reduce the running time of this solution further, by reducing the sub-solution set. Consider the case where it is possible to reduce the size of the sub-solution set maintained to ,
or
: this would result in algorithms with respective running times of
,
or
. By applying the currently defined algorithm, these sub-solution sets would no longer be able to guarantee optimal solutions for the fixed node ordering instance, but by applying a heuristic sampling technique in the selection of sub-solutions, it is possible that the respective algorithms may recover good solutions in many biological cases, much faster. This approach, although not guaranteeing optimal solutions, would in principle, be able to recover all optimal solutions, with high probability, provided sufficient repetitions are performed when applying random sampling. Therefore, it is believed that a trial within a meta-heuristic framework would be worthwhile.
Conclusion
This work presents a number of new optimisations for the existing node mapping algorithm proposed by Libeskind-Hadas and Charleston. It applies several techniques to reduce the time and space complexity of this well-known problem to provide the fastest implementation using node mapping to date to solve the cophylogeny reconstruction problem where the host tree’s node ordering is fixed. Along with an improved complexity performance for node mapping, this paper offers a reduction to the preprocessing complexity to for time and space complexity. Further, we have presented a formulation for a faster method for recovering cophylogeny mappings for phylogenetic networks. Finally, this work offers future avenues for improving the complexity of the node mapping algorithm, allowing for even faster reconstruction methods and proposes a method to apply this algorithm to the cophylogeny reconstruction problem where widespread parasite associations are permitted.
Availability and requirements
The newly proposed algorithm presented in this paper is available at http://sydney.edu.au/engineering/it/~mcharles/ and runs on any machine with Java 1.6 or higher.
Competing interests
The authors declare that they have no competing interests.
Funding
This work was supported by an Australian Postgraduate Award to BD and an Australian Research Council Grant [grant number DP1094891] to MAC.
References
- Bender MA, Farach-Colton M. 2004. The level ancestor problem simplified. Theor Comput Sci. 321:5–12.
- Charleston MA. 2003. Recent results in cophylogeny mapping. Adv Parasitol. 54:303–330.
- Charleston MA, Perkins SL. 2006. Traversing the tangle: algorithms and applications for cophylogenetic studies. J Biomed Inform. 39:62–71.
- Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithm Mol Biol. 5:16.
- Cruaud A, Cook J, Da-Rong Y, Genson G, Jabbour-Zahab R, Kjellberg F, Pereira RAS, Rnsted N, Santos-Mattos O, Savolainen V, et al. 2011. Fig-fig wasp mutualism: the fall of the strict cospeciation paradigm? In: Evolution of plant-pollinator relationships. Cambridge: Cambridge University Press; p. 68–102.
- Doyon J-P, Ranwez V, Daubin V, Berry V. 2011. Models, algorithms and programs for phylogeny reconciliation. Briefings Bioinf. 12:392–400.
- Doyon J-P, Scornavacca C, Yu Gorbunov K, Szöllősi GJ, Ranwez V, Berry V. 2011. An efficient algorithm for gene/species trees parsimonious reconciliation with losses duplications and transfers. In: Proceedings from the 14th International Conference on Research in Computational Molecular Biology (RECOMB-CG). Springer; p. 93–108.
- Hafner MS, Nadler SA. 1988. Phylogenetic trees support the coevolution of parasites and their hosts. Nature. 332:258–259.
- Harel D, Tarjan RE. 1984. Fast algorithms for finding nearest common ancestors. SIAM J Comput. 13:338–355.
- Hoyal Cuthill J, Charleston M. 2012. Phylogenetic codivergence supports coevolution of mimetic heliconius butterflies. PloS One. 7:e36464.
- Jackson AP. 2004. Cophylogeny of the ficus microcosm. Biol Rev. 79:751–768.
- Libeskind-Hadas R. 2010. Who is Jane? Available from: http://www.cs.hmc.edu/~hadas/jane/Jane1/index.html
- Libeskind-Hadas R. 2013. Jane 4. Available from: http://www.cs.hmc.edu/~hadas/jane
- Libeskind-Hadas R, Charleston MA. 2009. On the computational complexity of the reticulate cophylogeny reconstruction problem. J Comput Biol. 16:105–117.
- Merkle D, Middendorf M. 2005. Reconstruction of the cophylogenetic history of related phylogenetic trees with divergence timing information. Theor Biosci. 123:277–299.
- Merkle D, Middendorf M, Wieseke N. 2010. A parameter-adaptive dynamic programming approach for inferring cophylogenies. BMC Bioinf. 11:S60.
- Ovadia Y, Fielder D, Conow C, Libeskind-Hadas R. 2011. The cophylogeny reconstruction problem is np-complete. J Comput Biol. 18:59–65.
- Page RDM. 1994a. Maps between trees and cladistic analysis of historical associations among genes, organisms, and areas. Syst Biol. 43:58–77.
- Page RDM. 1994b. Parallel phylogenies: reconstructing the history of host-parasite assemblages. Cladistics. 10:155–173.
- Page RDM. 2002. Tangled trees: phylogeny, cospeciation, and coevolution. Chicago: University of Chicago Press.
- Paterson AM, Wallis GP, Wallis LJ, Gray RD. 2000. Seabird and louse coevolution: complex histories revealed by 12s rrna sequences and reconciliation analyses. Syst Biol. 49:383–399.
- Paterson AM, Palma RL, Gray RD. 2002. Drowning on arrival, missing the boat, and x-events: how likely are sorting events. In: Page RDM, editor. Tangled trees: phylogeny, cospeciation, and coevolution. Chicago: University of Chicago Press; p. 287–309.
- Percy DM, Page RDM, Cronk QCB. 2004. Plant–insect interactions: double-dating associated insect and plant lineages reveals asynchronous radiations. Syst Biol. 53:120–127.
- Refrégier G, Le Gac M, Jabbour F, Widmer A, Shykoff JA, Yockteng R, Hood ME, Giraud T. 2008. Cophylogeny of the anther smut fungi and their caryophyllaceous hosts: prevalence of host shifts and importance of delimiting parasite species for inferring cospeciation. BMC Evol Biol. 8:100.
- Ronquist F. 1997. Phylogenetic approaches in coevolution and biogeography. Zool Scr. 26:313–322.
- Ronquist F. 1998. Three-dimensional cost-matrix optimization and maximum cospeciation. Cladistics. 14:167–172.
- Ronquist F. 2002. Parsimony analysis of coevolving species associations. In: Page RDMTangled trees: phylogeny, cospeciation and coevolution. Chicago: Chicago University Press; p. 22–64.
- Sorenson MD, Balakrishnan CN, Payne RB. 2004. Clade-limited colonization in brood parasitic finches (vidua spp.). Syst Biol. 53:140–153.
- Yodpinyanee A, Cousins B, Peebles J, Schramm T, Libeskind-Hadas R. 2011. Faster dynamic programming algorithms for the cophylogeny reconstruction problem. HMC CS Technical Report. Claremont: Harvey Mudd College.
Appendix 1.
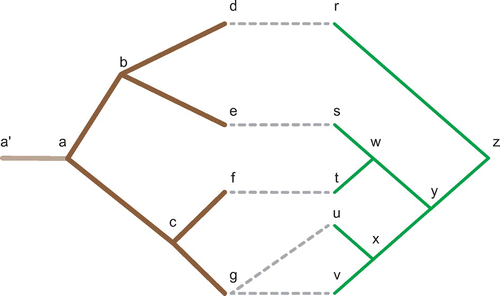
In the following appendix, the algorithm proposed by Libeskind-Hadas and Charleston and the newly proposed algorithm are manually run over a simple tanglegram, where each step is recorded and then compared to demonstrate the improvements offered by the new algorithm. Each algorithm is run on the tanglegram in Figure . The following definitions are used to describe the current state of the algorithms at each step.
Definition 3 An image is a possible mapping
for a particular parasite node
where each node
has a set of images
and where
.
Definition 4 is the ith image of the set of images
for the parasite node
which is written in the form
where
and
are the two children of
and
is the jth image of
and
is the kth image of
.
Definition 5 is the reduced set of images, which is the result of a filter applied by a Node Mapping algorithm such that
and
.
These three definitions allow us to describe each state of both dynamic programming algorithms. For each node we will generate a list of all possible images and will filter the set of recorded images using the processes defined for each algorithm to generate the set
, the states used to incrementally build the final mapping for the parasite tree.
When describing the state as an image, there are three events which can occur: a codivergence, duplication or a host switch. Depending on the location of these events, this may give rise to a number of loss events. The format we use for each event is – codivergence,
– duplication and
– host switch. In the case of a host switch, the edge
is the starting edge and
is the landing edge. Also each event is assigned an event cost. Codivergence is assigned a cost of 0, duplication and loss a cost of 1 and host switch events a cost of 2, in line with the Jungle cost scheme (Ronquist Citation2002). This leads to the following notation to describe the state of each algorithm for each iteration
.
The previous line shows the first image of the node generated from the first images of its two children
and
(
and
, respectively). The mapping is a host switch from the edge
to the edge
in the host tree, and which is assigned a cost of 2. As this is the first mapping of an internal edge, the cost vector only includes a host switch although this will be accumulated as we build up the solution. The form of the cost vector (0, 0, 1, 0) is the number of codivergence, duplication, host switch and loss events in the current image, respectively.
The results aim to adhere the following lemma which is derived from the implementation details of the new algorithm. This lemma is important as if this property is not preserved the new algorithm is unable to recover the optimal solution in .
Lemma 1 The newly proposed algorithm ensures that the size of is bound by the number of nodes in
. Proof in main text.
The following results show that both algorithms recover the optimal solution. The newly proposed algorithm, however, only requires 42 image states to be explored as opposed to the previous algorithm which requires 131. Further, this result confirms the assertion that the newly proposed algorithm, is able to recover the optimum solution while bounding the size of to the number of nodes in
which in this case is 7. This is shown by size of
for
and
which is 6 and 7, respectively, as opposed to the alternate algorithm where the size of the retained set
for both
and
are 12 and 13, respectively.
Solving Figure in
Figure 4. A coevolutionary relationship represented using the host and parasite phylogenies first shown in Figure . In this case, each node in the host and parasite tree is labelled to assist in the reconstruction described in Appendix 1. Of note node has been fixed to a timing interval before
which leads to the reconstruction described in Appendix 1 and an edge has been added to the root of
to map all parasite divergence events which occurred prior to this phylogeny.
Not all the images of are required for the computation of the images of
. In fact, the algorithm only the needs Pareto Set of mappings to compute the images of
. Therefore the final set (the retained set
) of images for
required for generating the mappings for
is
, which can easily be seen as the dominating images based on cost. Ties are broken by taking the first mapping with the lowest cost
.
Of this set of images for only
is retained where
is the cheapest solution.
Solving Figure in
Using the Newly Proposed Algorithm only the best mapping for each node in is required. Therefore, the set of retained images of
is
Of this set of images for only
is retained where
is the cheapest solution matching the mapping of
from the alternate node mapping algorithm.