
Abstract
Amyotrophic lateral sclerosis is the most common motor neuron disease of the adulthood. Genetic analyses performed on cases with sporadic ALS (sALS) and familial ALS (fALS) have revealed mutations most commonly in the genes C9orf72, SOD1, TARDBP, FUS, and UBQLN2. The aim of this study was to investigate the presence and incidence of these most common genomic alterations in these genes of 30 ALS cases. Increase in the number of hexanucleotide repeats within the gene C9orf72 was investigated using the fragment analysis method. A heterozygote c.-45 + 162_-45 + 163insGGGGCC alteration was observed in the first intron of the C9orf72 gene in only two sALS cases (6.6%), c.72 + 133C>T alteration was observed in the first intron of the SOD1 gene in six fALS and two sALS cases (26.6%), a c.169 + 41C>A alteration was observed in the second intron of the same gene in one case, a c.239 + 34A>C alteration was observed in the third intron of the same gene in three fALS cases (10%), and a c.714 + 67_714 + 68insG alteration was observed in the fifth intron of the TARDBP gene in six sALS cases (20%). Three different genomic alterations were detected in the FUS gene; two silent variants p.G49G (c.147C>A) in 3 cases (10%) and p.Y97Y (c.291C>T) in 25 cases (83%) and an intronic variant c.1067-61T˃C in 4 cases (13%). A novel missense variant, p.G457H (c.1371G>C), was observed in the UBQLN2 gene in one case with sALS. Our study has revealed three novel and previously reported genomic alterations in the ALS-related genes.
PUBLIC INTEREST STATEMENT
This study investigates genetic alterations of major ALS genes which are C9orf72, SOD1, FUS, TARDBP and UBQLN2 in familial and sporadic ALS cases.
1. Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and clinically heterogeneous neurodegenerative disease that may result in muscle weakness, paralysis, and death due to affected motor neurons within the cerebral cortex, brain stem, and spinal cord (Brooks, Citation1994). The incidence and prevalence of the disease have been reported to be approximately 2/100,000 and 5/100,000 worldwide. In the USA, approximately 25,000 patients with ALS have been reported and the disease has been classified as the “orphan” disease since it is below the limit of 200,000 (Rowland & Shneider, Citation2001). The diagnosis is made upon the electromyography (EMG) test used to investigate whether the motor neurons are clinically affected or not. The average survival of the patients with ALS is 27.5 months, and the 4-year survival rate has been reported as 40% (Robberecht & Philips, Citation2013). The cases' cause of death is due to respiratory distress developed as a result of weakness in respiratory muscles. It is known that both the familial and sporadic cases have the same clinical and pathological findings, and many mutations observed in familial ALS (fALS) cases have also been detected in sporadic ALS (sALS) cases. On the other hand, although ALS may be diagnosed clinically, the clinical differences between individuals suggest the presence of multiple mechanisms included in the pathophysiology of the disease. Autosomal dominant, autosomal recessive, and X-linked inheritance of the disease demonstrate the genetic heterogeneity of the disease among cases with familial ALS (Maruyama et al., Citation2010).
There are a limited number of studies investigating the causes of ALS and the predisposing genetic factors for the disease among the Turkish population. In one of these studies, an association between ATXN2 and SH2B3 genes has been observed (Lahut et al., Citation2012). In another study, alterations in EPHA3 gene copy number were investigated and their role in the pathogenesis of ALS was reported (Uyan et al., Citation2013).
The most comprehensive study investigating the genetic factors related to ALS in the Turkish population was published in 2015. In this study, in 116 familial ALS cases; C9orf72 (18.3%), SOD1 (12.2%), FUS (5%), TARDBP (3.7%), and UBQLN2 (2.4%); in 361 sporadic ALS cases, C9orf72 (3.1%) and UBQLN2 (0.6%) gene mutations were identified (Ozoguz et al., Citation2015). Additionally, according to the Amyotrophic Lateral Sclerosis Online Genetic Database (ALSoD) (Wroe, Wai-Ling Butler, Andersen, Powell, & Al-Chalabi, Citation2008), mutation frequencies for major ALS-related genes in Turkish population were C9orf72 (28.30%), SOD1 (4.18%), FUS (4.60%), TARDBP (4.63%), and UBQLN2 (4.44%) genes. The aim of our study was to investigate both the presence and the incidence of the most common mutations reported in the literature, which are observed in the genes C9orf72, SOD1, TARDBP, FUS, and UBQLN2 in the Mediterranean Region of the Turkish population.
2. Materials and method
The study was supported by the ethics committee of Akdeniz University Faculty of Medicine Clinical Research. The study included a total of 30 patients diagnosed with ALS at Akdeniz University Faculty of Medicine Department of Neurology. Among those, 12 had fALS from six different families and 18 had sALS. Genomic DNA isolation was performed from the peripheral blood samples by using salting out method (Miller, Dykes, & Polesky, Citation1988). The hexanucleotide repeat region of the first intron of C9orf72 gene was amplified by the triplet primed PCR method and analyzed via fragment analysis. Five exons of the SOD1 gene, including exon–intron junction sites, were amplified using five different primer pairs. FUS gene exons; 3, 4, 5, 6, 11, 12, 13, 14, and 15 which are carrying the most common mutations, and the exon–intron junction sites were amplified using six different primer pairs. All sequences coding the TARDBP gene and the exon–intron junction sites were amplified using five different primer pairs, UBQLN2 gene that has no introns were amplified using four different primer pairs.
The PCR products of these genes were checked using 2% agarose gel electrophoresis, and sequence analysis was performed using the ABI 3130 XL DNA sequencing device. The raw data were evaluated in the ABI Sequence Analysis v3.1 program.
The primers used for the amplification of C9orf72, SOD1, TARDBP, FUS, and UBQLN2 genes have been presented in Table .
Table 1. Fluorescent-labeled primers used for GGGGCC repeats of the C9orf72 gene and the primer sequences used for the DNA analysis of SOD1, TARDBP, FUS, and UBQLN2 genes
3. Results
Among 12 patients in six different families with ALS; 6 were male and 6 were female; and among 18 patients with sALS; 8 were female and 10 were male. The most extended family among fALS cases was family number I, which included 4 cases with the precise diagnosis (III-1, III-5, III-8, and IV-5), 2 pre-symptomatic individuals (IV-1 and IV-7), a 35-year-old male individual with affected father and brother (IV-6), a 50-year-old female individual whose brother had the diagnosis (III-2), and an 80-year-old individual being the oldest member of the family (II-1) (healthy control). A total of nine individuals were included in the study (Figure )).
Figure 1. (a) Pedigree of family I showing history of ALS for six cases. SOD1 and FUS genotype are shown corresponding symbols. The arrow denotes the proband of the family. The individuals' age of medical history taking is shown in the upper left corner. Pre-symptomatic individuals are shown with question mark. Square indicates male; circle female; slash deceased; double line consanguineous marriage. Black symbols indicate patients affected with ALS; white unaffected individuals.
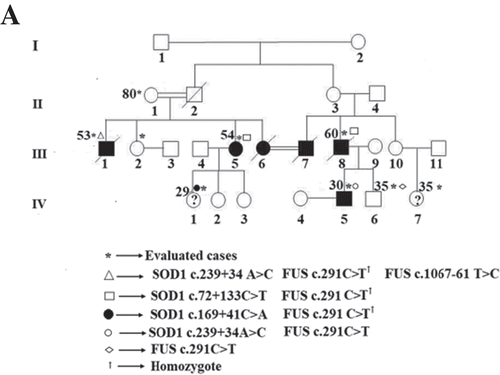
In family II, two of the three individuals with ALS were alive, and only the index individual agreed to participate in the study (III-5) (Supplementary Figure 1(a)).
In family III, one of two affected individuals was alive (III-2) (Supplementary Figure 1(b)); in family IV, one of two affected individuals was alive (III-1) (Supplementary Figure 1(c)); in family V, two siblings among three affected individuals were alive and one agreed to participate in the study (Supplementary Figure 1(d)).
In family VI, a 60-year-old male with ALS (III-5), who had lost her sister due to ALS at 50 years of age, was included in the study (Supplementary Figure 1(e)).
The genomic alterations observed in familial and sporadic cases have been presented in Table .
Table 2. The genomic alterations observed in fALS and sALS cases
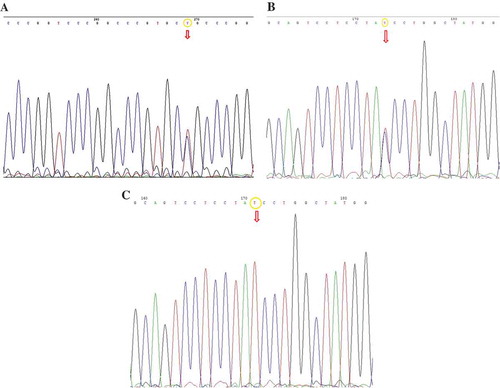
Three genomic alterations were detected in the SOD1 gene. c.72+133C>T alteration, which had previously been defined as rs17881180 in NCBI in the first intron of this gene was observed in individuals III-5 and III-6 in family number I in heterozygote form, in one sporadic case in heterozygote form, in one of six sporadic cases in homozygote form, making a total of eight cases (26.6%). The alteration has been displayed in Figure ).
Figure 2. (a) DNA sequencing analysis revealed the C to T transition in the heterozygote form on position 133 of intron 1 of the SOD1 gene (c.72+133C>T). (b) DNA sequencing analysis revealed the c.291C>T genomic alteration (predicting p.Y97Y) in the heterozygote form in the exon 4 of FUS gene. (c) DNA sequencing analysis revealed the c.291C>T genomic alteration (predicting p.Y97Y) in the homozygote form in the exon 4 of FUS gene.
c.169+41C>A heterozygote alteration, which was not previously defined in NCBI, was observed in the intron 2 of SODI gene. For the first time, the alteration was only determined in the 29-year-old, pre-symptomatic individual with the case number IV-1 in family I. The genomic alteration has been presented in Supplementary Figure 2(a).
Another alteration, c.239+34A>C, was detected in the intron 3 of the SODI gene, which had previously been defined in NCBI as rs2234694. This alteration was detected in individuals number III-1 and IV-5 in family I and the proband of family VI in the heterozygote form. This genomic alteration has been presented in Supplementary Figure 2(b).
Three different alterations were detected in the FUS gene. The first one was p.Y97Y (c.291C>T), which is located in the fourth exon and did not lead to an amino acid change in protein level. This alteration was previously defined in NCBI as rs1052352. It was observed in homozygote form in four individuals, and in heterozygote form in two members of family I with fALS. It was detected in heterozygote form in the index cases of families II, IV, and VI. Among 18 cases with sporadic ALS, this alteration was observed in 16 cases, 8 of which were heterozygote and 8 were homozygote. p.Y97Y (c.291C>T) was the most frequently observed genomic alteration in 25 of 30 cases in our study. This alteration has been presented in Figure .
The second alteration observed in the FUS gene was c.1067-61T>C, and was observed in the heterozygote form in individual number III-1 of family I, and in three individuals with sporadic form. This alteration has been presented in Supplementary Figure 2(c).
The third alteration observed in the FUS gene was p.G49G (c.147C>A), which is located in the third exon, and which did not lead to an amino acid change. This alteration was previously defined in NCBI as rs741810 and was detected in the heterozygote form in three different individuals with sporadic form. This alteration has been presented in Supplementary Figure 2(d).
Sequence analysis in the TARDBP gene revealed c.714+67_714+68insG alteration in the fifth intron in the homozygote form in six sporadic cases, which had previously been defined in NCBI as rs143130606. This alteration has been presented in Supplementary Figure 2. E.
Sequencing of Ubiquilin2 gene revealed p.G457H (c.1371G>C) heterozygote alteration that led to the transformation of the amino acid glycine in the 457th position into the amino acid histidine in only one case of sALS, which previously had not been defined in NCBI. This variant has been presented in Supplementary Figure 2. F.
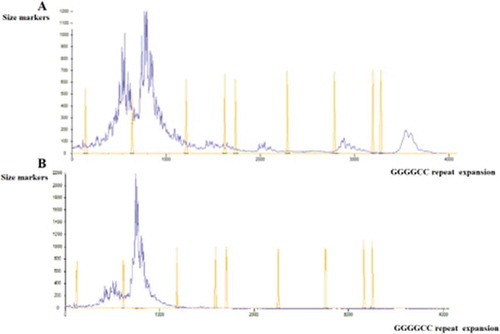
DNA fragment analysis of the C9orf72 gene revealed an increase in GGGGCC repeats in the first intron in the heterozygote form in only two sporadic ALS patients, which had previously been defined in NCBI as rs74180757. This alteration has been presented in Figure .
Figure 3. (a) Fragman analysis result showing expanded repeat numbers c.-45+162_-45+163insGGGGCC, (b) Fragman analysis result showing normal repeat numbers in intron 1 of C9orf72 gene.
4. Discussion
ALS is the third most frequent motor neuron disease worldwide (Boillée, Velde, & Cleveland, Citation2006). In the literature, it is indicated that C9orf72, SOD1, FUS, TARDBP, and UBQLN2 gene mutations are frequently observed in ALS cases. The frequency of these mutations in Turkish population is known to be 4–12% for the SOD1 gene, 2–4% for the UBQLN2 gene, and 3.1–28.30% for the C9orf72 gene (Ozoguz et al., Citation2015; Wroe et al., Citation2008). Although we screened the most probable genomic regions for the ALS, we could not identify any pathogenic mutation in the SOD1 gene and we found a novel mutation in the UBQLN2 gene in only one sporadic ALS. The reason for these results in SOD1 and UBQLN2 genes probably depends on our limited number of cases.
In 20% of familial cases and 2% of sporadic cases, SOD1 gene mutations are observed (Kwon et al., Citation2012). It has been reported in a study conducted in 2009 that the heterozygote c.358–304C >G alteration observed in the fourth exon of SOD1 gene in fALS cases led to the formation of a 43 bp pseudoexon, addition of seven new amino acids, and a subsequent stop codon. This cryptic exon was described to show co-segregation in fALS cases, and believed to be pathogenic (Valdmanis et al., Citation2009).
Starting at this point, the mutation analysis that we performed on the SOD1 gene revealed c.72+133C>T alteration in the first intron of the gene at a rate of 26.6% in six sporadic and two familial cases, which had a European incidence of 6% (dbSNP, Citation2010; https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ss.cgi?subsnp_id=32469335). The alteration was observed to be higher in the Turkish population.
The c.169+41C>A alteration observed in the second intron of the SOD1 gene in one case with fALS was not reported in the literature previously. However, it could not be possible to attribute the presymptomatic ALS phenotype to this variant without any further functional studies.
The c.239+34A>C genomic alteration observed in the third intron of the SOD1 gene in three cases with fALS was previously defined in NCBI. This change was first described as a polymorphism in the study conducted in 1994 and was observed in 7 of 157 fALS cases and 11 of 100 normal controls. They have reported that the A to C transversion of intron 3 leads to the formation of novel sites for Hae II, Hha I, and Hin PI restriction enzymes (Esteban et al., Citation1994). This polymorphism was also reported in a study in 2006 with 8/70 Italian patients with sALS and 11/181 control individuals. They have reported that this intronic alteration has not caused an aberrant transcript formation in the splicing mechanism by RT-PCR analysis (Corrado et al., Citation2006). This alteration was detected in two individuals in two different generations of fALS family I. One of these individuals had died, and the clinical presentation of the other was mild. On the other hand, when these two individuals were compared, the one with mild disease has a heterozygote p.Y97Y (c.291C>T) alteration in the fourth exon of the FUS gene, in addition to the SOD1 gene. The one who passed away was homozygote for this mutation and had heterozygote c.1067-61T>C alteration in the FUS gene, which was observed in three different sporadic cases in this study as well. It is yet not possible to comment on the possible reflections to clinics of this finding which was evaluated as a new intronic SNP by us.
The most common cause of the disease has been described as the increase in GGGGCC (G4C2) hexanucleotide repeats within the first intron of C9orf72 gene, which comprises 45–50% of familial cases and 5–10% of sporadic ALS (Cooper-Knock, Shaw, & Kirby, Citation2014). The pathogenic mechanism caused by the increase in the number of G4C2 repeats has not been clearly understood yet but is believed to cause the loss of C9orf72 protein function and in RNA toxicity (DeJesus-Hernandez et al., Citation2011). The extent of the increased number of repeats may be determined with the Southern blot analysis, which is known to be 20 in healthy individuals and 30 or more in those with the disease (Renton et al., Citation2011). In the study conducted by Renton et al. on the Finnish population (Citation2011), an increased number of hexanucleotide repeats was detected in 113 of 402 patients with ALS (46.4% among 52 fALS cases and 21% among 61 sALS cases), and in 2 of 478 individuals of the control group (0.4%). In this study investigating the genetic structure and new mutations of ALS in the Turkish population, an increased number of hexanucleotide repeats was observed in 11 of 361 cases with sporadic ALS (3.1%) (Ozoguz et al., Citation2015). In our study, no increase was observed in the number of repeat sequences of the C9orf72 gene among cases with fALS, whereas an increased number of hexanucleotide repeats was detected in 2 of 18 cases with sALS (11.1%). Our data support the findings in the previously reported Turkish literature that the most common genetic defect for the development of sALS was the increase in the number of repeats.
In 2006, pathologically ubiquitinated TDP-43 cytoplasmic inclusions were detected in patients with sporadic ALS and sporadic frontotemporal dementia (FTD) (Neumann et al., Citation2006). More than 40 mutations have been defined in the TARDBP gene, mainly composed of missense mutations, at a rate of 5% in cases with fALS and 1% in cases with sALS (Janssens & Van Broeckhoven, Citation2013). In our study, six of the cases with sALS included insertion of guanine nucleotide in the fifth intron of TARDBP gene at a rate of 20%, and this genomic alteration has been defined in NCBI previously. In a study conducted in the Finnish population in 2012, another guanine insertion (rs70977536, c.714 + 68_714 + 69insG) which was observed in the fifth intron of TARDBP gene in 46/50 Finnish ALS cases, indicates that this change is a common polymorphism. Also, in this study, pathogenic mutation in the TARDBP gene was not detected similar to our findings (Mentula et al., Citation2012).
In 2009, in fALS patients with mutations defined in the TARDBP gene, it was known to have structural and functional similarities to the FUS gene (Vance et al., Citation2009). More than 46 mutations were determined in the FUS gene, and among these, 4–6% belonged to fALS and 0.7–1.8% belonged to sALS (Millecamps et al., Citation2010). Up to now, 42 mutations have been defined within exons 3, 5, 6, 14, and 15 of the FUS gene. Individuals bearing a mutation in one of these exons have a typical ALS phenotype and cognitive dysfunction (Kwiatkowski et al., Citation2009). In our study, cognitive dysfunction was detected in one of the three cases with a genomic alteration in exon 3 in sALS.
In sequencing analysis of all 15 exons of the FUS gene in 2012, the previously known p.G49G (c.147C>A) and p.Y97Y (c.291C>T) synonymous variants were observed two fALS and it was shown that these single nucleotide polymorphisms do not affect the splicing mechanism by bioinformatic analysis (Zou et al., Citation2012). In the present study, in 16 of 18 sporadic cases and 9 of 12 familial cases, p.Y97Y (c.291C>T) synonymous variant was detected in the fourth exon of the FUS gene, which shows the polymorphic nature of this gene, which is also consistent with the findings in the literature. In family I, nine cases were screened and whilst five different variants were described, there was only one consistent variation (p.Y97Y, c.291C>T) in FUS gene in all affected family members (III-1; III-5; III-8, and IV-5). But this change, which was also found in an unaffected family member (IV-6), was also frequently seen in other fALS and sALS cases (25/30). Although there is no SNP data for the Turkish population in the literature, this situation might explain that the variation is not a susceptibility factor for ALS.
Additionally, the p.Y97Y (c.291C>T) genomic alteration observed in the fourth exon was also observed in the two sporadic cases with increased hexanucleotide repeats in the C9orf72 gene, which is the most common genetic defect observed in ALS. This suggests that this alteration is not a pathological variant; however, the clinical severity of the disease, age of cases, and region of involvement should also be considered.
ALS-related mutations in the Ubiquilin2 gene, which is localized on the X chromosome, are located in the region coding for the proline-rich PXX domain (Deng et al., Citation2011). In the study by Ozoguz et al., which was conducted on the Turkish population, genomic alterations were observed in the Ubiquilin2 gene in 116 cases with familial ALS at a rate of 2.4%, and in 361 cases with sporadic ALS at a rate of 0.6% (Ozoguz et al., Citation2015). In our study, the p.G457H (c.1371G>C) heterozygote alteration that led to the transformation of glycine in the 457th position into histidine was observed in only one case of sALS. In vivo studies are needed to demonstrate the functional importance of this amino acid change.
In conclusion, the increased number of hexanucleotide repeats detected in the C9orf72 gene in 2 of 30 cases with ALS supports the finding that this genomic alteration is the most important genetic defect for the occurrence of ALS.
Furthermore, oligogenic ALS cases that include more than one ALS-related variants have been reported (Van Blitterswijk et al., Citation2012). Genomic alterations were observed in 2 genes in 11 cases, in 3 genes in 5 cases, and in 5 genes in 1 case in our study. Our results might support the role of epistasis in ALS, which is caused by the interactions among multiple genes. On the other hand, since six of nine different genomic changes observed in this study were intronic variants, those are not very informative. It could be possible to evaluate these changes as benign variants but much more work and larger cohorts need to be done.
Declaration of interest statement
The authors report no declarations of interest.
Supplemental Material
Download Zip (1.5 MB)Supplementary material
Supplementary material for this article can be accessed here.
Additional information
Funding
Notes on contributors
Ciftci Vildan
Ciftci Vildan MSc., Biologist, PhD student. She studies on genetics of ALS in Turkish population.
Darbas Sule
Darbas Sule MSc., Biologist, PhD student. She has been working in Tissue Typing Laboratory and on ALS.
Bilgen Turker
Bilgen Turker Assoc. Prof. Dr. His doctoral thesis was over cancer biology and drug resistance. He also studied ß-thalassemia mutation screening in Turkish population.
Uysal Hilmi
Uysal Hilmi MD, Neurologist, Professor. He investigated monitoring of swallowing and respiration in ALS patients, electrophysiological findings of obstetric brachial plexus lesions and axonal excitability.
Karauzum Berker Sibel
Karauzum Berker Sibel Professor Dr. She is working in Genetic Diseases Diagnosis Center Laboratory. She is supervisor of Vildan Ciftci and she is coordinated projects on ALS and FSHD.
References
- Boillée, S., Velde, C. V., & Cleveland, D. W. (2006). ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron, 52, 39–10. doi:10.1016/j.neuron.2006.09.018
- Brooks, B. R. (1994). Escorial world federation of neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Journal of the Neurological Sciences, 124, 96–107. doi:10.1016/0022-510X(94)90191-0
- Cooper-Knock, J., Shaw, P. J., & Kirby, J. (2014). The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathologica, 127, 333–345. doi:10.1007/s00401-014-1251-9
- Corrado, L., D’alfonso, S., Bergamaschi, L., Testa, L., Leone, M., Nasuelli, N., … Mazzini, L. (2006). SOD1 gene mutations in Italian patients with sporadic Amyotrophic Lateral Sclerosis (ALS). Neuromuscular Disorders, 16, 800–804. doi:10.1016/j.nmd.2006.07.004
- dbSNP. (2010). “dbSNP Short Genetic Variations” NCBI. Retrived from https://www.ncbi.nlm.nih.gov/projects/SNP/snp_ss.cgi?subsnp_id=32469335
- DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., … Kouri, N. (2011, October 20). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron, 72, 245–256.
- Deng, H.-X., Chen, W., Hong, S.-T., Boycott, K. M., Gorrie, G. H., Siddique, N., … Siddique, T. (2011). Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature, 477, 211–215. doi:10.1038/nature10353
- Esteban, J., Rosen, D. R., Bowling, A. C., Sapp, P., McKenna-Yasek, D., O’Regan, J. P., … Brown, R. H., Jr. (1994). Identification of two novel mutations and a new polymorphism in the gene for Cu/Zn superoxide dismutase in patients with amyotrophic lateral sclerosis. Human Molecular Genetics, 3, 997–998.
- Janssens, J., & Van Broeckhoven, C. (2013). Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD–ALS spectrum disorders. Human Molecular Genetics, 22, R77–R87. doi:10.1093/hmg/ddt349
- Kwiatkowski, T. J., Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., … Valdmanis, P. (2009, February 27). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science, 323, 1205–1208.
- Kwon, M. J., Baek, W., Ki, C. S., Kim, H. Y., Koh, S. H., Kim, J. W., & Kim, S.H. (2012, May). Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiology of Aging, 33(1017), e1017–1023. doi:10.1016/j.neurobiolaging.2011.12.003
- Lahut, S., Omur, Ö., Uyan, O., Agım, Z. S., Ozoguz, A., Parman, Y., … Veitia, R. A. (2012). ATXN2 and its neighbouring gene SH2B3 are associated with increased ALS risk in the Turkish population. PloS one, 7, e42956. doi:10.1371/journal.pone.0042956
- Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., … Komure, O. (2010, May 13). Mutations of optineurin in amyotrophic lateral sclerosis. Nature, 465, 223–226.
- Mentula, H.-K., Tuovinen, L., Penttilä, S., Suominen, T., Udd, B., & Palmio, J. (2012). TARDBP mutations are not a frequent cause of ALS in finnish patients. Acta Myologica, 31, 134.
- Millecamps, S., Salachas, F., Cazeneuve, C., Gordon, P., Bricka, B., Camuzat, A., … Meininger, V. (2010, Aug). SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: Genotype-phenotype correlations. Journal of Medical Genetics, 47, 554–560. doi:10.1136/jmg.2010.077180
- Miller, S., Dykes, D., & Polesky, H. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16, 1215. doi:10.1093/nar/16.3.1215
- Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., … Lee, V. M.-Y. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science, 314, 130–133. doi:10.1126/science.1134108
- Ozoguz, A., Uyan, O., Birdal, G., Iskender, C., Kartal, E., Lahut, S., … Başak, A. N. (2015, Apr). The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiology of Aging, 36, 1764 e1769–1764 e1718. doi:10.1016/j.neurobiolaging.2014.12.032
- Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., … Kalimo, H. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron, 72, 257–268. doi:10.1016/j.neuron.2011.09.010
- Robberecht, W., & Philips, T. (2013, Apr). The changing scene of amyotrophic lateral sclerosis. Nature Reviews. Neuroscience, 14, 248–264. doi:10.1038/nrn3430
- Rowland, L. P., & Shneider, N. A. (2001). Amyotrophic lateral sclerosis. New England Journal of Medicine., 344, 1688–1700. doi:10.1056/NEJM200105313442207
- Uyan, O., Omur, Ö., Agım, Z. S., Ozoguz, A., Li, H., Parman, Y., … Cookson, M. R. (2013). Genome-wide copy number variation in sporadic amyotrophic lateral sclerosis in the Turkish population: Deletion of EPHA3 is a possible protective factor. PloS One, 8, e72381. doi:10.1371/journal.pone.0072381
- Valdmanis, P. N., Belzil, V. V., Lee, J., Dion, P. A., St-Onge, J., Hince, P., … Rouleau, G. A. (2009). A mutation that creates a pseudoexon in SOD1 causes familial ALS. Annals of Human Genetics, 73, 652–657.
- Van Blitterswijk, M., Van, E., Michael, A., Hennekam, E. A., Dooijes, D., Van Rheenen, W., … De Visser, M. (2012). Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Human Molecular Genetics, 21(17), 3776–3784. doi:10.1093/hmg/dds199
- Vance, C., Rogelj, B., Hortobágyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., … Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science, 323(5918), 1208–1211. doi:10.1126/science.1165942
- Wroe, R., Wai-Ling Butler, A., Andersen, P. M., Powell, J. F., & Al-Chalabi, A. (2008). ALSOD: The amyotrophic lateral sclerosis online database. Amyotrophic Lateral Sclerosis, 9, 249–250. doi:10.1080/17482960802146106
- Zou, Z. ‐. Y., Peng, Y., Feng, X. ‐. H., Wang, X. ‐. N., Sun, Q., Liu, M. ‐. S., … Cui, L. ‐. Y. (2012). Screening of the FUS gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. European Journal of Neurology : the Official Journal of the European Federation of Neurological Societies, 19(7), 977–983. doi:10.1111/j.1468-1331.2012.03662.x