
Abstract
A 61-year-old man with no previous record of autoimmune disease developed fever, polyarthralgia, purpura, and urticaria-like rash 2 weeks after the first dose of the Moderna mRNA-1273 vaccine, and symptoms deteriorated following the second dose. He presented reduced erythrocyte and platelet counts, hyperferritinemia, high sIL-2R levels, and severe hypocomplementemia. We diagnosed hypocomplementemic urticarial vasculitis (HUVS), and his symptoms as well as laboratory findings improved following treatment with mPSL 1000 mg/day for 3 days and PSL 40 mg/day. Twelve weeks following treatment initiation, the patient relapsed with fever, sore throat, pancytopenia, and hyperferritinemia when the PSL dose was reduced to 12.5 mg/day. Bone marrow biopsy and MRI presented fatty marrow and hemophagocytosis. The patient’s blood cells started recovering using ATG + CsA + EPAG therapy for hemophagocytic lymphohistiocytosis (HLH). This is the first case report of HUVS and HLH following SARS-CoV-2 mRNA vaccination. It is presumed that SARS-CoV-2 mRNA vaccine can induce the excessive production of certain types of cytokines, such as TNF-α, IL-1, IL-4, IL-5, IL-6, and IL-17 as a consequence of IL-6 Amplification (IL-6 Amp). SARS-CoV-2 mRNA-vaccines can cause disruption of immune homeostasis in healthy individuals. An extremely rare disease of HUVS complicated by HLH can be developed as a consequence.
1. Introduction
Immune-related manifestations are increasingly recognized conditions in patients with COVID-19 [1]. Although the inflammation caused by SARS-CoV-2 infection predominantly occurs in the respiratory system, some patients can develop an abnormal inflammatory reaction involving extrapulmonary tissues. The signs and symptoms associated with this excessive immune response are very diverse and can resemble some autoimmune or inflammatory diseases [Citation1].
On the other hand, whether SARS-CoV-2 vaccine can cause an abnormal inflammatory reaction has been unclear. SARS-CoV-2 vaccine-induced activation of immune response and its subsequent association with the development of autoimmune disease is beginning to be the focus of much attention. For example, Tang et al. reported a case of hemophagocytic lymphohistiocytosis (HLH) following SARS-CoV-2 mRNA vaccination [Citation2]. Dash et al. reported a case of normocomplementemic urticarial vasculitis syndrome following SARS-CoV-2 mRNA vaccination [Citation3]. We have experienced a case of hypocomplementemic urticarial vasculitis syndrome (HUVS) case with HLH following SARS-CoV-2 mRNA vaccination. We performed a comprehensive measurement of cytokines and chemokines using the patient’s serum and conducted a detailed study. As far as we know, this is the first case report of HUVS with HLH following SARS-CoV-2 mRNA vaccination.
2. Case presentation
A 61-year-old man with no previous record of autoimmune disease was admitted to our hospital with fever, polyarthralgia, purpura on both lower legs, and urticaria-like rash on both upper arms that had persisted for more than 2 months following his COVID-19 vaccination. We have obtained signed consent for publication from the patient.
The patient received the first dose of mRNA-1273 vaccine on 2 July X. Two weeks following the vaccination, low fever, arthralgia in both shoulders and knees, maculopapular purpura spread on both lower legs, and leg edema appeared. These symptoms deteriorated following the second vaccination of mRNA-1273 on 30 July. As the symptoms of fever, polyarthralgia, purpura on both lower legs, and urticaria-like rash on both upper arms persisted, the patient was admitted to our hospital on 23 September X.
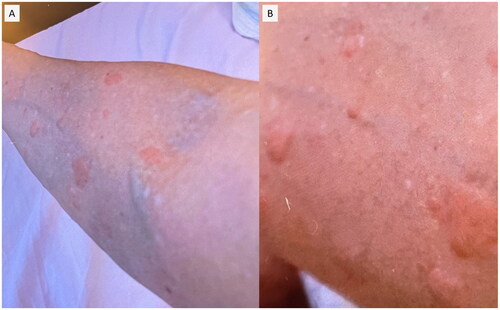
The patient had no autoimmune disease record, nor his blood relatives have previously any autoimmune disease. The patient had been receiving Lansoprazole 15 mg/day and Teprenone 1.5 g/day for duodenal ulcer before the onset of the disease, and his family doctor had prescribed him bepotastine besilate 20 mg/day for urticaria-like skin rash. The patient had a record of smoking 15 cigarettes/day and had no history of alcohol consumption, asthma, or allergies to any drugs or foods. During admission, the patient presented tachycardia (132/min) and tachypnea (26/min), but no other abnormal vital signs were noted. Scattered purpura was observed on both lower extremities; and was not resolved by acupressure, some of which had faded away leaving hyperpigmentation (). On both upper extremities, eruptions of 0.5–2 cm were noted that faded within 12–24 h (). No other findings of note were observed except for the skin rash on the extremities.
Figure 1. Scattered purpura was observed on both lower extremities; and was not resolved by acupressure, some of which had faded away leaving hyperpigmentation.
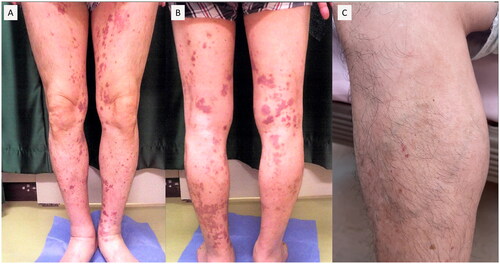
Figure 2. On both upper extremities, eruptions of 0.5–2 cm were noted that faded within 12–24 h.
Laboratory tests were performed on admission (); blood counts demonstrated reduced erythrocytes (erythrocyte 2.53 million/μL), hemoglobin (hemoglobin 7.7 g/dL), platelets (platelet 41,000/μL), as well as elevated eosinophils (eosinophil count 635/μL). Coagulation and microbiological tests did not reveal any abnormality while biochemical tests revealed reduced serum albumin (albumin 2.5 g/dL) and hyperferritinemia (ferritin 1045 μg/L), but no defects were observed in liver or renal function. Immunological examination demonstrated slightly increased C-reactive protein (C-reactive protein 0.99 mg/dL), high soluble IL-2 receptor (sIL-2R 3386 U/mL), high IgG4 (IgG4 753 mg/dL), and high IgE (IgE 1340 IU/mL). In addition, the complement system was considerably decreased; CH50 was below sensitivity, C3 22 mg/dL and C4 1.5 mg/dL. Antinuclear antibody test was found to be negative, anti-DNA antibody 12.0 IU/mL, C-ANCA and P-ANCA were below sensitivity, but rheumatoid factor was elevated (rheumatoid factor 281 IU/mL) and anti-CCP antibody was slightly increased (anti-CCP antibody 5.7 U/mL). Urinalysis revealed elevated urinary β2MG (urinary β2MG 1672 μg/L) and urinary NAG (urinary NAG 27.1 U/L). A CT scan of the head to pelvic region demonstrated enlarged lymph nodes in the right axilla, mediastinum, and bilateral external iliac regions, but no other findings of note. Bone marrow evaluation presented a considerable decrease in megakaryocytes (2/smear) and erythroblasts (0/smear) as well as a significant increase in mature eosinophils (39.6%). Skin biopsy of the rash area demonstrated noticeable infiltration of lymphocytes and eosinophils around small blood vessels in the dermis.
Table 1. Laboratory tests performed on first admission.
Following admission to our hospital, the patient was treated with methylprednisolone 1000 mg/day intravenously for 3 days as was suspected for HUVS, followed by oral prednisolone (PSL) 40 mg/day. The patient was discharged from the hospital on Day 26, as both symptoms and laboratory results were improving.
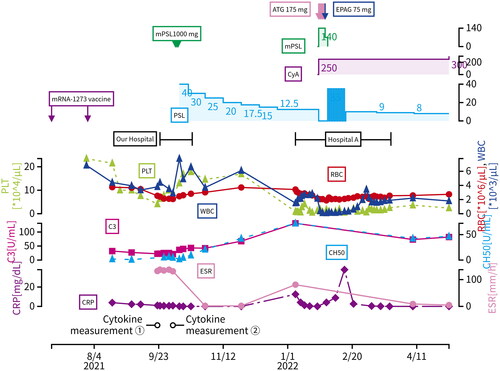
On 7 January, X + 1 (80 days after discharge), when the PSL dose was reduced to 12.5 mg/day, the patient presented to our emergency department with fever, sore throat, and scattered bleeding spots on the extremities. Blood tests revealed leukopenia (absolute neutrophil count 187/μL), erythrocytopenia (erythrocyte count 3.42 million/μL), thrombocytopenia (platelet count 9000/μL), so he was admitted in the same day to Hospital A, which has a hematology department. MRI revealed that his lumbar bone marrow turned into fatty marrow (), while his bone marrow puncture demonstrated multiple hemophagocytosis (more than 10 phagocytes/smear), leading to Grade IV bone marrow failure on Day 24 from the second admission with an absolute neutrophil number of 238 cells/μL, platelet count of 13,000/μL and reticulocyte count of 13,500/μL. Based on the above findings, the patient was diagnosed as aplastic anemia for the time being without close scrutiny because he was required immediate therapeutic intervention. According to the guidelines, anti-thymocyte globulin (ATG) combined with cyclosporin (CyA) and eltrombobag (EPAG) therapy has been prescribed to the patient to treat suspected of aplastic anemia, which was later found out to be rather HLH. The patient’s blood cells started to recover and he was discharged on Day 74 from the second admission. He has been then treated with prednisolone 8 mg/day and cyclosporine 250 mg/day. Nevertheless, even 18 weeks following discharge from Hospital A, patient’s blood cell numbers had not recovered to the levels before the disease onset ().
Figure 3. MRI T1 (18 January, X + 1) weighted image shows the patient’s lumbar spine turned into fatty bone marrow.
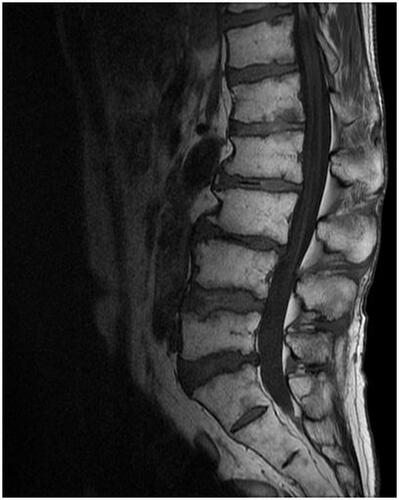
Figure 4. Clinical course.
3. Discussion
3.1. Differential diagnosis
We cited systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), IgG4-releated disease (IgG4-RD), eosinophilic granulomatosis with polyangiitis (EGPA), cryoglobulinemic vasculitis, HUVS, and HLH as differential diagnosis cases.
We suspected SLE because of fever, arthralgia, pancytopenia, and hypocomplementemia on admission. However, this patient did not have a clinical history of malar rash or photosensitivity. In addition, all of anti-nuclear antibody, anti-dsDNA antibody, and anti-Sm antibody were negative. These findings lowered the possibility of SLE as diagnosis.
We also suspected RA, especially Felty syndrome for fever, arthralgia, pancytopenia, and hypocomplementemia. The fact that both rheumatoid factor (RF) and anti-citrullinated cyclic peptide antibody (ACPA) were positive reminds us of RA. However, the titer of ACPA was too low (5.7 U/ml) to implicate RA. In addition, arthrography did not show the findings of arthritis. Also, RA could not explain the proliferation of eosinophils or elevated IgG4.
Another differential diagnosis was IgG4-RD based on the fact that this patient demonstrated enlarged lymph nodes, elevated IgG4 titer (753 mg/dL), increased β2-microgroblin (1672 μg/L), and increased N-Acetyl-β-d-glucosaminidase (NAG) (27.1 U/L) on urinalysis. Otherwise, a CT scan did not show any enlarged organ, mass, or hypertrophic lesion. IgG4-RD typically does not demonstrate fever, arthralgia, or elevated C-reactive protein, which was inconsistent with this case. No one has reported IgG4-RD with pancytopenia or hypocomplementemia.
EGPA was also suspected for the proliferation of eosinophils in serum and the infiltration of eosinophils at the perivascular site. Elevated IgE, IgG4, and CRP and increased erythrocyte sedimentation rate were supporting findings. However, anemia, thrombocytopenia, and negative MPO-ANCA were not consistent with EGPA.
Although increased erythrocyte sedimentation, elevated CRP, positive RF, and hypocomplementemia suggested cryoglobulinemic vasculitis, cryoglobulin was not detected. Also, cryoglobulinemic vasculitis could not explain the proliferation of eosinophils or elevated IgG4.
Another differential diagnosis is a hypereosinophilic syndrome (HES) masked by aplastic anemia based on the fact that eosinophilia was elevated and skin biopsy showed the infiltration of eosinophil on the first admission. According to the 1975 diagnostic criteria of Chusid et al. HES is diagnosed when peripheral blood eosinophils are increased (>1500/µl) for more than 6 months, parasitic infections, allergies, and other diseases are excluded and organ damage (heart, lung, CNS, skin) due to eosinophil infiltration is present. However, eosinophils on the first admission, in this case, was 635/μL. Moreover, HES complicated by aplastic anemia cannot explain severe hypocompementaemia in this case.
Chronic urticaria accompanied by hypocomplementemia implicated HUVS. HUVS was able to explain arthralgia, proliferation of eosinophils, and elevated IgE and IgG4 titer. Anti-C1q antibody level was not clear for anti-C1q antibody test was not available in Japan. Skin biopsy revealed significant infiltration of lymphocytes and eosinophils, which is consistent with HUVS. Otherwise, no one has reported HUVS accompanied by pancytopenia.
This case satisfied seven out of eight diagnostic criteria for secondary HLH (2004). However, HLH is not usually related to hypocomplementemia or proliferation of eosinophils. For those reasons, any of differential diagnosis case was not able to explain this case. Thus, we considered this case as HUVS complicated by HLH at last.
3.2. Definitive diagnosis
Based on the above findings, HLH was suspected based on pancytopenia and hemophagocytosis on bone marrow puncture. This case satisfied 7 out of 8 diagnostic criteria for secondary HLH (2004); the patient presented fever (maximum temperature 39.0 °C), splenomegaly (maximum diameter 11 cm), anemia (hemoglobin 7.7 g/dL, nadir 6.3 g/dL), thrombocytopenia (platelets 41,000/µL, nadir 0.4 million/µL), neutropenia (absolute number of neutrophils <90/µL), higher than 10 hemophagocytes per smear by bone marrow puncture, which was not revealed on the first admission, hyperferritinemia (ferritin 1045 μg/L), increased sIL-2R (sIL-2R 3386 U/mL), and hypertriglyceridemia (Triglycerides 266 mg/dL). The HScore was 160, 40–54% probability of HLH, and the case was consistent with HLH.
Urticarial vasculitis (UV) was then suspected due to severe hypocomplementemia and urticarial rash, which is extremely rare disease; morbidity rate is 9.5 per million [Citation4]. Urticaria-like skin lesions noticed in UV differed from urticaria as they occur as a chronic or recurrent course, with limited individual lesions (0.5–5 cm in diameter), lasting for more than 24 h, and leaving a dark red pigmentation [Citation5]. The urticarial rash observed in the present case was 0.5–2 cm in size and faded away leaving pigmentation, something that is consistent with the characteristics of the urticarial rash observed in UV patients. The urticaria-like rash observed in this patient faded within 12 to 24 h, which is not characteristic of the skin rash seen in UV, but it could result from bepotastine administration. UV patients are reported to present arthralgia, myalgia observed in most of these patients [Citation6], renal involvement is observed in 20% of them [Citation7], COPD was observed in 20 to 30% [Citation8], gastrointestinal symptoms in up to 30% [Citation9], as well as other systemic symptoms. This case also presented arthralgia, myalgia, and tubular disorders. Hypocomplementemia is a useful predictor [Citation8], while increased erythrocyte sedimentation and hypocomplementemia are the most common laboratory aberrations in UV patients. In the case described in the present study, the patient presented an increased erythrocyte sedimentation rate of 131 mm/h and acute hypocomplementemia; CH50 was below sensitivity measurement, C3 22 mg/dL, C4 1.5 mg/dL and complement transition reflected the disease status efficiently (). UV without hypocomplementemia is defined as normocomplementemic urticarial vasculitis (NUVS), while UV with hypocomplementemia is defined as HUVS, which is the more critical type [Citation9]. Despite the fact that anti-C1q antibodies could not be examined in the case presented herein due to technical difficulties. Autoantibodies, such as antinuclear antibodies are detected in 50% of HUVS patients [Citation10]. In the case presented herein, rheumatoid factor and anti-CCP antibodies were detected that were consistent with the characteristics of HUVS. In addition, Antinuclear antibodies were negative on admission to our hospital, although they were positive in the previous test at Hospital A (anti-nuclear antibodies titer 160).
Pathologic features of leukocytoclastic vasculitis are essential for UV diagnosis [Citation11]; a cellular infiltrate composed of neutrophils, eosinophils, and lymphocytes is noticed in the walls of small vessels and perivascular areas [Citation12]. The predominance of neutrophils and eosinophils changes to lymphocytes with time [Citation13]. It is also suggested that eosinophils may play a role in the pathogenesis of HUVS because the infiltration of these cells is significantly greater than that seen in urticaria [Citation14]. Skin biopsy of the rash in the case presented demonstrated significant infiltration of lymphocytes and eosinophils surrounding small blood vessels, suggesting that this pathology is similar to the pathology of UV patients after a short period since the disease onset. The patient’s blood counts showed a recovery trend after mPSL 1000 mg for three days following PSL 40 mg, but pancytopenia developed severely during PSL reduction 80 days following discharge from the hospital. Nevertheless, 18 weeks following discharge, the patient’s blood cell counts had not recovered to the levels before the onset of the disease.
HUVS could be treated using oral antimicrobials, colchicine, hydroxychloroquine, methotrexate, mycophenolate mofetil, azathioprine, cyclosporine, prednisone, intravenous immunoglobulin, rituximab, omalizumab, interleukin-1 inhibitors, but many cases experience relapse if treatment is not continued [Citation15]. In the case presented in the current study, mPSL 1000 mg/day and PSL 40 mg/day stabilized HUVS disease temporarily, but the disease relapsed 80 days following discharge when PSL dose was reduced to 12.5 mg/day. Treatment intensity at the time of initial admission was possibly inadequate while the rate of PSL dose reduction was possibly too fast.
It is well-known that HLH symptoms progress regarding their severity as the underlying disease progresses and bone marrow becomes markedly hypoplastic [Citation16]. In the case presented herein, the systemic bone marrow was fatty myelinated at the time of relapse with pancytopenia, indicating that PSL alone was insufficient to control the HLH disease course or disruption of immune homeostasis that may have underly HLH. An increased cyclosporine dose or the initiation of mycophenolate mofetil or azathioprine would be considered if the patient presented signs of relapse during PSL reduction.
Based on all of the above, the variety of symptoms and laboratory test results observed in this case were in accordance with HLH and HUVS and they could not be explained by either HLH or HUVS alone. This case was thus considered to be a combination of HLH and HUVS.
3.3. COVID-19 mRNA vaccine
The development of various immune system associated disorders linked with COVID-19 mRNA vaccine has been previously reported [Citation17]. Based on the fact that the patient did not have a record of autoimmune disease, he presented fever and other systemic symptoms 2 weeks after the first dose of COVID-19 mRNA vaccine, and the systemic symptoms deteriorated immediately after the second dose of COVID-19 vaccine, the COVID-19 mRNA vaccine presumably induced the development of HLH and HUVS in the patient.
The related UV pathogenesis factors include drugs, COVID-19 [Citation18], influenza [Citation19], SLE [Citation20], and IgG-4 related diseases [Citation21]. The patient did not initiate any medication therapy within 6 months from the disease onset and had no history of COVID-19 or influenza infection, no pre-existing or new autoimmune diseases, such as SLE or IgG-4-related disease. The patient received the initial dose of mRNA-1273 vaccine 2 weeks before the disease onset. The mRNA-1273 vaccine was presumed to be effectively implicated in the development of HUVS for the patient as skin rash, arthralgia, and fever symptoms were exacerbated directly after the second dose of mRNA-1273 vaccine. To the best of our knowledge, this is the first report of HUVS after COVID-19 vaccination, despite the fact that a case of NUVS after COVID-19 vaccination has been previously reported [Citation22].
HLH pathogenesis that is caused by an underlying disease can be broadly divided into primary and secondary. Primary HLH occurs in children, and familial hemophagocytic lymphohistiocytosis is an established example. In the case presented herein, no family history of HLH was reported, while the disease onset occurred at a relatively older age (60 years old). Primary HLH could thus be ruled out, although genetic testing was not conducted. The majority of adult-onset HLH is secondary and is triggered by underlying diseases, such as infections, malignancies, autoimmune diseases, or drugs, despite the fact that Wu et al. reported two HLH cases after COVID-19 vaccination [Citation23]. Compared to the previous report (), Wu et al. reported HLH after Pfizer-BioNTech vaccination in both cases, while the present case occurred following Moderna mRNA-1273 vaccination. The time from the first vaccination to symptoms occurrence was 6 and 52 days, respectively in the previous cases, while in the case presented herein was 13 days. As the above cases met the HLH 2004 criteria, it was suggested that the COVID-19 mRNA vaccine was also the underlying cause of HLH in the present case.
Table 2. Comparison of the previous report [Citation22] with the present case.
3.4. Cytokine storm syndrome caused by IL-6 amplification
It has been previously demonstrated that various immune-related diseases develop or deteriorate following vaccination with the COVID-19 mRNA vaccine, including the cytokine storm syndrome (CSS) that has been proposed to be caused by the disruption of immunological homeostasis triggered by the vaccine. HUVS and HLH observed in the case presented herein were considered to be a manifestation of CSS caused by the COVID-19 mRNA vaccine.
In the case presented herein, cytokine measurements were thoroughly conducted using serum on 2 days before (first measurement) and 10 days after (second measurement) admission (). Both cytokine measurements were conducted prior to the initiation of immunosuppressive therapy, excluding bepotastine besilate, at the period of greatest disease activity during the first hospitalization ().
Table 3. Cytokine measurements thoroughly conducted using serum 2 days before and 10 days after admission.
The first measurement was conducted using SRL kits. The second measurement was conducted by following the next steps. Serum samples were centrifuged at 3000 × g for 5 min, and the supernatants were collected and stored at −80 °C for a maximum of 90 days before analysis. A blinded multiplex cytokine bead assay was performed in parallel using the MILLIPLEX® MAP Human Cytokine/Chemokine Magnetic Bead Panel 1-Premixed 41 Plex (Millipore, Billerica, MA, USA) kits, according to the manufacturers’ instructions. Cytokines that were frequently found to be at non-detectable levels were excluded from the analysis. The control group (n = 118) was recruited from the staff at Nagasaki University and residents of the town of Saza in Nagasaki Prefecture, as previously described [Citation24].
The laboratory results for this study included TNF-α (61.5 IU/mL), IL-1α (38.6 IU/mL), IL 1β (65.9 IU/mL), IL-18 (242.5 IU/mL), IL-10 (60.1 IU/mL), MIP-1a (102.6 IU/mL), MIP-1β (55.6 IU/mL). TNF-α is produced by macrophages, NK cells and T cells. IL-1α and IL-1β are produced by macrophages and epithelial cells. IL-18 is produced by activated macrophages and Kupffer cells. Although we could not measure the activity of macrophages, these facts suggest the existence of activated macrophages. In addition, IL-10 strongly inhibits the activation of macrophages and MIP-1α and MIP-1β target macrophages, which may indirectly suggest the existence of activated macrophages.
A significant increase in IL-4 (18.0 IU/mL in the first measurement) and IL-5 (36.1 IU/mL) was observed. Nevertheless, the second measurement of IL-4 (2.3 IU/mL) did not vary from the median value in healthy subjects. With respect to the patient of the present study, bepotastine was administered the day before the second measurement, indicating that bepotastine might have affected test results. Despite the fact that the effect of bepotastine administration on cytokine activity has not been previously reported, many antihistamine H1 receptor antagonists have been reported to decrease IL-4, IL-5, IL-6, and TNF-α [Citation25]. Since the second IL-4 measurement for the patient of the present case was performed the day after the initiation of bepotastine administration, bepotastine could be possibly involved in the reduction of IL-4 levels.
Moreover, a significant increase in IL-17A (54.0 IU/mL) and IL-17F (32.5 IU/mL) was observed suggesting A significant increase in IL-10 (IL-10 60.1 IU/mL) was also observed, suggesting iTreg cell well activity. The activation of iTreg cells was assumed to be a compensatory response to the abnormal increase in inflammatory cytokines.
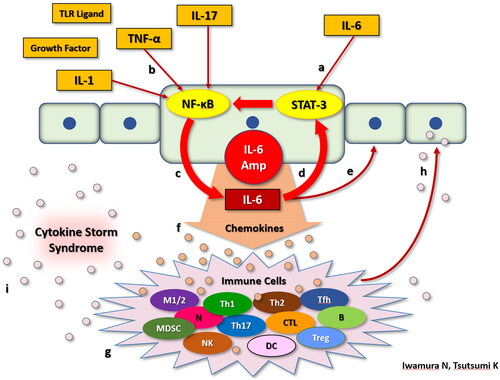
It is presumed that SARS-CoV-2 mRNA vaccine can induce the production of certain types of cytokines, such as TNF-α, IL-1, IL-4, IL-5, IL-6, and IL-17 by some mechanisms. The presence of excessive those cytokines is presumed to be involved in the development of HUVS with HLH. If that is so, we can explain the reason why the immunosuppressive therapy of mPSL, PSL, and CyA was successful in this case. A mechanism known as ‘IL-6 Amp’ has been suggested to mediate the pathogenesis of cytokine storms. IL-6 Amp was assumed to be involved in the case presented.
A group of cytokines secreted by activated CD4+ cells and other cells simultaneously activate the NF-κB and STAT3 pathways in non-immune cells, such as fibroblasts and endothelial cells. This secretion promotes the activation of excessive NF-κB signaling, resulting in the local production of excessive chemokines, growth factors, IL-6, as well as other inflammatory cytokines. This mechanism is known as ‘IL-6 Amp’ and the IL-6 produced by this pathway further activates STAT3, which supports the activation of IL-6 Amp. Various inflammatory cytokines, chemokines, as well as growth factors secreted by this pathway, induce the migration of various inflammatory cells and cytokines secretion. These cytokines stimulate local cell proliferation, and further increase the IL-6 Amp, leading to chronic inflammation and induction of cytokine storm [Citation26]. We demonstrate the detailed mechanism of IL-6 Amp (). It is noticeable that IL-6 Amp can take place under the circumstance of adequate existence of both IL-6 to activate STAT-3 and TNF-α, IL-1 or IL-17 to activate NF-κB. IL-6 levels of the patient presented herein were increased in both the first (11.2 IU/mL) and second (11.59 IU/mL) measurement. In addition, all of TNF-α (61.4 IU/mL), IL-1α (38.6 IU/mL), IL-1β (65.9 IU/mL), IL-17A (54.0 IU/mL), and IL-17F (32.4 IU/mL) were remarkably increased in the second measurement. These facts suggest that IL-6 Amp was presumably involved in the development of cytokine storm in this case.
Figure 5. IL-6 Amp mechanism. IL-6 stimulates an intracellular signaling pathway: STAT-3 (a). While TNF-α, IL-17, IL-1, TLR ligands, and growth factors stimulate another intracellular signaling pathway: NF-κB (b). In non-immunocompetent cells, when STAT-3 and NF-κB are stimulated at the same time, NF-κB becomes activated excessively, which results in excessive production of IL-6 (c). Large amount of IL-6 acts by way of autocline (d) and paracline (e), promoting much more production of IL-6 following the activation of STAT-3. We defined this positive feedback mechanism mediating IL-6 as “IL-6 Amp” in a narrow sense. Non-immunocompetent cells activated by IL-6 Amp product various kinds of chemokines (f). These chemokines stimulate immunocompetent cells, such as macrophages, T cells, and B cells (g). Various kinds of chemokines produced by these immunocompetent cells stimulate STAT-3 and NF-κB in non-immunocompetent cells again (h) which produce much more IL-6 and chemokines as a consequence. We defined this mechanism as “IL-6 Amp” in a broad sense. Thus, IL-6 amp can take place under the circumstance of adequate existence of both IL-6 to activate STAT-3 and TNF-α, IL-1 or IL-17 to activate NF-κB. In the course of IL-6 Amp, tremendous amounts of cytokines and chemokines are produced, which is called “cytokine storm syndrome” (i).
3.5. What we can learn from this case
One of what we can learn from this case is that HLH and HUVS might be triggered by SARS-CoV-2 mRNA vaccines and HUVS can be complicated by HLH. Of course, we cannot prove anything about the causality of SARS-CoV-2 mRNA vaccine and HUVS or HLH, but it is very likely that HLH and HUVS are triggered by SARS-CoV-2 mRNA vaccine based on the fact that the patient, in this case, developed the symptoms of HLH and HUVS immediately following the first vaccination and these symptoms got worse immediately following the second vaccination. No disease as far as we know can uniquely explain all his significant symptoms and laboratory findings. If we were to venture a diagnosis for this case, it would be HUVS complicated by HLH. As no one has reported a case of HUVS complicated by HLH, this case is the first report of HUVS complicated by HLH in that sense. The more important thing is the fact that SARS-CoV-2 mRNA-vaccines can cause disruption of immune homeostasis in healthy individuals. In the measurement of cytokine levels of this case, TNF-α, IL-1, IL-4, IL-5, IL-6, and IL-17 were remarkably elevated as presented. We speculated that those cytokines might be involved in the development of symptoms in this case. Given the fact that HUVS (morbidity rate; 9.5 per million) and HLH (morbidity rate; 1.3 per million) are rare diseases, HUVS complicated by HLH is extremely rare in normal circumstances. It is inferred that SARS-CoV-2 mRNA vaccine made the condition of HUVS complicated by HLH in way of disruption of immune homeostasis, in other words, cytokine storm syndrome. Moreover, we mentioned IL-6 Amp, which we thought to be one of the mechanisms of the development of cytokine storm syndrome in this case based on the results of cytokine measurement.
3.6. Limitations
This study has several limitations. Despite the fact that increased anti-C1q antibody is essential for the diagnosis of HUVS, C1q of the patient presented herein could not be measured using a commercial assay in Japan due to technical difficulties.
We can measure cytokines in serum only twice, but we have not seen changes in cytokines over time, because patient’s serum only 2 days before and 10 days after admission are available. Since the effect of oral medications, such as bepotastine cannot be overlooked and the effect of bepotastin on cytokines is not fully identified, interpretation of the observed findings should be performed with caution.
Currently, 69.4% of the world population has received at least one dose of SARS-CoV-2 vaccine as of 14 September, 2022. Background factors are known to be present in patients that are prone to present cytokine storm following SARS-CoV-2 vaccination. In addition, the reason why cytokine storms caused HUVS, which is extremely rare clinical manifestation, in this case study with no history of autoimmune diseases or allergies is also unknown. We believe that further case accumulation is required.
4. Conclusion
In the present study, we report a case of HUVS and HLH in a 61-year-old man with no history of autoimmune disease following vaccination with SARS-CoV-2 vaccine (mRNA-1273). HLH and HUVS might be triggered by SARS-CoV-2 mRNA vaccines and HUVS can be complicated by HLH. SARS-CoV-2 mRNA-vaccines can cause disruption of immune homeostasis in healthy individuals. Elevated TNF-α, IL-1, IL-4, IL-5, IL-6, and IL-17 might be involved in the development of HUVS complicated by HLH. It is inferred that SARS-CoV-2 mRNA vaccine made the condition of HUVS complicated by HLH possible in way of disruption of immune homeostasis. IL-6 Amp is thought to be one of the mechanisms of the development of cytokine storm syndrome in this case.
Authors’ contributions
IN substantially contributed to the study conceptualization, data curation, investigation, visualization, and writing original draft. Dr. EK substantially contributed to the study conceptualization, resources, supervision, and writing review and editing. TK substantially contributed to the investigation and visualization. Dr. KT substantially contributed to the investigation and writing review and editing. AT substantially contributed to resources. Drs. AT, TA, TK, and UY substantially contributed to supervision. All authors critically reviewed and revised the manuscript draft and approved the final version for submission.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Manuel R, Pilar B, Xavier M. Systemin and organ-specific immune-related manifestations of COVID-19. Nat Rev Rheumatol. 2021;15:80–96.
- Tang L, Hu Y. Haemophagocytic lumphohisitiocytosis after COVID-19 vaccination. J Hematol Oncol. 2021;14(1):87.
- Dash S, Behera B, Sethy M, et al. COVID-19 vaccine-induced urticarial vasculitis. Dermatol Ther. 2021;34(5):e15093.
- Sjöwall C, Mandl T, Skattum L, et al. Epidemiology of hypocomplementaemic urticarial vasculitis. Rheumatology. 2018;57(8):1400–1407.
- Wisnieski JJ. Urticarial vasculitis. Curr Opin Rheumatol. 2000;12(1):24–31.
- Koc E, Aksoy B, Taliparmak A. Urtiarial vasculitis [Internet]; 2017 [cited 2020 Jun 17]. Available from: https://www.intechopen.com/books/a-comprehensive-review-of-urticaria-and-angioedema/urticarial-vasculitis
- Boyer A, Gautier N, Comoz F, et al. Nephropathy associated with hypocomplementic urticarial vasculitis: a case report and literature. Nephrol Ther. 2020;16(2):124–135.
- Venzor J, Lee WL, Huston DP. Urticarial vasculitis. Clin Rev Allergy Immunol. 2002;23(2):201–216.
- Buck A, Christensen J, Mccarty M. Hypocomplementic urticarial vacuities syndrome: a case-report and literature review. J Clin Aesthet Dermal. 2012;5(1):36–46.
- Dincy CV, George R, Jacob M, et al. Clinicpathologic profile of normocomplementic and hypocomplementic urticarial vasculitis: a study from South India. J Eur Acad Dermal Venereol. 2020;34(10):e566–e568.
- Kolkhir P, Grakhova M, Bonnekoh H, et al. Treatment of urticarial vasculitis: a systematic review. J Allergy Clin Immunol. 2019;143(2):458–466.
- Zuberbier T, Maurer M. Urticarial vasculitis and schnitzler syndrome. Immunol Allergy Clin North Am. 2014;34(1):141–147.
- Damman J, Mooyaart AL, Seelen MAJ, et al. Dermal C4d deposition and neutrophil alignment along the dermal-epidermal junction as a diagnostic adjunct for hypocomplementic urticarial vasculitis (anti-C1q vasculitis) and underlying systemic disease. Am J Dermatopathol. 2020;42(6):399–406.
- Kamyab K, Ghodsi SZ, Ghanadan A, et al. Eosinophilic infiltration: an under-reported histological findings in urticarial vasculitis. Int J Dermatol. 2019;58(7):825–829.
- Stephanie L, Gu BS, Joseph L, et al. Urticarial vasculitis. Int J Womens Dermatol. 2021;7(3):290–297.
- Kumakura S, Ishikura H, Kondo M, et al. Autoimmune-associated hemophagocytic syndrome. Mod Rheumatol. 2004;14(3):205–215.
- Watad A, De Marco G, Mahajna H, et al. Immune-mediated disease flares or new-onset disease in 27 subjects following mRNA/DNA SARS-CoV-2 vaccination. Vaccines. 2021;9(5):435.
- de Perosanz-Lobo D, Fernandez-Nieto D, Burgos-Blasco P, et al. Urticarial vasculitis in COVID-19 infection: an a-vasculopathy-related symptom? J Eur Acad Dermatol Venereol. 2008;22(7):789–794.
- Gökce S, Dortkardesler BE, Aslan A. Normocomplementic urticarial vasculitis associated with a/H1N1 in a child case report. SN Comps Coin Med. 2020;25:1–3.
- Baigrie D, Bansal P, Crane JS. Leukocytoclastic vasculitis. Treasure Island (FL): StatPearls Publishing; 2020.
- Takao M, Hamada T, Kaji T, et al. Hypocomplementic urticarial vasculitis arising in a patient with immunoglobulin G4-relates disease. Int J Dermatol. 2016;55(4):430–433.
- Dash S, Behera B, Sethy M, et al. COVID-19 vaccine-induced urticarial vasculitis. Dermatol Ther. 2021;34(5):el5093.
- Wu V, Lopez CA, Hines AH, et al. Haemophagocytic lymphohistiocytosis following COVID-19 mRNA vaccination. BMJ Case Rep. 2022;15(3):e247022.
- Koga T, Sumiyoshi R, Furukawa K, et al. Interleukin-18 and fibroblast growth factor 2 in combination is a useful diagnostic biomarker to distinguish adult-onset still’s disease from sepsis. Arthritis Res Ther. 2020;22(1):108.
- Morimoto C. Effect of H1 histamine receptor antagonists on T cell functions. Arerugi. 2003;52(11):1039–1047.
- Murakami K, Houjo S, Tanaka K, et al. Cytokine storm and IL-6 amplifier. Inflam Immun. 2022;30(1):18–26.