Abstract
To clarify the interactions of liposomes with blood cells, this study examined the behaviour of liposomes of a range of compositions in the presence of purified human blood cells in buffer or plasma; or in whole blood, or in mice in vivo. Liposomes, labeled with the hydrophilic fluorochrome, carboxy fluorescein (CF), or with membrane‐sequestering R18 or FITC‐labeled phospholipids, were mixed with blood cells and the appearance of the fluorochromes in the blood cell population was monitored by flow cytometry. Irrespective of composition, with or without poly(ethylene glycol), all types of liposomes were found to interact rapidly and dose‐dependently with red cells, leukocytes and platelets, both in vitro and in vivo. This took place equally in the presence and the absence of plasma proteins and functional enzyme cascades, suggesting that the prime facie interaction is opsonization‐independent and is consistent with liposome‐blood cell fusion.
Introduction
Liposomes have long been used as drug delivery systems and as a means of reducing the toxicity of the drugs incorporated within them. The fact that they can be manufactured from totally synthetic lipids (Barenholz and Amselem, [Citation1984]) and can be stored easily (Crowe and Crowe, [Citation1988]) contributed to their appeal. However, while the liposomes' composition appears to have a major impact on their behaviour in vitro, where liposomes are made and tested, other factors predominate in vivo where liposomes interact with blood and tissue components (Levchenko et al., [Citation2002]; Scherphof and Kamps, [Citation2001]). Although some formulations appear to last longer than others in the circulation there is not one overarching factor that is responsible for improvement of circulatory residence. Many studies have addressed various liposome aspects such as size (Hsu and Juliano, [Citation1982]), net charge (Loughrey et al., [Citation1990]) cholesterol content & lipid composition (Gabizon and Papahadjopoulos, [Citation1988]; Gabizon et al., [Citation1990]) phospholipid chain length and saturation (Reinish et al., [Citation1988]) and surface grafted poly(ethylene glycol) (PEG) (Vermehren et al., [Citation1999]; Woodle, [Citation1993]; Zalipsky et al., [Citation1995] etc) and have shown incremental, subtle improvements. Still, residence time in the circulation is less than desired in most cases: the liposomes disappear quite rapidly and liposome‐associated lipid or aqueous radiolabel subsequently reappear predominantly in the liver and the spleen (Allen, [Citation1998]; Hsu and Juliano, [Citation1982]; Litzinger et al., [Citation1994]; Torchilin et al., [Citation1996]). This fast removal of liposomes ostensibly by the reticuloendothelial system (RES) creates clinical opportunities because entrapment in capillary beds allows passive targeting of drugs to the liver (Koning et al., [Citation2001]), spleen (Laverman et al., [Citation2000a]), tumours (Poste, [Citation1983]; Storm et al., [Citation1984]), and sites of infection (Andreopoulos and Kasi, [Citation1997]) and inflammation (Yu et al., [Citation1999]). Active targeting of liposomes is still elusive because the half‐life of even ”long circulating” liposomes is such that they are cleared from the circulation before their targeting molecules can deliver them effectively.
Liposomes carrying surface grafted PEG are thought to be able to elude the RES (Allen, [Citation1998]; Klibanov et al., [Citation1990]; Woodle, [Citation1993]; Zalipsky et al., [Citation1995]) due to the ability of the PEG to reject plasma proteins (Bergstrom et al., [Citation1992]; Blume and Cevc, [Citation1993]; Chonn and Cullis, [Citation1995]; Lee et al., [Citation1997]). However, this filtering ability is defined for high concentrations (brush regime) (Kenworthy et al., [Citation1995]; Needham et al., [Citation1997]; Noppl‐Simpson and Needham, [Citation1996]) of long‐chain PEGs on stable surfaces (Brooks and Muller, [Citation1996]; Chaikof et al., [Citation1992]). A wealth of biological evidence is accumulating that shows that PEG‐bearing liposomes or lipid complexes can sequester in the same manner as PEG‐less liposomes (Bulte et al., [Citation1999]; Laverman et al., [Citation2000a]; Laverman et al., [Citation2001a], Citation[b]) and come into intimate contact with a variety of cell membranes and induce fusion (Chams et al., [Citation1999]; Higashi et al., [Citation1996]). Whether the resulting membrane fusion is accomplished by lipid monomer transfer (Silvius and Zuckerman, [Citation1993]), local destabilization (Regen et al., [Citation1989]) and dehydration by PEG (Silvander et al., [Citation1998]) and consequent lateral movement of the phospholipids to which the PEG are tethered creating PEG lateral pressures that can make the phospholipid bilayer unstable (Evans and Rawicz, [Citation1997]), or membrane curvature effects (Talbot et al., [Citation1997]; Yang et al., [Citation1997]), is still the subject of intense discussion (Lee and Lentz, [Citation1997]; Papahadjopoulos et al., [Citation1990]).
A single type of liposome formulation can interact with different cell types to different degrees (McLean et al., [Citation1997]), therefore it is safe to assume that different formulations vary in their extent of interactions with the same cell type (Dalencon et al., [Citation1996]; Okumura et al., [Citation1994]). Such varied interaction may explain the apparent persistence in the circulation of label (radio or fluorescent) rather than of intact, unchanged liposomes. To clarify the interactions of liposomes with blood cells, this study examined the behaviour of liposomes of a range of compositions in the presence of purified blood cells, blood cells in plasma or whole blood or in vivo. Liposomes, irrespective of composition, were found to interact rapidly with all the cellular components of blood. This took place both in the presence and the absence of plasma proteins and functional enzyme cascades, suggesting that the prime facie interaction is opsonization‐independent and most probably consists of liposome‐blood cell fusion.
Materials and Methods
Materials
5‐Carboxyfluorescein (CF) and octadecyl rhodamine B chloride (R18) were purchased from Molecular Probes (Eugene, OR, USA). 1,2 dipalmitoyl‐sn3‐phosphocholine (DPPC) was obtained from Northern Lipids Inc. (Vancouver, BC, Canada) and cholesterol from Sigma‐Aldrich (Oakville, ON, Canada). The other lipids, 1,2‐dimyristoyl‐sn‐glycero‐3‐[phospho‐rac‐(1‐glycerol)] (DMPG), 1,2‐dipalmitoyl‐sn‐glycero‐3‐[phospho‐rac‐(1‐glycerol)] (DPPG), 1,2‐dioleoyl‐sn‐glycero‐3‐[phospho‐rac‐(1‐glycerol)] (DOPG), 1,2‐dimyristoyl‐sn‐glycero‐ phosphocholine (DMPC), 1,2‐dipalmitoyl‐sn‐glycero‐phosphocholine (DPPC), 1,2‐distearoyl‐sn‐glycero‐phosphocholine (DSPC), 1,2‐dioleoyl‐sn‐glycero‐phosphocholine (DOPC), 1,2‐dioleoyl‐sn‐glycero‐phosphate (DOPA), L–lecithin‐(phosphatidylcholine) from egg (EPC), and phosphatidyl serine (PS) from brain, were from Avanti Polar Lipids (Alabaster, AL), as were the fluorescent head‐group labeled phosphatidyl ethanolamines (PE): N‐(7‐nitro‐2‐1,3‐benzoxadiazol‐4yl)‐PE (NBD‐PE), N‐(lissamine rhodamine B sulfonyl)‐PE (R‐PE) and fluorescein‐PE (FITC‐PE). DSPE modified with PEG2000 was from Shearwater/NEKTAR (Huntsville, AL). Antibodies against platelet GPIb, (anti‐CD42b) and the leukocyte common antigen CD45, both labeled with phycoerythrin (R), and CD45 labeled with phycoerythrin‐cyanin 5.1 (PC5) were bought from Beckman‐Coulter (Mississauga, ON, Canada).
Mice, for in vivo experiments were cared for and handled according to the ethical standards of the University of British Columbia (protocol number A00‐0130). They were white, inbred, CCAT 2, ∼ 3 month old females weighing ∼ 20 g.
Human blood was obtained from healthy laboratory personnel who had signed an “informed consent form” according to the ethical standards and procedures of the University of British Columbia.
Liposome Preparation
Lipids were solubilized in chloroform. Aliquots of stock solutions were mixed in appropriate volumes to give a molar ratio of 70:30 DPPC/cholesterol unless otherwise noted. The organic solvent was removed by freeze‐drying the samples for 12 hours. Head‐group labeled phospholipids were included at 1 mol‐%, and the relative concentration of the phospholipid was reduced by the same amount. To prepare liposomes (L) containing carboxyfluorescein (CF), a carboxyfluorescein (0.4‐2.5 mM) solution in buffer (10 mM HEPES, 150 mM NaCl, pH 6.8) was used to hydrate the dry lipids. For injection into a mouse, a higher concentration, 30 mM, of CF was used in the liposomes having a formulation of DSPE‐PEG2000/DPPC/Chol. (3.5:66.5:30). The suspension, with a 25 mM final lipid concentration, was submitted to 5 freeze‐thaw cycles using liquid nitrogen (Reinish et al., [Citation1988]). Finally, to obtain liposomes of 250 to 400 nm diameter, the sample was extruded 3 to 5 times through polycarbonate membranes with 400 nm pores (Costar Nuclepore, Toronto, ON, Canada), under nitrogen pressure (100‐500 lb/in2) (Hope et al., [Citation1985]) using an extruder (Lipex Biomembranes, Vancouver, BC). The homogeneity and mean diameter of the liposomes were detected by quasi‐elastic light scattering (Nicomp Submicron Particle Sizer System, Model 370, Santa Barbara, CA, USA). To prepare homogeneously labeled liposomes containing either 1 mol‐% head‐group‐labeled phospholipids or 2 mol‐% R18, the marker (the former solubilized in chloroform, the latter in ethanol) was added in an appropriate volume to the mixture of lipids in chloroform (Hoekstra et al., [Citation1984]). The rest of the procedure was as described above except that the buffer used to hydrate the dry lipid mixture of e.g. DPPC:Cholesterol:R18 (68:30:2) (mol‐%) was 10 mM HEPES, 150 mM NaCl, pH 6.8 (HEPES buffer).
Gel Filtration
In order to remove any non‐encapsulated marker, liposome suspensions (0.5 mL) labeled with CF (Ravoo et al., [Citation1999]), or R18 were passed through a G75 Sephadex gel filtration column (1.5 × 20 cm), at room temperature (RT) with HEPES buffer as eluent.
Quantitation of Phospholipids, R18 and CF
The cleaned liposomes were analyzed for phospholipid content using the phosphate assay (Fiske and Subbarow, [Citation1925]). The concentration of R18 and CF in the cleaned liposomes was determined by their absorbance at 560 nm and 492 nm respectively. The liposomes were lysed using Triton X‐100 (final concentration 0.5 % v/v) and the samples were cleared by centrifugation for 30 min. at 2400 × g. Absorbance of the supernatant was measured for each sample and compared to a standard curve based on known marker concentrations.
Preparation of Platelets and Plasma
Human blood (4 mL) was drawn into tubes containing 0.105 M sodium citrate buffer (0.5 mL) and centrifuged at 20 min. at 500 × g to recover the plasma (supernatant). Platelet rich plasma (PRP) was prepared by centrifugation at 200 × g for 15 min. The supernatant (PRP) was diluted ½ with ACD buffer (11.5 mM citric acid, 88.5 mM trisodium citrate, 111 mM dextrose, pH 6). The new suspension was centrifuged at 15 min. at 500 × g. The supernatant was removed and the pellet containing the platelets was washed once with 1 mL HEPES buffer. The washed platelets were resuspended in 2 mL HEPES and counted on a hematology analyzer (Beckman Coulter MAXM, Hialeah, FL) and diluted to the appropriate concentration (108 to109 platelets/mL). The buffer pH of 6.8 was chosen to prevent overt platelet activation.
Interaction of Platelets with Liposomes
Interactions in buffer: Various volumes, 1 to 70 µL, of column purified liposomes (1 mM lipid) labeled with CF were mixed with 10µL platelet (108/mL) suspension in HEPES buffer. The suspensions were diluted to 1 mL in buffer and incubated at room temperature for 1 to 2 hours. The platelet‐liposome samples were analyzed by flow cytometry (EPICS XL‐MCL, Beckman Coulter). The program was set to acquire counts for 20 seconds and the platelet population and the liposome population were gated based on forward (FS) and side scatter (SS) information displayed on a log scale. Between 6% to 15% of the liposome population was found to overlap the analyzed platelet region and the results were expressed in arbitrary units of fluorescence (FITC or PE). Samples containing only platelets, only labeled liposomes or platelets with free marker (with the same concentration of free marker that could be released from the labeled liposomes) were used as controls and their fluorescence was compared to the platelet sample incubated with liposomes.
Interactions in the presence of plasma: PRP platelets (in plasma) were counted and diluted to ∼ 2 × 108 platelets/mL with autologous plasma. Washed CF‐liposomes (6.6 mM lipid) were diluted with plasma to contain 1 mM liposome lipid. In order to identify the platelets in the mixture, they were pre‐incubated for 30 min. at room temperature with anti‐CD42b‐R antibody. Washed liposomes, (2 to 50 µL; 1 mM lipid) and 106 pre‐labeled platelets were mixed and incubated (in 0.1 mL plasma) for 2 hours. Prior to flow cytometric analysis, the samples were diluted to 1 mL with HEPES buffer. To have a good comparison, the experiment was repeated in buffer. Samples with CF‐liposomes with anti‐CD42b‐R, anti‐CD42b‐R antibody alone, platelets alone and platelets labeled with anti‐CD42b‐R in buffer, and buffer‐10% (vol.) plasma were used as controls. As described above, the flow cytometer program was gated on the platelet population (A), the liposome population (B) and the sum of both populations (C). Also it was set to stop automatically after the acquisition of 10,000 events in gate C. Control samples were run a second time and stopped manually at the same elapsed time (equivalent volume) as the corresponding test sample. Flow cytometric analysis of the control samples indicated that the plasma contained some particles or cell fragments that also had the same characteristics as liposomes. The presence of these particles did not influence the results because they were negative for the CD42b antigen and they were detected in the liposome gate. Changes of the % fluorescence of the platelet population (A) were considered indicative of platelet‐liposome interaction. A dose response curve was generated to express the platelet‐liposome interaction. Platelet‐liposome interactions in buffer, plasma and in the presence or absence of antibody were assessed and compared.
For liposomes of other compositions, containing 1 mol‐% FITC‐PE, 2 µL of PRP was mixed with an equal volume of 10 mM liposomes and 40 µL Hepes buffer and incubated at RT for 30 min. Three samples were set up for each liposome type: PRP and liposomes; PRP liposomes and CD42b‐R (3 µL); and PRP liposomes and IgG isotype control‐R (3 µL).
Mouse blood, drawn from the tail vein into 10mM EDTA was centrifuged to obtain PRP as described above, for human blood. Equal volumes of PRPmouse and 10 µL of liposomes DOPC:DOPG:FITC‐PE, (49.5:49.5:1) at a lipid concentration of 10 mM were mixed and allowed to interact for 5 minutes at RT, and a minimum of 5000 signals in the platelet bitmap were assessed for fluorescence.
Interaction of Liposomes with Blood Cells in Whole Blood
Whole blood (WB) (5 µL) was mixed with 2 to 50 µL CF‐liposomes (1mM lipid), made to a final volume of 100 µL with HEPES buffer, and incubated for 2 h at room temperature (RT). The samples were then incubated for an additional 30 min. RT, with anti‐CD45‐R in order to label the leukocytes. Control samples were prepared containing identical concentrations of CF‐liposomes; CF‐liposomes and anti‐CD45‐R; anti‐CD45‐R antibody alone; and whole blood in HEPES buffer. Finally, each sample was diluted with buffer to 1 mL and analysed by flow cytometry using two programs. The first program acquired counts on the FS sensor, and was used to analyze the interaction between leukocytes and CF‐liposomes. The flow cytometer was stopped when 600 events were collected in the leukocyte gate. The control sample containing CF‐liposomes and anti‐CD45‐R was negative for the presence of antibody on the liposomes' surface. The second program was set to acquire fluorescent events only and was used to analyze the red cell‐CF‐liposome and platelet‐CF‐liposome interactions. Gates were set around the platelet population (A), the liposome population (B) and the red cell population (C). Signal acquisition was stopped at 2000 events in the platelet gate (∼ 20 sec). During that time, approximately 40,000 events were collected in the red cell gate. When the liposome control sample (50 µM lipid) was run, 1000 events were recorded in the liposome gate in 20 sec. Also, liposomes were found not to overlay the leukocyte gate and only 1% to 2% of events were counted in the other blood cell gates. The percent fluorescence of the platelet population, red cell and leukocyte populations was recorded. A dose‐response curve was constructed to express and compare the interaction of liposomes with each type of blood cell. A time‐course experiment compared the interaction of R18‐liposomes (2:68:30 R18/DPPC/Chol.), CF‐liposomes (7:63:30 DPPE/DPPC/Chol.) and CF‐liposomes (3.5:66.5:30 DSPE‐PEG2000/DPPC/Chol). Whole blood (50 µL) was mixed at with 400 µL labeled liposomes (0.5 mM lipid). At the indicated times, 45 µL samples of the mixture were diluted to 1 mL with Hepes buffer and assessed by flow cytometry. The acquisition time for each run was 10 sec and the fluorescence of the platelet and red cell population was analyzed.
For liposomes of other compositions the assay was carried out as described for PRP.
Separation of Liposomes and Platelets
Washed platelets 130 µL (1.5 × 109/mL) were incubated with 320 µL CF‐liposomes (10.5 mM lipid, 35 µM CF) for 2 hours, at RT. The sample was passed through a Sephadex G75 column (1.5 × 20 cm) using HEPES buffer at a flow of 0.71 mL/min. The collected fractions developed peaks containing a liposome‐platelet complex, the free species or both.
The experiment was repeated, under similar conditions using R18‐liposomes or a 1:1 (molar equivalent) of CF‐liposomes and R18‐liposomes. The flow rates were 0.72 mL/min and 0.82 mL/min respectively. Control samples containing platelets alone or liposomes alone were passed through the column and the chromatography pattern of each species was detected. All the fractions were tested by flow cytometry and the platelets were counted. The fractions were evaluated by flow cytometry with programs gated on the platelet and liposome populations. Using the control, each gate was set to contain a maximum of 15% crossover from the other population. Automatic acquisition was set to stop at 20 sec. or 10,000 events counted in both populations. Finally, the fluorescence (PE or FITC, arbitrary units) of each population was analyzed and the background, based on unlabelled platelets, was set at 0.3. Fractions with the same eluted volume were compared.
Interaction of Liposomes with Blood Cells In Vivo
Mice were bled by puncturing the tail vein and recovering ∼ 50 µL of blood into ACD for a negative control. Thereafter, ∼ 100 µL of 10 mM 70:30 DPPC/cholesterol liposomes labeled with CF or R18 in bicarbonate buffer (20 mM NaHCO3, 150 mM NaCl, pH 7.4) were injected separately or together by the intra‐venous route. One mouse received 200 µL of 2.25 mM (lipid) liposomes of 3.5:66.5:30 DSPE‐PEG2000/DPPC/CHOL labeled with CF. At the indicated times, the contra‐lateral tail vein was punctured, ∼ 50 µL of blood was recovered into ACD. The samples were diluted in buffer to approximately the same concentration and blood cell fluorescence was assessed by flow cytometry, as described above. Mouse and human blood cells are of similar scatter parameters, which permitted the use of the same flow cytometer programs that were already defined for human cells.
Results
Platelet‐Liposome Interactions in Buffer
The results obtained by flow cytometry indicate that the presence of labeled liposomes strongly affects platelet fluorescence. Although the analysis of washed platelets pre‐incubated with CF in buffer indicated that the free marker does not become associated with the platelets, to prevent any potential interaction with non‐encapsulated marker, the CF‐liposomes were separated from free marker by column chromatography. Controls of CF‐liposomes (lipid 70 nmol/mL, CF 0.3 nmol/mL) alone gave a count rate of around 10 events/sec while the platelets alone (10e6/mL) registered as 500‐1000 events/sec. When the two were mixed, the count rate was almost identical to the count of the platelets alone: at the same time, 90% of the platelet population became fluorescent. Platelets (10e6/mL) incubated with free CF 0.3 nmol/mL gave a fluorescence intensity level 17 times less than when they were incubated with CF‐liposomes containing the same amount of encapsulated marker (see ). This shows that in the platelet‐CF‐liposome mixture, the hydrophilic marker contained in the aqueous core of the liposomes (Tardi et al., [Citation2001]) becomes associated with the platelet, while the free marker does not. Washing the platelets after incubation with their liposomes did not significantly alter the amount of platelet fluorescence nor did the presence of EDTA, or pre‐incubation of the platelets with a metabolic poison cocktail.
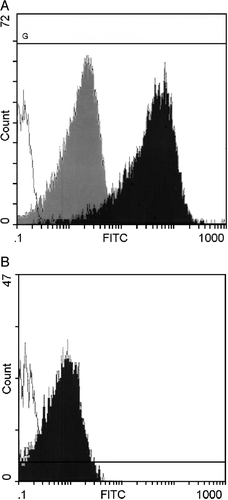
Figure 1. Interaction of liposomes and platelets detected by flow cytometry. A) CF‐liposomes interacted with washed platelets in buffer. Green fluorescence intensity is shown on the log scale of the x–axis; cell numbers on the y‐axis. The open histogram is the control platelet population; the dark histogram shows the level of fluorescence of the platelet population incubated with CF‐liposomes; the degree of fluorescence conferred by an equal amount of free CF is the gray histogram. B) Fluorescence of mouse platelets in plasma in response to FITC‐PE labeled liposomes is shown. The open histogram is the control platelet population; the dark histogram is the fluorescence of mouse PRP interacted with lipid‐labeled liposomes.
Identification of Platelets Using Anti‐CD42b‐R Antibody
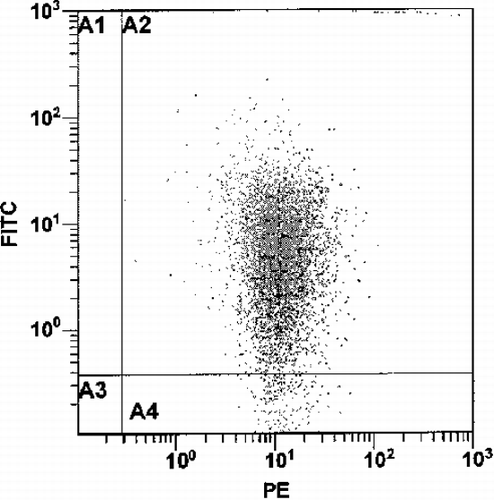
The fluorescent events found in the region of the platelet population could possibly be aggregates of CF‐liposomes having the same size characteristics, therefore the platelets were identified with anti‐CD42b‐R antibody. Control samples showed that 99% of the particles in the platelet gate were positive for CD42b‐R. CF‐liposomes treated with anti‐CD42b‐R were negative. For a mixture of platelets (106/mL) and CF‐liposomes (lipid 10 nmol/mL, CF 17 pmol/mL) in buffer, 57% of the events counted in the platelet gate contained green fluorescence. When the mixture of anti‐CD42b‐R labeled platelets and CF‐liposomes was tested, 99% of the events in the platelet gate were red labeled, and had the same percent and mean fluorescence intensity as the control sample of anti‐CD42b‐R labeled platelets without liposomes. Because the antibody does not bind to liposomes, the events detected in the platelet gate were indeed platelets. Of these PE‐labeled‐platelets, 37% percent also fluoresced in the green, CF range (see ). Comparison with the control sample of unlabeled platelets incubated with CF‐liposomes indicated that the green fluorescence level was reduced by ∼ 20% in the presence of the antibody on the platelet membrane. This suggests that the interaction between the liposomes and platelets was partially inhibited by the presence of the anti‐platelet antibody. Also, the interaction with liposomes did not affect platelet integrity (Male et al., [Citation1992]) because the number of events detected in the platelet gate was similar for antibody labeled platelets in presence or absence of CF‐liposomes.
Figure 2. The fluorescence pattern of 106/mL platelets labeled with anti‐CD42b‐R in the presence of CF‐liposomes (lipid 10 nmol/mL, CF 17 pmol/mL) in buffer. The x–axis quantitates red phycoerythrin (PE) florescence and the y‐axis, green CF fluorescence, on a log scale in arbitrary units.
Analysis of Fluorochrome Location by Exclusion Chromatography
The analysis of the eluted fractions of pure platelets and labeled liposomes using exclusion chromatography indicated a shorter retention time for platelets than for liposomes. Although the difference between two peaks was around 1.5 mL eluted volume (2 fractions), and thus the peaks overlapped when mixtures were applied to the column, the initial platelet fractions contained pure platelets. shows the results obtained by flow cytometric analysis of the fraction where the purified platelets eluted from the column (eluted volume ∼ 10 mL).
Table 1. Analysis of Fluorescence Associated with the Platelet and Liposome Fractions Separated by Gel Exclusion Chromatography
In each population there were two types of events: non‐fluorescent and fluorescent (red or green). The first column of gives the initial composition of the suspension separated on the gel as well as the rate of events (relative concentration) detected by the flow cytometer. Generally, ∼ 7‐fold fewer events/s were detected in the platelet fraction when only liposomes were passed through the column than when platelets only, or liposome‐platelet mixtures were chromatographed. The data is expressed as a percentage of the sum of fluorescent events in the platelet and liposome gates. Analysis of the platelet fraction separated from platelet and CF‐liposome mixtures indicated that 91% of the detected events were CF‐positive platelets. In this fraction, the non‐interacting platelets represented only 7% of the population, and the free liposomes 2%. A similar analysis of platelets and R18‐liposomes showed an even higher level of interaction (98.5%) between the platelets and the liposomes. Free R18‐liposomes represented only 1.5% of the population and nearly all of the platelets became R18 labeled. When both R18‐liposomes and CF‐liposomes were mixed with platelets, the platelet fraction contained only 0.6% free CF‐liposomes and no R18‐liposomes. However, the platelets were labeled with both markers: 88.2% with CF and 31% with R18. These results indicate an interaction between the platelets and liposomes that is at least sufficient for both hydrophobic and hydrophilic marker transfer; concurrent with the disappearance of detectable liposomes, and confirm that platelets are the recipients of that transfer.
Dose‐Response of the Liposome‐Platelet Interaction
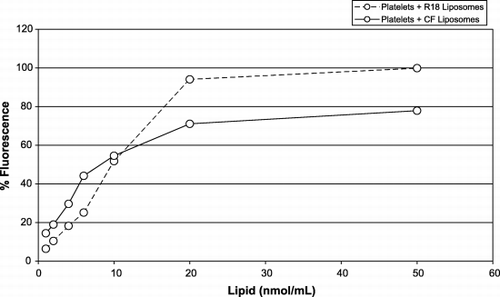
To determine the maximum amount of liposomes that can interact with platelets in vitro, 106 platelets/mL were incubated with either CF‐liposomes or R18‐liposomes at a range of liposome‐lipid ratios. The resulting percentage of fluorescent platelets is shown on the dose‐response curves of . Although the R18 marker sequesters to the liposome membrane while CF is encapsulated in the aqueous compartment, both curves reached a plateau at around ∼ 10 to 20 fmol lipid per platelet. This indicates that platelets have a limited capacity to take up liposome lipids. Even a small amount of labeled liposomes can have a visible effect on platelets: 1 fmol lipid/platelet causes a fluorescence increase of 7 to 15%.
Figure 3. The dose‐response curve of platelets (106/mL) in buffer with liposomes labeled with: CF (solid line) (molar ratio of lipid:CF, 760:1); R18 (broken line) (molar ratio of lipid:R18, 135:1). The x‐axis represents the amount of liposomes (lipid nmol/mL); the y‐axis represents the percentage of fluorescent (red or green) labeled platelets.
Comparison of the Platelet‐Liposome Interaction in Plasma and Buffer
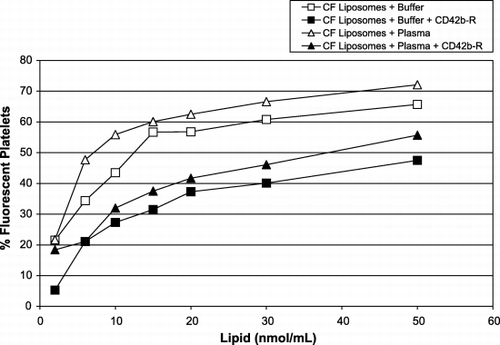
To better understand the probable liposome‐platelet interactions in vivo, experiments were set up in vitro to compare the effects of plasma and buffer on this interaction. As described in the Methods section, the interaction of CF‐liposomes with unlabeled platelets and with platelets pre‐treated with anti‐CD42b‐R antibody was studied. Liposomes incubated in filtered plasma, in the absence of blood cells, had a tendency to form microaggregates (not shown). On microscopic examination all formulations, with PEG or without, formed such clumps. This is what prompted the concurrent staining of platelet‐sized flow cytometer signals with anti‐CD42b‐R to distinguish clumped liposomes from true platelets. shows the effect of the CF‐liposome dose taken up by platelets (106/mL) during 2 h of incubation. In general, the interaction of liposomes with platelets in plasma and buffer were similar regardless of whether they were pre‐treated with anti‐CD42b‐R, or not. They reached a plateau at the same lipid concentration, but plasma appeared to enhance the process such that a higher proportion of the platelet population became fluorescent. The anti‐CD42b antibody bound to the extra‐cellular domain of the GPIb integrin had a negative effect on this interaction. shows that the percentage of fluorescent platelets in buffer or plasma is decreased by ∼ 20% in the presence of antibody on the platelet surface. As well, somewhat higher amounts of lipids were necessary to saturate the platelets (∼ 30 nmol/mL). This was also the case for liposome formulations 1 and 3 (from ). As well, all of the formulations interacted with activated platelets in plasma, but the degree of interaction was unaffected by the level of platelet activation, however, liposome interaction with platelets did not, per se cause platelet activation. Mouse platelets in plasma were also tested for interaction with liposomes. shows that in vitro mouse platelets acquired fluorescence from liposomes in the same manner as do human platelets.
Table 2. Liposomes of a Range of Compositions Interact with Platelets in Plasma or Whole Blood
Figure 4. Interaction of platelets and liposomes in plasma (triangles) or buffer (squares) and in the presence of anti‐CD42b‐R on platelets’ surface in plasma (solid symbols) or in buffer (open symbols). The x‐axis represents the amount of CF‐liposomes, lipid nmol/mL (molar ratio of lipid:CF, 760:1); the y‐axis represents the percentage of fluorescent platelets (106/mL).
Interaction of Liposomes with Whole Blood in Vitro
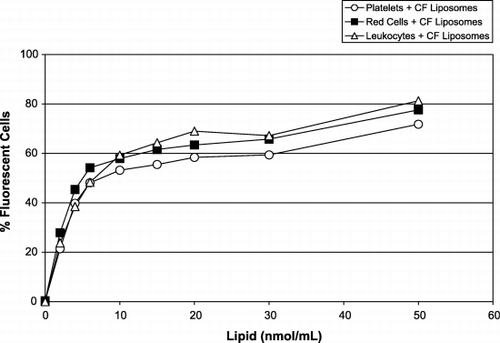
Although platelets interact strongly with liposomes, they do not constitute the only kind of blood cell membrane that may interact with the liposomes in whole blood. The interactions of red cells, platelets and leukocytes with CF‐liposomes are shown in . Post‐labeling with anti‐CD45‐R antibodies (see Methods) identified the leukocytes. It can be seen in that all three types of blood cells interact with liposomes. In each case, the pattern of the dose‐response curves for platelet ‐ liposome and red cell – liposome interaction is similar, but the leukocyte ‐ liposome curve differs somewhat. Although the difference is small, the leukocytes seem to be the most active cells in this interaction, producing the highest proportion of (∼ 81%) of green labeled cells. The red cell population achieved ∼ 77% fluorescence while the platelets reached ∼ 72%. This result clearly indicated that the liposomes have the capacity to interact with most blood cells.
Figure 5. Fluorescence of cells in WB induced by the presence of increasing concentrations of CF‐liposomes (molar ratio lipid/CF, 760:1): leukocytes (triangles); red cells (squares); and platelets (circles). The x‐axis represents the amount of CF‐liposomes, lipid nmol/mL (molar ratio of lipid:CF, 760:1); the y‐axis represents the percentage of fluorescent blood cells.
A range of liposome compositions was prepared using head‐group labeled phospholipids (1 mol‐% FITC‐PE) rather than the R‐18 fatty acid or the hydrophilic CF, and allowed to interact with PRP platelets or platelets in anticoagulated whole blood (WB). Using the flow cytometer, the platelet area, defined by forward (FS) and side scatter (SS) characteristics was interrogated for fluorescence. Signals that were positive (98% to100%) for CD42b‐R became rapidly positive for green, FITC fluorescence. summarizes two experiments showing that at a uniform lipid concentration, the majority of platelets rapidly became fluorescent in the presence of different formulations of liposomes. Because the platelet donors were different, as were their platelet counts, as well as and the degree of fluorescence obtained from each formulation, it is remarkable that the percentages are so similar. In general, interactions with different individuals' platelets by different liposome formulations but at this defined lipid concentration, gave an average of 47% fluorescence (+ /‐SD 15.7%) of platelets in PRP. Formulation 3 at the same concentration also produced 47% fluorescence of mouse PRP. Thus the differences between formulations are not statistically significant.
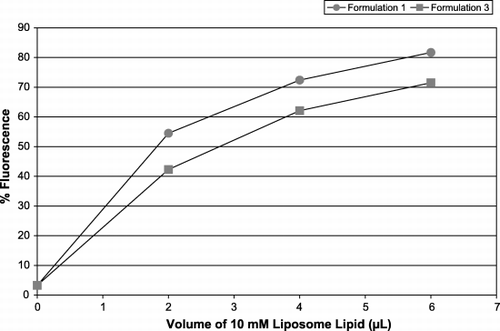
Formulations 1 and 3 () were chosen for further experiments because they gave the highest and lowest levels of platelet fluorescence consistently. Diluting the amount of liposomes offered to the same population of platelets showed that platelet fluorescence diminished: a smaller proportion of the population became fluorescent and also to a lesser degree. However, for the same lipid concentration, formulation 1 did consistently render the platelets more fluorescent than formulation 3 (data not shown but vide infra). These two formulations were compared for their ability to interact with both leukocytes () and leukocyte‐platelet aggregates () in WB. Both the general leukocyte population, as well as those leukocytes attached to platelets were positive for the presence of liposomes (selected first by the red platelet marker CD42b in the leukocyte bitmap, associated with the purple CD45‐PC5 and interrogated for green) and this was dependent on lipid dose. Preliminary observations indicate that as the concentration of liposomes increased, so did the number of leukocyte‐platelet aggregates.
Figure 6. Leukocyte fluorescence in WB. Increasing concentrations of liposome formulations 1 (circles) and 3 (squares) were added and the leukocytes were identified by anti‐CD45‐PC5. The x‐axis represents L of 10 mM liposome lipid added to a platelet population; the y‐axis shows the percentage of the leukocyte population positive for FITC‐PE.
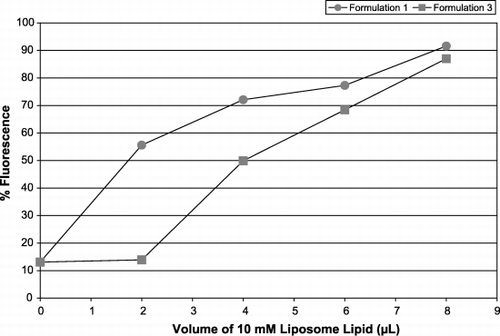
Figure 7. Fluorescence of leukocyte‐platelet aggregates in WB. Increasing concentrations of liposome formulations 1 (circles) and 3 (squares) were added and the leukocyte‐platelet aggregates were selected by anti‐CD45‐PC5, interrogated for CD42b‐R then assessed for the presence of FITC‐PE. The x‐axis represents L of 10 mM liposome lipid added to a platelet population; the y‐axis shows the percentage of the leukocyte population positive for FITC‐PE.
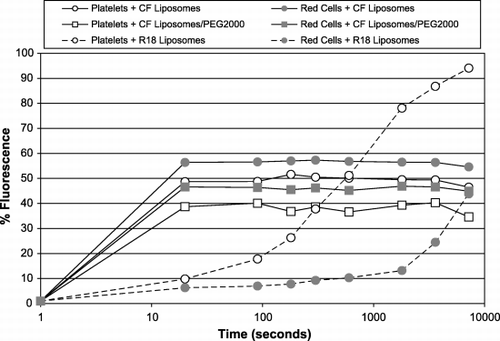
The interaction of liposomes with blood cells is rapid. shows that liposomes of the same basic formulation, containing the hydrophilic marker CF, with or without 3.5 mol‐% PEG2000 interact with the same kinetics. This is true for both red cells and platelets. The interaction is essentially complete by the time the flow cytometer can measure it, in less than one minute, though in some cases, the intensity of the fluorescence continues to increase. Fluorescence associated with the fatty acid marker R18, takes somewhat longer to appear in the blood cells, with platelets being loaded much faster than red cells.
Figure 8. Time course of liposome‐blood cell interaction in WB. Liposomes labeled with CF (solid lines) or R18 (broken lines) were added to whole blood and samples were removed at the indicated intervals. Red cells (closed symbols) and platelets (open symbols) were assessed for fluorescence. Liposomes with PEG2000 (squares) or without (circles) were compared. The x‐axis represents time in seconds; the y‐axis shows the % fluorescence of the cell population.
Interaction of Liposomes with Whole Blood In Vivo
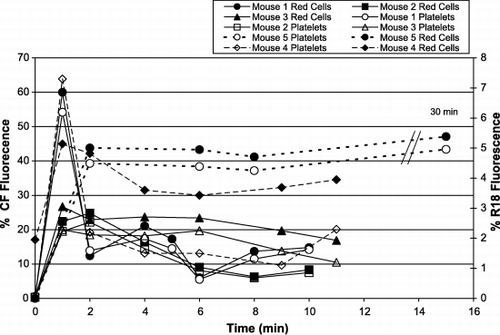
Mice were injected intravenously with different lots of the 70:30 DPPC‐CHOL liposome preparations carrying differing amounts of fluorescent label. shows that immediately after injection of the liposomes, both the CF and R‐18 markers become associated with both platelets and red cells. Depending on the injection speed and the final amount of lipid in the circulation, the kinetics of the appearance of the fluorescence varies slightly during the first few minutes. However, after an initial increase of the fluorescence in both cell types, the percentage of the population that fluoresces decreases somewhat as the fluorescent and non‐fluorescent cells in the circulation equilibrate. Interestingly, in each case, more relative fluorescence is present in the red cell population than the corresponding population of platelets. Although in this case only ∼ 10 min were assessed, from other experiments we know that blood cell fluorescence persists for at least 2 hours, irrespective of the material used to label the liposomes. It is interesting to note that the liposomes carrying PEG2000 associate with blood cells as rapidly as do related formulations without PEG, but the fluorescence they transfer to blood cells does not decay as rapidly.
Figure 9. Intravenous injection of fluorescent liposomes into mice. Different lots of liposomes containing CF (solid lines) or R18 (broken lines) were injected into mice and the fluorescence of platelets and red cells was monitored. The x‐axis shows the times of blood samples taken after the injection; the y‐axes are % florescence of the cell populations: left y‐axis ‐ CF fluorescence; right ‐ R18. The platelet populations are shown by open symbols, and the red cells by closed symbols.
Discussion
This study brings together both the in vitro and in vivo picture of the interactions of liposomes with blood cells. Liposomes, irrespective of the characteristics of their lipid formulations, or the presence or absence of PEG on their surface have a tendency to irreversibly interact with blood cells. While it could be argued that only the fluorescent marker, in the absence of other liposome components, is transferred to the recipient cells, the use of a hydrophilic marker encapsulated in the aqueous core of the liposomes, as well as labeled phospholipids and fatty acids that are membrane associated, would argue against that possibility. Indeed simultaneous addition of both CF‐ and R18‐containing liposomes resulted in platelets acquiring both fluorochromes (data not shown). Similarly, the disappearance of the counted number of signals in the liposome gate of the flow cytometer, and the appearance of fluorescent marker associated with e.g. the platelets, without an increase of platelet numbers would also support the blood cell‐liposome fusion hypothesis (see ).
The fact that liposome‐blood cell interactions take place as well in the presence or absence of plasma components argues against the initiation of liposome clearance being predominantly plasma protein dependent. Plasma does enhance the interaction between platelets and liposomes () and it is known that a variety of plasma components do become deposited on liposomes in vitro (Devine et al., [Citation1994]; Ishida et al., [Citation2001]; Oja et al., [Citation1996]; Semple et al., [Citation1996]). While it is not unlikely that opsonization has an enhancing role in liposome engulfment by phagocytic cells in vivo (Ishida et al., [Citation2001]) the mechanism of action of the plasma proteins in vitro may be more prosaic, and similar to the charge‐masking phenomenon ascribed to PEG (Levchenko et al., [Citation2002]). The similar rapid kinetics of the association of liposomes and blood cells, both in buffer and in vivo also supports this possibility. The small amount of inhibition caused by antibody on the platelets' surface was the same in plasma and buffer: in the presence of antibody on the platelets a smaller number of liposomes arrive at the platelets surface and consequently the interaction is reduced. This also implicates fusion indirectly: steric hindrance of the liposomes' approach to the platelets can prevent some fusion while metabolic inhibitors and chelators are unable to do so. In whole blood, each blood cell population became fluorescent to a different degree: this was not proportional to cell size, but may reflect the membrane to which the liposomes have the most immediate access.
Thus evidence is accruing that blood cells in general and platelets in particular can be responsible for liposomes' disappearance from the circulation. Others have noted that an intravenous injection of liposomes, irrespective of formulation, is accompanied by a rapid reduction of platelet (and sometimes leukocyte) counts in the blood, followed by a slow recovery (Doerschuk et al., [Citation1989]; Goins et al., [Citation1997]; McLean et al., [Citation1997]; Reinish et al., [Citation1988]). Furthermore, experiments have shown that liposome lipids can be taken up and retained by platelets (Male et al., [Citation1993]). Although this could be due to endocytosis (Behnke, [Citation1992]) there is evidence of lipid mixing (Nishiya et al., [Citation1995]), or complete fusion (Nishiya and Sloan, [Citation1996]) with platelets. As well, a transfer of platelet membrane proteins to the liposomes (Dalencon et al., [Citation1996]; Okumura et al., [Citation1994]) can be accomplished by a variety of liposome formulations. Red cells are also able to do this (Sato et al., [Citation1990]; Suzuki and Okumura, [Citation2000]) as can leukocytes (Devoisselle et al., [Citation2001]; Gerber et al., [Citation2001]). In many cases platelet‐liposome fusion is not detrimental to platelets (Male et al., [Citation1992]): but may (Juliano et al., [Citation1983]) or may not cause (Nishiya and Sloan, [Citation1996]; Stensrud et al., [Citation1999]) overt changes of platelet function such as aggregation or hemostasis.
It is not impossible that liposome‐platelet interactions cause lipid bilayer changes and consequent exposure of inner‐leaflet PS. Platelet surface PS acts as both an activation (Briede et al., [Citation1999]) and senescence marker (Berger et al., [Citation1998]) of platelets and causes platelet retention in the liver and spleen (Pereira et al., [Citation1999]). The accumulation there of label/liposome may be the result of it being transported within the platelet (or other blood cell) membrane, or a shed vesicle (Okumura et al., [Citation1994]), whose membrane asymmetry was perturbed by lipid mixing, fusion, and/or loss of membrane proteins (Dalencon et al., [Citation1996]; Okumura et al., [Citation1994]) and was consequently ‘sent’ to the spleen (or liver & bone marrow).
Fusion could explain the lack of enhanced platelet aggregation with or without ADP shown by RGD liposomes (Rybak and Renzulli, [Citation1993]) and GPIIbIIIa‐loaded liposomes (Sloan et al., [Citation2000]). Similarly, it could offer an alternative explanation for the effectiveness and localization of synthetic phospholipids that promote procoagulant activity on damaged vessels (Galan et al., [Citation1998]), the clot accumulation of plasminogen‐bearing liposomes (Heeremans et al., [Citation1998]) and the improved thrombolysis of streptokinase‐containing liposomes (Perkins et al., [Citation1997]). Finally, liposomes are found to accumulate in splenic cords (Shibazaki et al., [Citation1998]) and in the liver, both of which are sites of normal platelet sequestration (Peters et al., [Citation1985]) and in the case of opsonized platelets, their destruction, in splenic cords (Luk et al., [Citation1980]).
If lipid disturbances, such as PS exposure on the membrane's outer leaflet, have an influence on a cell's localization (Pereira et al., [Citation1999]) blocking of that recognition system may perturb PS‐dependent localization. We hypothesize that blood cells that absorb PEGylated liposomes may incorporate the PEG‐carrying phospholipids in their membrane so to expose PEG on the cell's surface. Such surface PEG may interfere with that cell's being recognized as having a lipid disturbance and exposed PS. This, in turn, may prevent sequestration e.g. in the spleen thereby allowing the cell to remain in the circulation for the length of its natural half‐life (Bradley et al., [Citation2002]). We are currently testing this hypothesis.
In conclusion, it appears that liposome formulations are rapidly absorbed by blood cells both in vitro and in vivo. Such absorption can explain both the rapid disappearance of detectable liposomes from the circulation and the passive targeting to sites of high blood pool activity (Laverman et al., [Citation2000b]), as well as the persistence and slow release of the drugs that they encapsulate.
Acknowledgments
The authors wish to acknowledge the financial support of the Canadian Blood Services Research and Development Program – Grant # XV00004 as well as the technical expertise of Miss Jill Miners, B.Sc.
References
- Allen T. M. Liposomal drug formulations. Rationale for development and what we can expect for the future. Drugs 1998; 56: 747–756
- Andreopoulos D., Kasi L. P. 99m‐Tc‐labelled diphytanoyl phosphatidylcholine liposomes: in vitro and in vivo studies. J. Microencapsul 1997; 14: 427–436
- Barenholz Y., Amselem S. Quality control assays in the development and clinical use of liposome‐based formulations. Liposome Technology I, G. Gregoriadis. CRC Press, Boca Raton 1984; 527–616
- Behnke O. Degrading and non‐degrading pathways in fluid‐phase (non‐adsorptive) endocytosis in human blood platelets. J. Submicrosc. Cytol. Pathol. 1992; 24: 169–178
- Berger G., Hartwell D. W., Wagner D. D. Platelet death: an apoptotic–like process?. Blood 1998; 92: 347a
- Bergstrom K., Holmberg K., Safranj A., Hoffman A. S., Edgell M. J., Kozlowski A., Hovanes B. A., Harris J. M. Reduction of fibrinogen adsorption on PEG‐coated polystyrene surfaces. J. Biomed. Mat. Res. 1992; 26: 779–790
- Blume G., Cevc G. Molecular mechanism of the lipid vesicle longevity in vivo. Biochim. Biophys. Acta 1993; 1146: 157–168
- Bradley A. J., Murad K. L., Regan K. L., Scott M. D. Biophysical consequences of linker chemistry and polymer size on stealth erythrocytes: size does matter. Biochim. Biophys. Acta 2002; 1561: 147–158
- Briede J. J.M., Heemskerk J. W., Hemker H. C., Lindhout T. Heterogeneity in microparticle formation and exposure of anionic phospholipids at the plasma membrane of single adherent platelets. Biochim. Biophys. Acta 1999; 1451: 163–172
- Brooks D. E, Muller W. Size‐exclusion phases and repulsive protein‐polymer interaction. J. Mol. Recognit. 1996; 9: 697–700
- Bulte J. W.M., de Cuyper M., Despres D., Frank J. A. Short‐ vs long‐circulating magnetoliposomes as bone marrow‐seeking MR contrast agents. J. Magn. Reson. Imaging 1999; 9: 329–335
- Chaikof E. L, Merrill E. W., Callow A. D., Connolly R. J., Verdon S. L., Ramberg K. PEO enhancement of platelet deposition, fibrinogen deposition and complement C3 activation. J. Biomed. Mater. Res. 1992; 26: 1163–1168
- Chams V., Bonnafous P., Stegmann T. Influenza heamagglutinin mediated fusion of membranes containing poy(ethylene‐glycol) grafted lipids: new insights into the fusion mechanism. FEBS Lett. 1999; 448: 28–32
- Chonn A., Cullis P. R. Recent advances in liposomal drug‐delivery systems. Curr. Opin. Biotechnol. 1995; 6: 698–705
- Crowe J. H., Crowe L. M. Factors affecting the stability of dry liposomes. Biochim. Biophys. Acta 1988; 939: 327–334
- Dalencon F., Rosilio V., Puisieux F., Baszkin A., Wautier J. L. Liposomes bearing platelet proteins: a model for surface function studies. Biochim. Biophys. Acta 1996; 1302: 241–248
- Devine D. V., Wong K., Serrano K., Chonn A., Cullis P. R. Liposome‐complement interactions in rat serum: implications for liposome survival studies. Biochim. Biophis. Acta 1994; 1191(1)43–51
- Devoisselle J. M., Begu S., Tourne‐Peteilh C., Desmettre T., Mordon S. In vivo behaviour of long‐circulating liposomes in blood vessels in hamster inflammation and septic shock models—use of intravital fluorescence microscopy. Luminescence 2001; 16: 73–78
- Doerschuk C. M., Gie R. P., Bally M., Cullis P. R., Reinish L. W. Platelet distribution in rabbits following injection of liposomes. Thromb. Haemost. 1989; 61: 392–396
- Evans E., Rawicz W. Elasticity of fuzzy membranes. Phys. Rev. Lett. 1997; 79: 2379–2382
- Fiske C., Subbarow Y. The calorimetric determination of phosphorus. J. Biol. Chem. 1925; 66: 375–389
- Gabizon A., Papahadjopoulos D. Liposome formulations with prolonged circulation time in blood and enhanced uptake by tumours. Proc. Natl. Acad. Sci. U. S. A. 1988; 85: 6949–6953
- Gabizon A., Price D. C., Huberty J., Bresalier R. S., Papahadjopoulos D. Effect of liposome composition and other factors on the targeting of liposomes to experimental tumours: biodistribution and imaging studies. Cancer Res. 1990; 50: 6371–6378
- Galan A. M., Hernandez M. R., Bozzo J., Reverter J. C., Estelrich J., Roy T., Mazzarra R., Ordinas A., Escolar G. Preparations of synthetic phospholipids promote procoagulant activity on damaged vessels: studies under flow conditions. Transfusion 1998; 38: 11–12
- Gerber C. E., Bruchelt G., Falk U. B., Kimpfler A., Hauschild O., Kuci S., Bachi T., Niethammer D., Schubert R. Reconstitution of bactericidal activity in chronic granulomatous disease cells by glucos‐oxidase‐containing liposomes. Blood 2001; 99: 3097–3105
- Goins B., Phillips W. T., Klipper R., Rudolph A. S. Role of complement in rats injected with liposome‐encapsulated hemoglobin. J. Surg. Res. 1997; 68: 99–105
- Heeremans J. L.M., Prevost R., Feitsma H., Kluft C., Crommelin D. J.A. Clot accumulation characteristics of plasminogen‐bearing liposomes in a flow system. Thromb. Haemost. 1998; 79: 144–149
- Higashi N., Yamauchi M., Okumura Y., Nakanishi M., Sunamoto J. Fusion between Jurkat cell and PEO‐lipid modified liposomes. Biochim. Biophys. Acta 1996; 1285: 183–191
- Hoekstra D., de Boer T., Kllape K., Wilschut J. Fluorescence method for measuring the kinetics of fusion between biological membranes. Biochemistry 1984; 23(24)5675–5681
- Hope M. J., Bally M. B., Webb G., Cullis P. R. Production of large unilamellar vesicles by a rapid extrusion procedure. Characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophis. Acta 1985; 812: 55–65
- Hsu M. J., Juliano R. L. Interactions of liposomes with the reticuloendothelial system. II: Nonspecific and receptor‐mediated uptake of liposomes by mouse peritoneal macrophages. Biochim. Biophys. Acta 1982; 720: 411–419
- Ishida T., Harashima H., Kiwada H. Interactions of liposomes with cells in vitro and in vivo: opsonins and receptors. Curr. Drug Metab. 2001; 2: 397–409
- Juliano R. L., Hsu M. J., Peterson D., Regen S. L., Singh A. Interactions of conventional or photopolymerized liposomes with platelets in vitro. Exp. Cell Res. 1983; 146: 422–427
- Kenworthy A. K., Hristova K., McIntosh T. J., Needham D. Range and magnitude of the steric pressure between bilayers containing lipids with covalently attached polyethylene glycol. Biophys. J. 1995; 68: 1921–1936
- Klibanov A. L., Maruyama K., Torchilin V. P., Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990; 268: 235–237
- Koning G. A., Morselt H. W., Gorter A., Allen T. M., Zalipsky S., Kamps J. A., Scherphof G. L. Pharmacokinetics of differently designed immunoliposome formulations in rats with or without hepatic colon cancer metastases. Pharm. Res. 2001; 18: 1291–1298
- Laverman P., Brouwers A. H., Dams E. T., Oyen W. J., Storm G., van Rooijen N., Corstens F. H., Boerman O. C. Preclinical and clinical evidence for disappearance of long‐circulating characteristics of polyethylene glycol liposomes at low lipid dose. J. Pharmacol. Exp. Ther. 2000a; 293: 996–1001
- Laverman P., Zalipsky S., Oyen W. J., Dams E. T., Storm G., Mullah N., Corstens F. H., Boerman O. C. Improved imaging of infections by avidin‐induced clearance of 99mTc‐biotin‐PEG liposomes. J. Nucl. Med. 2000b; 41: 912–918
- Laverman P., Carstens M. G., Boerman O. C., Dams E. T., Oyen W. J., van Rooijen N., Corstens F. H., Storm G. Factors affecting the accelerated blood clearance of polyethylene glycol‐liposomes upon repeated injection. J. Pharmacol. Exp. Ther. 2001a; 298: 607–612
- Laverman P., Boerman O. C., Oyen W. J.G., Corstens F. H.M., Storm G. In vivo applications of PEG liposomes: unexpected observations. Crit. Rev. Ther. Drug Carr. Syst. 2001b; 18: 551–566
- Lee J., Lentz B. R. Evolution of lipidic structures during model membrane fusion and the relation of this process to cell membrane fusion. Biochemistry 1997; 36: 6251–6259
- Lee J. H., Jeong B. J., Lee H. B. Plasma protein adsorption and platelet adhesion onto comb‐like PEO gradient surfaces. J. Biomed. Mater. Res. 1997; 34: 105–114
- Levchenko T. S., Rammohan R., Lukyanov A. N., Whiteman K. R., Torchilin V. P. Liposome clearance in mice: the effect of a separate and combined presence of surface charge and polymer coating. Int. J. Pharm. 2002; 240: 95–102
- Litzinger D. C., Buting A. M., van Rooilen N., Huang L. Effect of liposome size on the circulation time and intraorgan distribution of amphipathic poly(ethylene glycol)‐containing liposomes. Biochim. Biophys. Acta 1994; 1190: 99–107
- Loughrey H. C., Bally M. B., Reinish L. W., Cullis P. R. The binding of phosphatidylglycerol liposomes to rat platelets is mediated by complement. Thromb. Haemost. 1990; 64: 172–176
- Luk S. C., Musclow E., Simon G. T. Platelet phagocytosis in the spleen of patients with idiopathic thrombocytopenic purpura (ITP). Histopathology 1980; 4: 127–136
- Male R., Vannier W. E., Baldeschwieler J. D. Phagocytosis of liposomes by human platelets. Proc. Natl. Acad. Sci. U. S. A. 1992; 89: 9191–9195
- Male R., Moon D. G., Garvey J. S., Vannier W. E., Baldeschwieler J. D. Organ distribution of liposome‐loaded rat platelets. Biochem. Biophys. Res. Commun. 1993; 195: 276–281
- McLean J. W., Fox E. A., Baluk P., Bolton P. B., Haskell A., Pearlman R., Thurson G., Umemoto E. Y., McDonald D. M. Organ‐specific endothelial cell uptake of cationic liposome‐DNA complexes in mice. Am. J. Physiol. 1997; 273: H387–H404
- Needham D., Stoicheva N., Zhelev D. V. Exchange of monooleoylphosphatidylcholine as monomer and micelle with membranes containing poly(ethylene glycol)‐lipid. Biophys. J. 1997; 73: 2615–2629
- Nishiya T., Sloan S. Interaction of RGD liposomes with platelets. Biochem. Biophys. Res. Commun. 1996; 224: 242–245
- Nishiya T., Lam R. T.T., Enf F., Zerey M., Lau S. Mechanistic study on toxicity of positively charged liposomes containing stearylamine to blood. Artif. Cells Blood Substit. Immobil. Biotechnol. 1995; 23: 505–512
- Noppl‐Simpson D. A., Needham D. Avidin‐biotin interaction at vesicle surfaces: adsorption and binding, cross‐bridge formation, and lateral interactions. Biophys. J. 1996; 70: 1391–1401
- Oja C. D., Semple S. C., Chonn A., Cullis P. R. Influence of dose on liposome clearance: critical role of blood proteins. Biochim. Biophis. Acta 1996; 1281(1)31–37
- Okumura Y., Ishitobi M., Sobel M., Akiyoshi K., Sunamoto J. Transfer of membrane proteins from human platelets to liposomal fraction by interaction with liposomes containing artificial boundary lipid. Biochim. Biophys. Acta 1994; 1194: 335–340
- Papahadjopoulos D., Nir S., Duzgunez N. Molecular mechanisms of calcium‐induced membrane fusion. J. Bioenerg. Biomembranes 1990; 22: 157–179
- Pereira J., Palomo I., Ocqueteau M., Soto M., Aranda E., Mezzano D. Platelet aging in vivo is associated with loss of membrane phospholipid asymmetry. Thromb. Haemost. 1999; 82: 1318–1321
- Perkins V. R., Vaughan D. E., Plavin S. R., Daley W. L., Rauch J., Lee L., Janoff A. S. Streptokinase entrapment in interdigitation‐fusion liposomes improves thrombolysis in an experimental rabbit model. Thromb. Haemost. 1997; 77: 1174–1178
- Peters A. M., Saverymuttu S. H., Bell R. N., Lavender J. P. The kinetic of short‐lived indium‐111‐radiolabelled platelets. Scand. J. Haematol. 1985; 34: 137–145
- Poste G. Liposome targeting in vivo: problems and opportunities. Biol. Cell 1983; 47: 19–38
- Ravoo B. J., Weringa W. D., Engberts J. B.F.N. Membrane fusion in vesicles of oligomerizable lipids. Biophys. J. 1999; 76(1)374–386
- Regen S. L., Jayasurya N., Fabianowski W. Supramolecular surfactants: amphiphilic polymers designed to disrupt lipid membranes. Biochem. Biophys. Res. Commun. 1989; 159: 566–571
- Reinish L. W., Bally M. B., Loughrey H. C., Cullis P. R. Interactions of liposomes and platelets. Thromb. Haemost. 1988; 60: 518–523
- Rybak M. E.M., Renzulli L. A. A liposome based platelet substitute with hemostatic efficacy. Artif. Cells Blood Substit. Immobil. Biotechnol. 1993; 21: 101–118
- Sato Y., Nakajimaya K., Suzuki Y. Interaction of liposomes with human erythrocytes. Chem. Pharm. Bull. 1990; 38: 2228–2232
- Scherphof G. L., Kamps J. A. The role of hepatocytes in the clearance of liposomes from the blood circulation. Prog. Lipid Res. 2001; 40: 149–166
- Semple S. C., Chonn A., Cullis P. R. Influence of cholesterol on the association of plasma proteins with liposomes. Biochemistry 1996; 35(8)2521–2525
- Shibazaki M., Nakamura M., Nitta Y., Endo Y. Displacement of platelets from blood to spleen following intravenous injection of liposomes encapsulating dichloromethylene bisphosphonate. Immunopharmacology 1998; 39: 1–7
- Silvander M., Johnsson M., Edwards K. Effects of PEG‐lipids on permeability of phosphatidylcholine/cholesterol liposomes in buffer and in human serum. Chem. Phys. Lipids 1998; 97: 15–26
- Silvius J. R., Zuckerman M. J. Interbilayer transfer of phospholipid‐anchored macromolecules via monomer diffusion. Biochemistry 1993; 32: 3153–3161
- Sloan S. M., Brown E. B., Liu Q., Frojmovic M. M. Glycoprotein IIb‐IIIa‐liposomes bind fibrinogen but do not undergo fibrinogen‐mediated aggregation. Platelets 2000; 11: 99–110
- Stensrud G., Passi S., Larsen T., Sandset P. M., Smistad G., Monkkonen J., Karlsen J. Toxicity of gamma irradiated liposomes. In vitro interactions with blood components. Int. J. Pharm. 1999; 178: 33–46
- Storm G., Oussoren C., Peeters P. A.M., Berenholz Y. Tolerabilty of liposomes in vivo. Liposome Technology III, G. Gregoriadis. CRC Press, Boca Raton 1984; 345–383
- Suzuki K., Okumura Y. Mechanism of selective release of membrane proteins from human erythrocytes in the presence of liposomes. Arch. Biochem. Biophys. 2000; 379: 344–352
- Talbot W. A., Zheng L. X., Lentz B. R. Acyl chain unsaturaturation and vesicle curvature alter outer leaflet packing and promote poly(ethylene glycol)‐mediated membrane fusion. Biochemisty 1997; 36: 5827–5836
- Tardi C., Drechsler M., Bauer K. H., Brandl M. Steam sterilisation of vesicular phospholipid gels. Int. J. Pharm. 2001; 217: 161–172
- Torchilin V. P., Narula J., Halper E., Khaw B. A. Poly(ethylene glycol)‐coated anti‐cardiac myosin immunoliposomes: factors influencing targeted accumulation in the infarcted myocardium. Biochim. Biophys. Acta 1996; 1279: 75–83
- Vermehren C., Jorgensen K., Frokjaer S. Influence of lipopolymer concentration on liposome degradation and blood clearance. Int. J. Pharm. 1999; 183: 13–16
- Woodle M. C. Surface‐modified liposomes: assessment and characterization for increased stability and prolonged blood circulation. Chem. Phys. Lipids 1993; 64: 249–262
- Yang Q., Guo Y., Li L., Hui S. W. Effects of lipid headgroup and packing stress on poly(ethylene glycol) – induced phospholipid vesicle aggregation and fusion. Biophys. J. 1997; 73: 277–282
- Yu R. Z., Geary R. S., Leeds J. M., Watanabe T., Fitchett J. R., Matson J. E., Mehta R., Hardee G. R., Templin M. V., Huang K., Newman M. S., Quinn Y., Uster P., Zhu G., Working P. K., Horner M., Nelson J., Levin A. A. Pharmacokinetics and tissue disposition in monkeys of an antisense oligonucleotide inhibitor of Ha‐ras encapsulated in stealth liposomes. Pharm. Res. 1999; 16: 1309–1315
- Zalipsky S., Puntambekar B., Boulikas P., Engbers C. M., Woodle M. C. Peptide attachment to extremities of liposomal surface grafted PEG chains: preparation of the long‐circulating form of laminin pentapeptide. YIGSR. Bioconjug. Chem. 1995; 6: 705–708