Abstract
Ischemic renal injury is associated with defects in transport functions of the proximal tubules and urinary concentration ability. To determine whether alterations in expression of various transporter genes contribute to an impairment in renal functions, the expression of various solute transport genes was analyzed in renal cortex and medulla of rabbits with ischemic acute renal failure. Rabbits were subjected to 60 min of renal pedicle clamping followed by 24, 48, or 72 h of reperfusion. Urine volume and glomerular filtration rate were markedly decreased, which were accompanied by an increase in serum creatinine level and fraction Na+ excretion. Glucosuria and phosphaturia were evident during reperfusion periods. These alterations in renal functions were persisted to 72 h after reperfusion. The Na+-dependent uptakes of glucose and phosphate by brush border membrane vesicles were inhibited by 24 h of reperfusion. mRNA levels for Na+-glucose, Na+-phosphate, and Na+-succinate cotransporter analyzed by RT-PCR were not changed by 60 min of ischemia alone, but were significantly reduced by 24 h of reperfusion. mRNA levels for apical Na+-K+-2Cl− cotransporter, NaCl cotransporter, and turea transporter in the medulla were not changed during reperfusion. Protein levels for AQP2 in the medulla, but not AQP1 in the cortex, analyzed by Western blot were significantly reduced at 24 h after reperfusion.
These results suggest that reductions in expression of Na+-cotransporter genes in the proximal tubules may be important factors in the impairment in Na+-dependent reabsorption of solutes and that decrease in AQP2 protein may be involved in defect in urinary concentration ability in rabbits with ischemic acute renal failure.
INTRODUCTION
Acute renal ischemia occurring during severe blood loss such as traumatic injury or a sharp reduction in blood pressure stands as a major cause of acute renal failure Citation[1-3]. Renal ischemia produces significant changes in structure Citation[4-5] and function Citation[[6]] of the proximal tubule, a major site where organic compounds such as glucose, amino acids and Krebs cycle intermediates in the glomerular filtrate are mostly reabsorbed Citation[[7]]. It would be thus expected that an impairment in the reabsorption of various substances may occur in the ischemic kidneys. Indeed, renal ischemia has been reported to cause functional impairment such as reduction in proximal tubule fluid, Na+, phosphate, and glucose reabsorption Citation[[5]], Citation[8-10]. However, the underlying mechanisms have not been fully defined.
In previous studies, we observed in rabbits that uptakes of Na+-dependent transport systems such as glucose, phosphate, succinate, and glutamate by brush-border membrane vesicles (BBMV) are not altered by ischemia (60 min) or ischemia followed by 60 min of reperfusion, suggesting that the impairment in proximal tubular reabsorption of Na+-dependent transport systems is not resulted from a direct defect in the function of the membrane transport system Citation[[9]].
Recently, we observed a marked difference in renal function between the early phase (within 2 h) and the late phase (after 24 h) of reperfusion following 60 min of ischemia in rabbits Citation[[11]]. During the early phase of reperfusion, urine volume was increased and GFR was not significantly altered. But during the late phase both urine volume and GFR were markedly reduced. These data suggest that renal function is severely impaired during the late phase of reperfusion. However, alterations in Na+-dependent transport systems during the late phase of reperfusion remains unclear.
The present study was therefore undertaken to characterize alterations in function of membrane transporters during the late phase of reperfusion and to examine whether an impairment in Na+-dependent reabsorption of solutes is associated with changes in gene expression of these transporters.
MATERIALS AND METHODS
Induction of Ischemia-Reperfusion and Clearance Experiments
New Zealand White rabbits were anesthetized by intramuscular injection of xylazine (2.5 mg/kg) and ketamine (15 mg/kg) and was maintained by sustaining doses (ketamine 5 mg/kg and xylazine 1 mg/kg) at intervals of approximately 30 min. A polyethylene catheter was introduced into the carotid artery for recording blood pressure and for blood sampling, and the ear vein was catheterized for infusion for solution. Animals were placed on a heating pad to maintain their body temperature at a constant level. The kidneys were exposed and nontraumatic vascular clamps were applied across the pedicles of both kidneys. Completeness of ischemia was verified by the blanching of the kidneys, indicating cessation of blood flow. Blood flow to the kidneys was reestablished after 1 h by removal of the clamps. The animal was allowed to recover. Urine samples were collected with metabolic cages at an interval of 24 h and blood samples were taken at the same interval. The animals were sacrificed at 24, 48, or 72 h after reperfusion, and the kidneys were removed. Cortex and medulla were separated and snap-frozen. For control, kidneys were removed at the time of initial surgery without clamping. Urine and blood samples were analyzed for glucose, creatinine, and phosphate Citation[[12]]. Glomerular filtration rate (GFR) was calculated from endogenous creatinine clearance.
Preparation of Plasma Membrane Vesicles
Animals, in which changes in renal function by ischemia-reperfusion were evident, were euthanized at 24, 48, and 72 h. Brush-border membrane vesicles (BBMV) were isolated by Mg2+ precipitation method, as previously described Citation[[13]]. The vesicles were suspended in the vesicle buffer, adjusted to yield a protein concentration of 6 mg/mL, and stored at −70°C until use. The composition of vesicle buffer is given in the figure legends. Protein was determined according to Bradford Citation[[14]] using γ-globulin as a standard.
Transport Studies in Menbrane Vesicles
The uptake of substrates by vesicles was measured by a rapid filtration technique. Briefly, the reaction was initiated by adding membrane vesicles to the incubation medium (a 1:10 dilution of membrane vesicle suspension) containing the appropriate concentration of 14C-labeled substrate at 25°C. The composition of the incubation medium is given in the figure legends. At the designated times, 100 μl aliquots were taken and quickly filtered under vacuum through Millipore filters (HAWP, 0.45 μm pore size) which had been soaked overnight in distilled water. The filters were then washed with 5 mL of ice-cold stop solution comprising the identical composition to the incubation medium but without substrate and dissolved in 1.0 mL of methoxyethanol. After addition of 1 mL of scintillation cocktail, the amount of radioactivity taken up by vesicles was determined by liquid scintillation spectrophotometry. Non-specific binding of radioactive substrate to the plasma membrane was determined by incubating vesicles in transport buffer containing 0.1% deoxycholate and radiolabeled substrates. All uptake data were corrected for nonspecific binding. All the radioactive compounds were purchased from the Amersham International (Amersham, UK).
RNA Isolation and RT-PCR
Total cellular RNA was extracted from renal cortex or medulla using RNAzol B (Tel. Test. Inc, Friendswood, TX). RNA was quantified by spectrophotometry and formaldehyde gel electrophoresis, and stored at −80°C. Total RNA was reverse transcribed into first stand cDNA using oligo dT primer, and amplified by 35 cycles (94°C, 1 min; 52°C, 1 min; 72°C, 1 min) of polymerase chain reaction (PCR) using 20 pmole of specific primers. On completion of the PCR reaction, products were examined on 2% agarose gel. β-actin primers were used as an internal standard. The primers used for PCR are as follows:
Western Blot Analysis
Membrane fractions were prepared from the kidneys as previously described Citation[[15]]. Briefly, the outer cortex and inner medulla of kidneys were isolated, minced finely, and homogenized in 10 mL of dissecting buffer (0.3 M sucrose, 25 mM imidazole, 1 mM EDTA, 8.5 μM leupeptin, and 1 mM phenylmethylsulfonyl fluoride, pH 7.2) with a motor-driven Potter-Elvehjem homogenizer. This homogenate was centrifuged in a Beckman L8M centrifuge at 4,000 g for 15 min at 4°C to remove nuclei and mitochondria. The supernatant was centrifuged at 200,000 g for 1 h. The resultant pellet, containing a mixture of plasma membranes and intracellular vesicles, was resuspended in ∼100 μL of dissecting buffer and assayed for protein concentration Citation[[14]].
Samples of membrane fractions from kidneys (100 μg/lane) were run in duplicate on 12% polyacrylamide minigels (Bio-Rad Mini Protean II). One gel was Coomassie stained to ensure that loading in the lanes was consistent, while the other was subjected to immunoblotting. After transfer by electroelution to nitrocellulose membranes, blots were blocked with 5% milk in PBS-T (80 mM Na2HPO4, 20 mM NaH2PO4, 100 mM NaCl, and 0.1% Tween 20, pH 7.5) for 1 h and incubated with affinity-purified anti-AQP2 (40 ng IgG/L). The labeling was visualized with horseradish peroxidase (HRP)-conjugated secondary antibody (diluted 1: 1,000; Amersham International) using an enhanced chemiluminescence system (ECL, Amersham International).
RESULTS
Alterations in Renal Function in Postischemic Kidneys
summarized alterations in renal function in postischemic kidneys. When kidneys were reperfused for 24 h following 60 min ischemia, urine volume and GFR were markedly decreased, which were accompanied by an increase in serum creatinine level and fractional Na+ excretion. These data are consistent with those observed in our previous studies Citation[[11]]. These changes were persisted even when reperfusion was prolonged to 72 h (data not shown).
Table 1. Alterations in renal function in control and postischemic kidneys
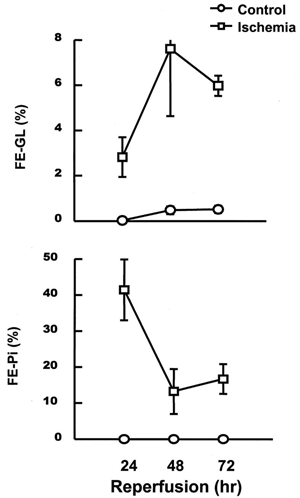
depicts changes in reabsorption of glucose and phosphate in kidneys subjected to 24–72 h reperfusion following ischemia. After 24 h of reperfusion, the fractional excretion of glucose and phosphate was significantly increased, and such changes were continued even after 72 h of reperfusion.
Figure 1. Alterations in fractional excretion of glucose (FEGl) and phosphate (FEPi). These changes were measured at 24–72 h of reperfusion following 60 min of ischemia in rabbits. Control samples were taken from sham-operated rabbits. Data are mean ± SE of 4 animals in each group.
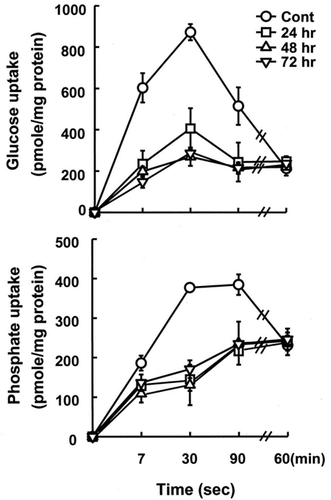
Alterations in Uptakes of Glucose and Phosphate by BBMV in Postischemic Kidneys
In order to examine if impairment in reabsorption of glucose and phosphate in postischemic kidneys was resulted from a direct defect of the transport systems in the brush border membrane of the proximal tubule, glucose and phosphate uptake by BBMV was measured. The time course of glucose uptake by BBMV isolated from the control and ischemic kidneys was determined in the presence of an inwardly-directed Na+ gradient. As shown in A, the uptake in the control vesicles increased rapidly during the initial period, then declined to equilibrium value, showing a characteristic ‘overshoot’ phenomenon of a Na+-gradient-dependent transport process. However, when kidneys were subjected to 24–72 h of reperfusion following ischemia, glucose uptake was significantly lower than that of control kidneys, and the overshoot phenomena were disappeared. The initial uptake in the absence of extravesicular Na+ were not altered by reperfusion (data not shown). Similar results were observed in phosphate uptake (B). These data indicate that 24 h of reperfusion causes a direct impairment in Na+-dependent transport systems for glucose and phosphate on the brush-border membrane.
Figure 2. Alterations in uptakes of D-glucose and phosphate in the presence of an inwardly-directed Na+ gradient by brush-border membrane vesicles isolated from control and postischemic kidney. Postischemic kidneys were subjected to 24–72 h of reperfusion following 60 min of ischemia. Membrane vesicles were suspended in a buffer containing 100 mM mannitol, 100 mM KCl, and 20 mM Hepes/Tris (pH 7.5), and were incubated in a buffer containing 100 mM mannitol, 100 mM NaCl, and 20 mM Hepes/Tris (pH 7.5). The concentration of substrate was 50 μM. Data are mean ± SE of 4 animals in each group.
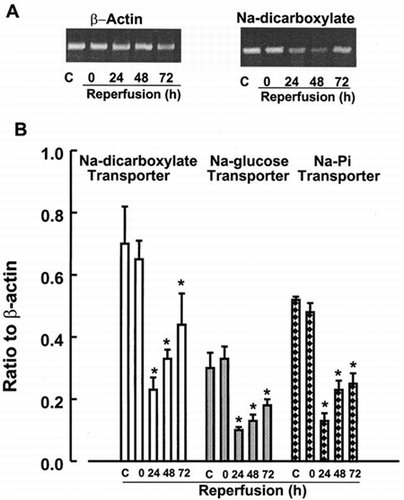
Alteratioans in Expression of Transporters and Aquaporins in Postischemic Kidneys
Expression of the solute transporters in kidney cortex was examined by RT-PCR analysis. Expression levels of Na+-glucose, Na+-dicarboxylate and Na+-phosphate transporters were not changed by 60 min of ischemia alone. Na+cotransporters mRNA levels decreased at 24 h after reperfusion and recovered gradually, but remained suppressed at 72 h of reperfusion ().
Figure 3. RT-PCR analysis of expression of Na+-cotransporters in kidney cortex. Transcript levels of Na+-dicarboxylate, Na+-glucose and Na+-Pi were analyzed in cortex from control kidneys or kidneys subjected to 0 (ischemia), 24, 48, and 72 h of reperfusion following 60 min of ischemia. Data represent ratio to b-actin signals and are mean ± SE of 3 separate experiments. *p<0.05 compared with the control.
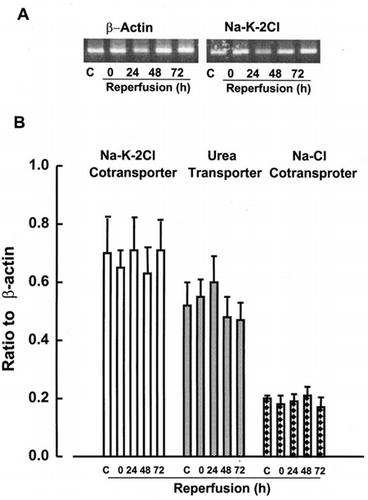
Apical Na+-K+-2Cl cotransporters are involved in Na+ reabsorption in the ascending limb of Henle Citation[[16]]. Na+ reabsorption in the distal convoluted tubule and collecting duct is mediated predominantly via Na+-Cl− cotransporter Citation[[17]]. Urea transporter (UT2) is located in descending thin limbs of Henle's loop, and participates in urinary recycling processes, which minimize urea escape from the inner medulla Citation[[18]]. We examined effect of reperfusion on these transport systems. As shown in , by contrast with significant reduction of Na+-dependent transporters in the kidney cortex, expressions of Na+-K+-2Cl− cotransporter, Na+-Cl− cotransporter, and UT2 mRNAs in the medulla were not affected by ischemia itself or ischemia-reperfusion injury. The expression of Na+-K+-2Cl and Na+-Cl− cotransporters in the cortex was not affected by ischemia-reperfusion (data not shown).
Figure 4. RT-PCR analysis of Na+-K+-2Cl, urea, and NaCl transporters in renal medulla. Transcript levels of these transporters were analyzed in medulla at control kidneys or kidneys subjected to 0 (ischemia), 24, 48, and 72 h of reperfusion following 60 min of ischemia. Data represent ratio to β-actin signals and are mean ± SE of 3 separate experiments.
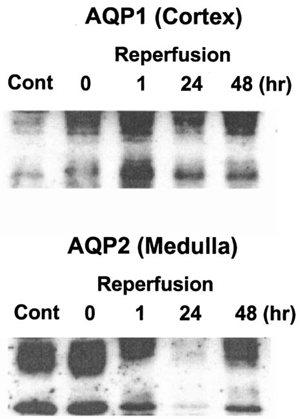
In the last series of experiments, we examined changes in expression levels of aquaporins during reperfusion following ischemia by Western blot analysis using rabbit polyclonal AQP1 and AQP2 antibodies. Membrane fractions were isolated from the cortex and the inner medulla from kidneys. Expression of AQP1 protein, that are expressed in the proximal tubule and the descending limb of Henle's loop Citation[[19]], did not change during reperfusion. Although the amount of AQP2 protein was not affected by 60 min ischemia itself, it was markedly reduced by 24 h of reperfusion after ischemia and its reduction was persisted even after 48 h of reperfusion ().
Figure 5. Western analysis of AQP1 and AQP2. Kidneys were subjected to 0 (ischemia), 24, 48, and 72 h of reperfusion following 60 min of ischemia. Cortex and inner medulla of the kidneys were separated and membrane fractions were prepared as described in “Methods and Materials”. Polyclonal anti-rabbit antibodies for AQP1 and AQP2 were used for immunoblot analysis.
DISCUSSION
The present study demonstrates that during the late phase (24 h) of ischemic acute renal failure in rabbits oliguria and reduction in GFR were observed. Such changes are different from those during the early phase (within 2 h) of reperfusion following 60 min of ischemia Citation[[9]], and consistent with the results of our previous studies Citation[[11]].
In the present study, reabsorption of glucose and phosphate was markedly impaired at 24 h of reperfusion and was not recovered up to 72 h after reperfusion (), indicating a significant defect in the proximal tubular function during reperfusion following ischemia. Since the brush-border membrane plays an important role in proximal tubular reabsorption processes of organic solutes, an impaired reabsorption of glucose and phosphate could result from a direct alteration of brush-border membrane. Such a mechanism has been suggested for the reduction in proximal reabsorption of glucose in rats subjected to ischemia Citation[[20]]. Indeed, the present study showed that Na+-dependent uptakes of glucose and phosphate by BBMV were significantly reduced in rabbits subjected to 24 h of reperfusion following ischemia and these changes were persisted to 72 h (), similarly to the clearance data on reabsorption of these solutes. These results are different from the data observed at 60 min of reperfusion following ischemia Citation[[9]]. In that study, glucosuria and phosphaturia were not resulted from alterations in uptakes of these solutes by BBMV. Taken together,
The present study also demonstrates that reduction in Na+-dependent uptakes of glucose and phosphate by BBMV were accompanied by the reduction in mRNA levels for these cotransporters (), suggesting that the decrease in the uptake may partly be related to reduction in protein synthesis of these transporters. These results are consistent with the finding that an impairment in tetraethylammonium Citation[[21]] and glucose Citation[[20]] uptake by BBMV isolated from ischemic kidney are associated with a decrease in their Vmax. However, the molecular mechanism of the decrease in expression of Na+-solutes cotransporter genes during reperfusion injury is not clear. One possibility is that severe ischemic injury in the proximal tubule causes a general decrease in transcriptional activity, which in turn leads to suppression of mRNA levels for membrane transporters, including Na+ cotransport systems. However, this possibility could be excluded by the data that AQP1 in the cortex is not affected in rabbits subjected to reperfusion ().
One of the main functional changes in experimental animals with ischemic acute renal failure is a defect in urine concentration ability Citation[[22]]. The urine concentrating ability is determined by the osmotic gradient between the medullary interstitium and tubular fluid and the water permeability of the tubular membrane. Since the major factors that contribute to the osmotic gradient are active NaCl and passive urea transport, a defect of urine concentration ability in ischemic acute renal failure could be resulted from reduction in expression of these transporter genes. In fact, reduction in expression of NHE-3, Na+/H+ exchanger, and apical Na+-K+-2Cl− cotransporter was reported in the rat kidney subjected to reperfusion following 30 min of ischemia Citation[[23]]. However, in the present study, any changes in expressions of Na+-K+-2Cl− cotransporter, urea transporter, and NaCl cotransporter were not observed in rabbits subjected to reperfusion (). Although the reason of this discrepancy is not clear from the present study, the rat kidney may be more vulnerable to ischemic damage than the rabbit kidney. Similar difference was also observd in glucose transport in BBMV. In rat only 15 min ischemia decreased sodium-dependent glucose uptake in BBMV Citation[[20]], whereas in rabbit 1 h ischemia did not affect it Citation[[9]].
A recent study has made it clear that osmotic water permaebility across the tubular epithelium is chiefly dependent on aquaporin water channels Citation[[24]]. At least six aquaporins are expressed in the kidney. The archetypal member of the aquaporin family, AQP1 Citation[[25]], is highly abundant in the proximal tubule, the descending thin limb of Henle, and the vasa recta Citation[[19]]. AQP2 is abundant in the apical membrane and apical vesicles in the collecting duct principal cells Citation[[26]] and is the primary target for vasopressin regulation of collecting duct water permeability Citation[[27]]. Changes in AQP2 expression were associated with various water balance disorders Citation[[24]]. The data in this study showed that AQP2 expression, not AQP1, was significantly reduced from 24 h after reperfusion, suggesting that reduction of AQP2 expression may be, in part, responsible for the concentration defect in rabbits with ischemic acute renal failure.
ACKNOWLEDGMENT
This study was supported by a Grant from the Korea Science and Engineering Foundation (96-0403-10-01-3).
REFERENCES
- Anderson R J, Schrier R W. Acute tubular necrosis. Acute renal failure, R Schrier, C Gottschalk. Little, Brown and Co., Boston, Toronto 1988; 1413–1446
- Bonventre J V. Mediators of ischemic renal injury. Annu Rev Med 1988; 39: 531–544
- Bonventre J V. Mechanisms of ischemic renal failure. Kidney Int 1993; 43: 1160–1178
- Hostetter T H, Brenner B M. Renal circulatory and nephron function in experimental acute renal failure. Acute renal failure, B M Brenner, J M Lazarus. Churchill Livingstone, New York 1988; 67–89
- Stein J H, Lifschitz M D, Barnes L D. Current concepts on the pathophysiology of acute renal failure. Am J Physiol 1978; 234: F171–F181
- Oliver J, MacDowell M, Tracy A. The pathogenesis of acute renal failure associated with traumatic and toxic injury. J Clin Invest 1951; 30: 1307–1314
- Berry C A, Rector F C, Jr. Renal transport of glucose, amino acid, sodium, chloride and water. The kidney, B M Brenner, F C Rector, Jr. Saunders Company, Philadelphia, London, Toronto 1991; 245–282
- Hanley M J. Isolated nephron gegments in a rabbit model of ischemic acute renal failure. Am J Physiol 1980; 239: F17–F23
- Kim Y K, Woo J S, Kim Y H, Jung J S, Kim B S, Lee S H. Effect of renal Ischemia on organic compound transport in rabbit kidney proximal tubule. Pharmacol Toxicol 1995; 77: 121–129
- Spiegel D M, Wilson P D, Molitors B A. Epithelial polarity following ischemia: a requirement for normal cell function. Am J Physiol 1989; 256: F430–436
- Kim S Y, Lim Y T, Kim B S, Cho S I, Woo J S, Jung J S, Kim Y K. Mechanism of reduced GFR in rabbits with ischemic acute renal failure. Renal Failure, (in press), 1999
- Fiske C H, SubbaRow Y. The colorimetric determination of phosphorus. J Biol Chem 1925; 66: 375–400
- Jung J S, Kim Y K, Lee S H. Characteristics of tetraethylammonium transport in rabbit renal plasma-membrane vesicles. Biochem J 1989; 259(2)377–383
- Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248–254
- Maples D, Christensen B M, Frokiaer J, Knepper M A, Nielsen S. Dehydration reverse vasopressin antagonist-induced diuresis and aquaporin-2 down regulation in rat. Am J Physiol 1998; 275: F400–F409
- Xu J C, Lytle C, Zhu T T, Payne J A, Benz E, Jr, Forbush B. Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci USA 1994; 91: 2201–2205
- Gamba G, Miyanoshita A, Lombardi M, Lytton J, Lee W S, Hediger M A, Hebert S C. Molecular cloning, primary structure and characterization of two members of the mammalian eletroneutral sodium-(potassium)-chloride cotransporter family expressed in kidney. J Biol Chem 1994; 269: 17713–17722
- Tsukaguchi H, Shayakul C, Berger U V, Hediger M A. Urea transporters in kidney: molecular analysis and contribution to the urinary concentrating process. Am J Physiol 1998; 275: F319–324
- Nielsen S, Smith B L, Christensen E I, Knepper M A, Agre P. Water channels are localized in constitutively water-permeable segments of the nephron. J Cell Biol 1993; 120: 371–383
- Molitoris B A, Kinne R. Ischemia induces surface membrane dysfunction. Mechanism of altered Na+-dependent glucose transport. J Clin Invest 1987; 80: 647–654
- Kim Y K, Kim Y H, Jung J S, Lee S H. Effect of renal ischemia on organic anion and cation transport in rabbit proximal tubule. Kidney Blood Press Res 1996; 19: 332–339
- Nihei H, Honda N, Suzuki K, Nagase M, Yoshitoshi Y. Renal hemodynamics and Medullary osmolal gradient in ischemic acute renal failure in rabbits. Jpn Heart J 1975; 16: 44–56
- Wang Z, Rabb H, Haq M, Shull G E, Soleimani M. A possible molecular basis of natriuresis during ischemic-reperfusion injury in the kidney. J Am Soc Nephrol 1998; 9: 605–613
- Nielsen S, Kwon T H, Christensen B M, Promeneur D, Frokiaer J, Marples D. Physiology and pathophysiology of renal aquaporins. J Am Soc Nephrol 1999; 10: 647–663
- Agre P, Preston G M, Smith B L, Jung J S, Raina S, Moon C, Guggino W B, Nielsen S, Aquaporin CHIP. The archetypal molecular water channel. Am J Physiol 1993; 265: F463–F476
- Nielsen S, DiGiovanni S R, Christensen E I, Knepper M A, Harris H W. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA 1993; 90: 11663–11667
- Knepper M A. Molecular physiology of urinary concentrating mechanism: Regulation of aquaporin water channels by vasopressin. Am J Physiol 1997; 272: F3–F12