Abstract
We describe a 29-old-year Japanese man with autosomal recessive polycystic kidney disease who was frequently hypoglycemic. Insulinoma as a cause of hypoglycemia was denied because the ratio of plasma immunoreactive insulin to glucose was low. Adrenal insufficiency was diagnosed because of the low urinary excretion of 17-hydroxycorticosteroids, and both blunted responses of plasma cortisol to an intravenous injection of adrenocorticotropin and of plasma adrenocorticotropin to an intravenous injection of human corticotropin releasing hormone were observed, although basal plasma concentrations of cortisol and adrenocorticotropin were normal. The elusion profile of plasma sample from our patient chromatographed on a Sephadex G-75 column showed two peaks of (1-39)-ACTH and β-lipotropin, with no evidence of high molecular weight form of ACTH. The plasma concentrations of thyroid stimulating hormone and growth hormone were within the normal range. These findings indicated that this patient with autosomal recessive polycystic kidney disease was associated with adrenal insufficiency due to isolated adrenocorticotropin deficiency.
Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is a rare inherent disorder characterized by chronic renal failure associated with medullary ductal ectasia, hepatic fibrosis, and portal hypertension.Citation[[1]], Citation[[2]], Citation[[3]], Citation[[4]] This disorder clinically manifests during early childhood. The prognosis is usually poor for patients with severe disease, but in milder form, variable renal failure develops even in later childhood or adolescence. Intracranial aneurysms have been reported to be consistently associated with autosomal dominant polycystic kidney disease (ADPKD), whereas only a few case reports describe the abnormalities associated with ARPKD other than hepatic fibrosis and portal hypertension.Citation[[5]], Citation[[6]], Citation[[7]]
We present here an adult with ARPKD who had adrenal insufficiency due to isolated adrenocorticotropin (ACTH) deficiency.
Case Report
A 29-old-year Japanese man with ARPKD was admitted because of diarrhea and fever to Hamamatsu University School of Medicine on September 17, 1998. At the age of 10, he had undergone esophageal transection and splenectomy to treat the rupture of esophageal varices complicated with ARPKD. At surgery, liver and kidney biopsies revealed ARPKD. None of his siblings were similarly affected. In 1987 and 1996, he underwent sclerotherapy for esophageal varices. His renal function had gradually declined over time.
On physical examination, blood pressure was 130/76 mmHg and his pulse rate was 102 beats per minute with regular. Body temperature was 38.5°C. He appeared well except for fever. Bowel sound was activated. The rest of the physical examination was unremarkable.
Laboratory data on the day of admission were as follows: hematocrit, 34.3%; hemoglobin, 11.3 g/dL; white blood cell count, 21,500/mm3 (segments 46%, bands 39%, lymphocytes 12%, eosinophils 0%, basophils 1%, monocytes 2%); platelets, 27.5 × 104/mm3; blood urea nitrogen, 42.4 mg/dL; creatinine, 5.1 mg/dL; sodium, 133 mEq/L; potassium, 6.3 mEq/L; chloride, 100 mEq/L; calcium, 4.9 mEq/L; phosphorus, 2.4 mg/dL; albumin, 3.4 g/dL; GOT, 247 IU/L; GPT, 148 IU/L, γ-GTP, 168 IU/L; total bilirubin, 0.9 mg/dL; total cholesterol, 139 mg/dL; choline esterase, 0.71 U/L; CRP, 17.0 mg/dL. The urinary protein value was 2+, and 20 red cells per high power field and no white cells were found. Arterial blood gas analysis revealed moderate metabolic acidosis with a normal anion gap and mild respiratory alkalosis (pH of 7.38, Pco2 of 26.7 mmHg, bicarbonate of 16.3 mmol/L).
Acute cholangitis and acute colitis were diagnosed, on the basis of the abdominal ultrasonography findings of edematous walls of the gallbladder and the colon. The right lobe was atrophic and the left and caudate lobes were enlarged in the liver. The spleen was undetectable, and no mass was found in the pancreas. Echogenicity of the renal parenchyma was increased with poor differentiation between the cortex and the medulla. This patient was prescribed cefoperazone sodium for acute cholangitis and acute colitis, and recovered from fever and diarrhea, accompanied by normalization of the serum transaminase levels within 7 days.
Since October 19, frequent syncope attacks lasted for a few minutes particularly before lunch. Computed tomography of the brain and electroencephalogram were unremarkable. Syncope attacks were presumed to be caused by hypoglycemia because the plasma glucose concentrations were below 50 mg/dL at every syncope attack. Endocrinological evaluations were undertaken to elucidate the cause of hypoglycemia.
Methods
A rapid ACTH stimulation test using 250 µg of intravenous synthetic corticotropin (Cortrosyn®, Daiichi Pharmaceuticals, Tokyo, Japan) was performed. Plasma samples were assayed for cortisol and aldosterone at 0, 30, and 60 min after injection. Human corticotropin releasing hormone (CRH) (Corticorelin®, Mitsubishi Tokyo, Tokyo, Japan) was administered as an intravenous bolus injection at a dose of 1 µg/kg. Plasma samples were assayed for cortisol and ACTH at 0, 30, 60, 90, and 120 min after CRH injection.
To analyze the molecular size of the circulating ACTH, gel chromatography was performed, as described previously.Citation[[8]] Briefly, acidified plasma was chromatographed on a Sephadex G-75 column equilibrated with 1% formic acid.
Hormone-Assay Methods
Plasma glucose was measured by the hexokinase method. Plasma concentrations of cortisol and aldosterone were determined by a radioimmunoassay (reference ranges: 5.3–11.0 µg/dL and 4.7–13.1 ng/dL, respectively). Plasma concentrations of ACTH and growth hormone (GH) were determined by an immunoradiometric assay (reference ranges: 4–48 pg/mL and 1.4–4.2 ng/mL, respectively). Plasma concentrations of immunoreactive insulin (IRI) and thyroid stimulating hormone (TSH) were determined by a chemiluminescent enzyme immunoassay (reference ranges: 5–19 and 0.5–4.1 µU/mL, respectively). All blood samples were collected in tubes on ice and centrifuged immediately. Plasma was stored at −20°C until assay.
Results
shows the results of the day profile of plasma concentrations of glucose and IRI. The plasma concentration of IRI was 17 µU/mL before breakfast when that of glucose was 68 mg/dL. The fasting ratio of plasma IRI to the glucose concentration were between 0.13 and 0.26.
Table 1. Day profile of fasting plasma levels of glucose, insulin, and ratio of insulin to glucose
Urinary excretion levels of 17-hydroxycortisteroids (17-OHCS) and 17-hydroxyketosteroids (17-KS) were 1.57 and 1.91 mg/g creatinine/24 h, respectively. Although basal plasma concentrations of cortisol and ACTH were normal (8.1 µg/dL and 38 pg/mL, respectively), the response of the plasma cortisol concentration to ACTH injection was considered subnormal (8.1 to 14.0 µg/dL) compared with the normal response. On the other hand, the response of the plasma aldosterone concentration to ACTH injection was normal as shown in (3.8 to 11.0 ng/dL). shows that the response of plasma ACTH to the CRH injection was blunted compared with the normal response because it did not fully increase within 30 min thereafter (62 to 72 pg/mL). The elusion profile of plasma sample from our patient chromatographed on a Sephadex G-75 column showed two peaks of (1-39)-ACTH and β-lipotropin, with no evidence of high molecular weight form of ACTH as shown in . Plasma concentrations of TSH and GH were within the normal range (1.61 µU/mL and 7.6 ng/mL, respectively).
Figure 1. Response of plasma concentrations of cortisol and aldosterone to 250 µg of synthetic adrenocorticotropin (ACTH) injection. The response of plasma cortisol to ACTH injection is subnormal compared with normal response. The response of plasma aldosterone concentration is normal.
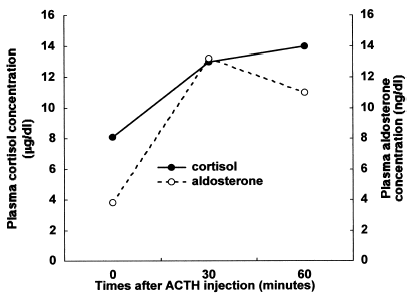
Figure 2. Response of plasma adrenocorticotropin (ACTH) concentration to 1 µg/kg of human corticotropin releasing hormone (CRH) injection. Response of plasma ACTH to CRH is blunted compared with normal response.
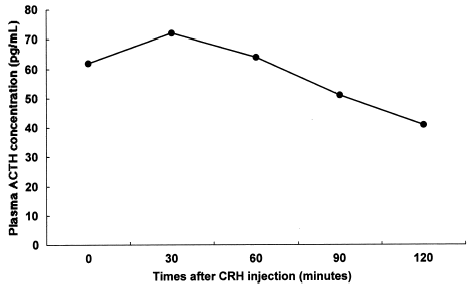
Figure 3. Gel filtration of plasma from our patient on a Sephadex G-75 column. The elusion profile of plasma sample from our patient chromatographed on a Sephadex G-75 column showed two peaks of (1-39)-ACTH and β-lipotropin, with no evidence of high molecular weight form of ACTH.
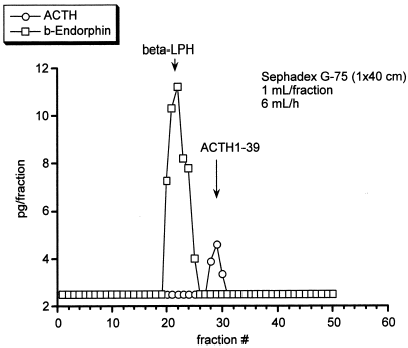
Discussion
A patient with ARPKD in this case report developed frequent syncope attacks caused by fasting hypoglycemia. Although hypoglycemia is associated with several conditions or diseases,Citation[[9]], Citation[[10]] ARPKD was not been identified as one of them. Fasting hypoglycemia can be caused by severe liver disease through inadequate gluconeogenesis,Citation[[9]] but our patient did not appear to have liver disease severe enough to cause hypoglycemia since serum choline esterase level was within the normal range.
The ratio of plasma IRI to glucose in patients with insulinoma is usually above 0.3 and it increases with fasting.Citation[[11]] Thus, we considered it unlikely that fasting hypoglycemia in our patient was caused by insulinoma because the ratio of fasting plasma IRI to glucose concentration were between 0.13 and 0.26.
Urinary excretion of 17-OHCS, an integrated measure of daily cortisol synthesis, was below the normal range, suggesting adrenal insufficiency.Citation[[12]] The response of the plasma ACTH concentration to CRH was blunted compared with the normal response, suggesting disturbed ACTH secretion. The plasma concentrations of TSH and GH were within the normal range. These findings suggest that our patient had adrenal insufficiency due to isolated ACTH deficiency. The secretion of ACTH sometimes becomes subtly impaired in patients with an isolated ACTH deficiency that undergoes procedures designed to increase ACTH secretion even though basal plasma cortisol and ACTH occasionally are normal.Citation[[12]], Citation[[13]]
Yamakita et al. have reported a patient with corticotropinoma showing no Cushingoid state whose plasma ACTH level measured with an immunoradiometric assay was extremely high and plasma cortisol level was within the normal range.Citation[[14]] The elusion profile of plasma sample from the patient chromatographed on a Sephadex G-75 column showed two peaks of ACTH, one of which was compatible with normal (1-39)-ACTH and another with high molecular weight ACTH. In addition, two another cases of the hypersecretion of high molecular weight ACTH without biological function have been reported.Citation[[15]], Citation[[16]] However, the circulating ACTH from our patient was (1-39)-ACTH, with no evidence of high molecular weight form.
The response of plasma cortisol to the stimulation of ACTH in our patient was sluggish compared with the normal response. The adrenal response to ACTH injection is likely to be sluggish when the adrenal gland has not been chronically stimulated by adequate circulating concentration of ACTH.
Isolated ACTH deficiency is rare and usually diagnosed during adulthood.Citation[[12]] However, the pathogenesis of the disease is usually uncertain. Several symptoms, including hypotension, weakness, weight loss, episodes of fever, abdominal pain, nausea, vomiting, and hypoglycemia are frequent in isolated ACTH deficiency. Among them, hypoglycemia is the most common biochemical abnormality.Citation[[12]]
Hypoglycemia is a relatively frequent problem among hospitalized patients.Citation[[10]] Approximately half are related to renal insufficiency caused by nondiabetic, as well as diabetic nephropathy. Hypoglycemia in diabetic patients is mostly related to insulin therapy. However, several mechanisms including reduced renal glucogenesis, decreased caloric intake and an impaired counter regulatory mechanism appear to account for the development of hypoglycemia observed in renal insufficiency caused by nondiabetic nephropathy.Citation[[10]] Thus, in addition to adrenal insufficiency, renal failure caused by ARPKD probably may exaggerate the development of hypoglycemia in our patient.
The linkage between ARPKD and isolated ACTH deficiency remains to be elucidated and is likely to be coincidental. In murine ARPKD model, the mislocalization of epidermal growth factor (EGF) receptors to the apical surfaces of the collecting tubule epithelium from the basolateral surface, and the tyrosine kinase activity of the EGF receptor are involved in collecting tubular cyst formations.Citation[[17]] On the other hand, EGF directly elevates plasma ACTH concentrations in sheep,Citation[[18]] although enhanced pituitary ACTH secretion by EGF is mediated through hypothalamic CRH secretion.Citation[[19]] Thus, a disturbance of the EGF receptor and subsequent tyrosine kinase activation in the pituitary gland may contribute to isolated ACTH deficiency in patients with ARPKD.
In conclusion, we describe a patient with ARPKD that was manifested as frequent syncope attacks due to hypoglycemia caused by adrenal insufficiency, which was ascribed to isolated ACTH deficiency. The linkage between ARPKD and isolated ACTH deficiency remains to be investigated.
References
- Zerres K., Rudnik-Schöneborn S., Steinkamm C., Mücher G. Autosomal recessive polycystic kidney disease. Nephrol. Dial. Transplant. 1996; 11(suppl 6)S29–S33
- Zerres K., Rudnik-Schöneborn S., Mücher G. Autosomal recessive polycystic kidney disease: clinical features and genetics. Adv. Nephrol. 1996; 25: 147–157
- Fick G.M., Gabow P.A. Hereditary and acquired cystic disease of the kidney. Kidney Int. 1994; 46: 951–964
- Shaikewitz S.T., Chapman A. Autosomal recessive polycystic kidney disease: issues regarding the variability of clinical presentation. J. Am. Soc. Nephor. 1993; 3: 1858–1862
- Bates C.M., Baum M., Hunchik M., Quan A. Acute vision loss in children with autosomal recessive polycystic kidney disease. Am. J. Kidney Dis. 1999; 34: 1125–1128
- Neumann H.P., Krumme B., van Velthoven V., Orszagh M., Zerres K. Multiple intracranial aneurysms in a patient with autosomal recessive polycystic kidney disease. Nephrol. Dial. Transplant. 1999; 14: 936–939
- Revai T., Harmos G. Steroid-sensitive nephrotic syndrome and minimal-change nephropathy associated with autosomal-recessive polycystic kidney disease. Clin. Nephrol. 1999; 52: 65
- Ratter S.J., Lowry P.J., Besser G.M., Rees L.H. Chromatographic characterization of adrenocorticotrophin in human plasma. J. Endocrinol. 1980; 85: 359–369
- Service F.J. Hypoglycemic disorders. N. Engl. J. Med. 1995; 332: 44–1152
- Fischer K.F., Lees J.A., Newman J.H. Hypoglycemia in hospitalized patients: causes and outcomes. N. Engl. J. Med. 1986; 315: 1245–1250
- Dizon A.M., Kowalyk S., Hoogwerf B.J. Neuroglycopenic and other symptoms in patients with insulinomas. Am. J. Med. 1999; 106: 307–310
- Stacpoole P.W., Interlandi J.W., Nicholson W.E., Rabin D. Isolated ACTH deficiency: a heterogenous disorder. Critical review and report of four new cases. Medicine 1982; 61: 13–24
- Steer P., Marnell R., Werk E.E., Jr. Clinical alcohol hypoglycemia and isolated adrenocorticotropic hormone deficiency. Ann. Intern. Med. 1969; 71: 343–348
- Yamakita N., Murai T., Kawamura S., Teramachi H., Matsuhisa T., Hirata T., Ikeda T., Morita H., Mune T., Yasuda K. High molecular weight corticotropin measured with immunoradiometric assay in a patient with asymptomatic pituitary corticotropinoma. Endocr. J. 1999; 46: 563–571
- Reincke M., Allolio B., Saeger W., Kaulen D., Winkelman W. A pituitary adenoma secreting high molecular weight adrenocorticotropin without evidence of Cushing's disease. J. Clin. Endocrinol. Metab. 1987; 65: 1296–1300
- Hashimoto K., Kaneda T., Nagano I., Asaba K., Takeda K., Takao T. Pituitary adenoma showing intermittent secretion of high molecular weight adrenocorticotropin without evidence of Cushing's disease. Horm. Res. 1999; 52: 39–44
- Richards W.G., Sweeney W.E., Yoder B.K., Wilkinson J.E., Woychik R.P., Avner E.D. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J. Clin. Invest. 1998; 101: 935–939
- Polk D.H., Gore-Ervin M., Padbury J.F., Lam R.W., Reviczky A.L., Fisher D.A. Epidermal growth factor acts as a corticotropin-releasing factor in chronically catheterized lambs. J. Clin. Invest. 1987; 79: 984–988
- Luger A., Calogero A.E., Kalogeras K., Gallucci W.T., Gold P.W., Loviaux D.L., Chrousos G.P. Interaction of epidermal growth factor with the hypothalamic-pituitary-adrenal axis: potential physiologic relevance. J. Clin. Endocrinol. Metab. 1988; 66: 334–337