Abstract
Objectives
Reactive oxygen species (ROS) are involved in many physiological and pathological processes. In the present study, we analysed whether the synthetic glucocorticoid dexamethasone induces oxidative stress in cultured pancreatic islets and whether the effects of dexamethasone on insulin secretion, gene expression, and viability can be counteracted by concomitant incubation with N-acetylcysteine (NAC).
Methods
ROS production was measured by dichlorofluorescein (DCFH-DA) assay, insulin secretion by radioimmunoassay, intracellular calcium dynamics by fura-2-based fluorescence, gene expression by real-time polymerase chain reaction analyses and cell viability by the MTS assay.
Results
Dexamethasone (Dexa) increased ROS production and decreased glucose-stimulated insulin secretion after 72 hours incubation. Intracellular ROS levels were decreased and the insulin secretion capacity was recovered by concomitant treatment with Dexa + NAC. The total insulin content and intracellular Ca2+ levels were not modulated in either Dexa or Dexa + NAC groups. There was a decrease in the NAD(P)H production, used as an indicator of viability, after dexamethasone treatment. Concomitant incubation with NAC returned viability to control levels. Dexa also decreased synaptotagmin VII (SYT VII) gene expression. In contrast, the Dexa + NAC group demonstrated an increased expression of SYT VII compared to controls. Surprisingly, treatment with NAC decreased the gene expression of the antioxidant enzyme copper zinc superoxide dismutase soluble.
Discussion
Our results indicate that dexamethasone increases ROS production, decreases viability, and impairs insulin secretion in pancreatic rat islets. These effects can be counteracted by NAC, which not only decreases ROS levels but also modulates the expression of genes involved in the secretory pathway and those coding for antioxidant enzymes.
Introduction
Increased oxidative stress and free-radical damage have been proposed to be implicated in diabetes onset and development.Citation1 In type 1 diabetes, reactive oxygen species (ROS) participate in beta-cell death triggered by the autoimmune reaction, a process that involves the mobilization of pro-inflammatory mediators, especially cytokines.Citation1–Citation3 In type 2 diabetes, excessive ROS production impairs insulin secretion, leading to pancreatic islet dysfunction and finally beta-cell death.Citation4,Citation5 On the other hand, controlled ROS production play an important physiological role, signalling for appropriate insulin synthesis and secretion in beta cells.Citation6 Pancreatic beta cells are particularly vulnerable to ROS toxicity, due to their low antioxidant capacity and especially due to their low capacity to detoxify hydrogen peroxide, as these cells possess very low levels of catalase and glutathione peroxidase (GPx).Citation4,Citation7
The diabetogenic action of glucocorticoids (GC) has been extensively studied. GC induces insulin resistanceCitation8 and impairs insulin secretion from pancreatic beta cells.Citation9 Additionally, high levels of GC also lead to an increase in hepatic gluconeogenesis,Citation9,Citation10 which contributes to the diabetic status. However, despite extensive studies, the molecular mechanisms involved in GC-induced diabetes are only partially known.
Recently, our group demonstrated that some of the deleterious effects of dexamethasone (Dexa, a synthetic GC) on insulin-producing cells are mediated by ROS generationCitation7 and hence it is evident that ROS play an important role in GC signalling. However, these effects have not yet been proven in normal growing tissues. In addition, potential therapeutic strategies have not yet been tested so-far in the context of GC toxicity in pancreatic islets. Hence, our objectives in the present study were to extend the results obtained in insulin-producing RINm5F cell line by analysing whether dexamethasone can induce oxidative stress also in cultured pancreatic islets and to test whether the dexamethasone effects on insulin secretion, gene expression, and cellular viability can be counteracted by concomitant incubation with N-acetylcysteine (NAC), a well-established antioxidant. Using this approach we found evidence that dexamethasone increases ROS, and that the use of NAC can ameliorate the lower viability and impaired insulin secretion induced by the corticoid.
Materials and methods
Materials
Dextrose, NaCl, KCl, CaCl2, MgCl2, and NaHCO3 were from Mallinckrodt Baker, Inc (Phillipsburg, NJ, USA). Ethanol, methanol, chloroform, and isopropyl alcohol were from Synth (Diadema, SP, Brazil). Trizol reagent, polymerase chain reaction (PCR) primers and all other real-time PCR reagents were from Invitrogen Corp. (Carlsbad, CA, USA) and MTS ((3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was from Promega Corporation (Madison, WI, USA). Dexamethasone (D4902, first diluted at high concentration in ethanol), NAC (A9165), Hepes, albumin, activated charcoal, dextran, and all other chemicals were from Sigma (St Louis, MO, USA), unless otherwise indicated.
Islet isolation and dexamethasone incubation
Ninety-days-old Wistar rats were provided by the Animal Breeding Centre of the University of Campinas. All experimental procedures were conformed to The Guiding Principles for the Care and Use of Animals (DHEW Publication, NIH 80–23) and were approved by the Ethics Committee of the University of Campinas (CEEA/IB/UNICAMP, protocol number: 1140–1). For each set of experiments, pancreatic islets from adult male Wistar rats were obtained by collagenase digestion of pancreata, isolated by centrifugation over a Ficoll gradient and subsequently hand-collected. After isolation, islets were cultured in RPMI 1640 medium, supplemented with glucose (at desired concentrations), 10% (v/v) foetal calf serum, penicillin, and streptomycin in a humidified atmosphere at 37°C and 5% CO2. Thereafter, cells were exposed for 72 hours to 1 µmol/l of dexamethasone (Dexa group), or dexamethasone plus 1 mmol/l of NAC (Dexa + NAC group). For the insulin secretion and MTS experiments, cells were also exposed to NAC alone. Control islets were kept in the absence of the compounds (Control group). The medium was renewed every other day.
Determination of intracellular ROS by dichlorofluorescein fluorescence
Control and treated islets were washed with Krebs–Hepes buffer. Thereafter islets were placed in black-coloured Costar plates and incubated in Krebs–Hepes buffer containing 25 mmol/l of Glucose and 10 µmol/l of dichlorofluorescein (DCFH-DA) for 30 minutes at 37°C. Fluorescence emission from DCF was detected at 460/530 nm excitation/emission using a fluorometer (Fusion, PerkinElmer Life Science, Waltham, MA, USA). Protein concentration was measured by Bradford. The resulting fluorescence was expressed as ΔUF/μg of protein.
Insulin secretion and total insulin measurements
Groups of five islets were first incubated for 45 minutes at 37°C in Krebs-bicarbonate buffer containing 5.6 mmol/l glucose and equilibrated with 95% O2 – 5% CO2, pH 7.4. The solution was then replaced with 1 ml of fresh Krebs-bicarbonate buffer and the cells were incubated for 1 hour with medium containing 2.8 or 25 mmol/l of glucose. The incubation medium contained: 115 mmol/l NaCl, 5 mmol/l KCl, 24 mmol/l NaHCO3, 2.6 mmol/l CaCl2, 1 mmol/l MgCl2, and 0.3% (w/v) BSA. At the end of the incubation period, aliquots of the medium were collected for insulin determination by radioimmunoassay (RIA) using rat insulin as standard. Samples were normalized by total protein content. For total insulin content, groups of five islets were homogenized by sonication (15 seconds) and centrifuged to remove debris. Supernatants were collected and insulin measured by RIA.
MTS viability assay
In all sets of experiments, the viability of islets was determined after a 4-hour incubation period using a microplate-based MTS kinetic assay, following the instructions contained in the manual (CellTiter 96 Non-Radioactive Cell Proliferation Assays, Promega Corporation, Madison, WI, USA). The principle of the method rely on the measurement of reducing equivalents, i.e. NAD(P)H, produced from the metabolism in viable cells. The results were expressed as area under the curve normalized to total protein content followed by calculation of control percentage.
[Ca2+]i measurement
After 72 hours in culture, groups of cultured control, Dexa- and Dexa plus NAC-treated islets were transferred to plates containing Krebs-bicarbonate buffer with 5.6 mmol/l glucose and 1% albumin, continuously gassed with 95% O2 and 5% CO2, and incubated for 1 hour at 37°C. Thereafter, 5 µmol/l fura2/AM (Invitrogen) was added and the islets incubated for an additional period of 1 hour. Islets were then transferred to a thermostatically regulated open chamber (37°C), placed on the stage of an inverted microscope (Nikon UK, Kingston, UK), and perifused with Krebs-bicarbonate buffer without albumin at a flow rate of 1.5 ml/minute. The solution was continuously gassed with 95% O2 and 5% CO2. A ratio image was acquired approximately every 5 seconds with a CCD camera (Photon Technology International Charge Coupled Device camera, PCO AG, Kelheim, Germany), equipped with 340 and 380 nm, 10 nm bandpass filters. Data were obtained using the ImageMaster3 software (Photon Technology International, Birmingham, NJ, USA).
Real-time PCR
Total cellular RNA was extracted from groups of 500 islets using Trizol reagent. The cDNA was obtained from 3 µg of total RNA using the high-capacity cDNA kit (Invitrogen), according to the manufacturer's instructions. Real-time reactions were made in duplicate using the Fast SYBR Green technology (Invitrogen) in a total volume of 15 µl. Samples were first denatured at 94°C for 10 minutes followed by 30 PCR cycles. Each PCR cycle comprised melting at 95°C for 3 seconds, and annealing-extension at 60°C for 30 seconds. The purity of the amplified PCR products was verified by melting curves. The mRNA expression of selected genes was normalized against the mRNA expression levels of the housekeeping gene, Gapdh. There were no differences in the expressions of Gapdh among all groups when normalized to the beta-actin gene expression (not shown). A list of primers is provided in .
Table 1. Sequences of the primers used for the real-time RT–PCR gene expression quantification
Statistics
All data are expressed as means ± SE. Statistical analyses for each group were performed using one-way analysis of variance (ANOVA) followed by Bonferroni (Prism analysis program, Graphpad, San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
Effects of dexamethasone on ROS generation
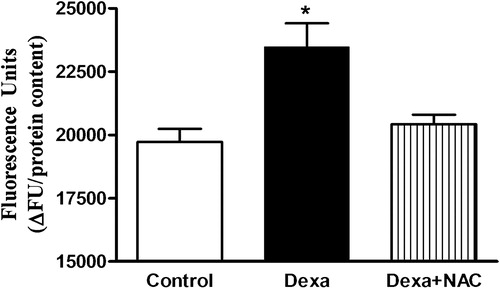
Dexamethasone (1 µmol/l) exposure for 72 hours increased ROS production by 20% compared to control islets, as measured by DCFH-DA oxidation (). Concomitant incubation of pancreatic islets with dexamethasone plus NAC suppressed excessive ROS production.
Figure 1. Effects of Dexa and NAC treatment on ROS generation. Total ROS production was measured by DCFH-DA oxidation. Islets were treated with 1 µmol/l of dexamethasone (Dexa) for 72 hours in the presence or absence of 1 mmol/l of NAC (Dexa + NAC) and assayed in the presence of 25 mmol/l glucose. N = 5–8. *P < 0.05, compared to control; ANOVA followed by Bonferroni.
Effects of dexamethasone on insulin release
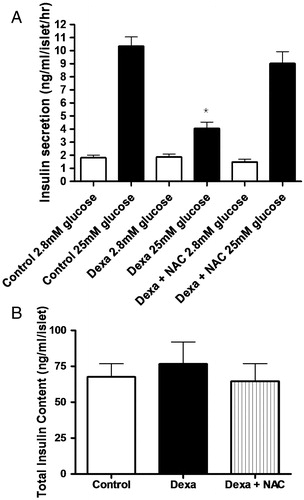
At a non-stimulatory concentration of glucose (2.8 mmol/l), there were no differences in insulin release among control, Dexa, and Dexa plus NAC groups (A). Using stimulatory concentrations of glucose (25 mmol/l), insulin secretion was significantly lower in the Dexa group, compared to control islets (4.1 ± 0.5 and 10.3 ± 0.7 ng/ml/islet/μg of protein, respectively). Concomitant incubation of pancreatic islets with dexamethasone plus NAC recovered insulin secretion capacity of pancreatic islets near to control levels at stimulatory conditions (9.0 ± 0.9 ng of insulin/ml/islet/μg of protein). Of note, treatment with NAC alone did not change insulin secretion (1.25 ± 0.2 and 11.4 ± 1.2 ng/ml/islet/μg of protein, for basal and glucose-stimulated insulin secretion, respectively, n = 12). The total insulin content after 72 hours of exposure to dexamethasone was not different either in Dexa or Dexa plus NAC (B) compared to control islets. Treatment with NAC alone also did not change total insulin content (65.2 ± 7.8 ng of insulin/ml/islet, n = 17). All the values were normalized to total protein content.
Figure 2. (A) Effects of Dexa and NAC treatment on insulin secretion. Islets were treated with 1 µmol/l of dexamethasone (Dexa) for 72 hours in the presence or absence of 1 mmol/l of NAC (Dexa + NAC). After the culture period, groups of five islets were pre-incubated in Krebs-bicarbonate buffer containing 5.6 mmol/l of glucose and then incubated with 2.8 mmol/l of glucose or 25 mmol/l glucose. After 1 hour of incubation, supernatants were collected and insulin was measured by RIA. N = 20–30. *P < 0.05; ANOVA followed by Bonferroni. (B) Effect of Dexa and NAC on total insulin content in cultured rat islets. After 72 hours of culture, groups of five islets were homogenized and the total insulin content was measured by RIA (N = 18).
Effects of dexamethasone treatment on pancreatic islet viability
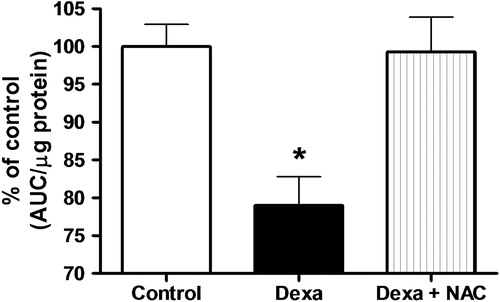
Exposure of pancreatic islets to 1 µmol/l of dexamethasone for 72 hours decreased viability by 20% compared to control cells, as measured by the NAD(P)H production using the MTS test (100 ± 2.9 and 79 ± 3.8% for control and Dexa group, respectively, P < 0.05, N = 5) (). Concomitant incubation of pancreatic islets with dexamethasone plus NAC completely recovered pancreatic islets viability to control levels (99 ± 4.6% of control) (). Although not shown in the picture, treatment with NAC alone slightly but significantly increased NAD(P)H levels (115 ± 2.0% of controls, n = 5, P < 0.05).
Figure 3. Effects of Dexa and NAC treatment on pancreatic islet cell viability. Islets were treated with 1 µmol/l of dexamethasone for 72 hours in the presence or absence of 1 mmol/l of NAC. NAD(P)H production, as an indicator of viability, was accessed using the MTS method. N = 5. *P < 0.05; ANOVA followed by Bonferroni.
Effects of dexamethasone on [Ca2+]i dynamics
[Ca2+]i measurements were accessed using the fura-2 method. The rise in the cytosolic calcium in response to glucose is shown in . No differences were found in glucose-induced calcium changes neither in Dexa nor Dexa plus NAC groups, compared to the control group (). The bars represent mean values for the frequency of oscillations, maximal amplitude, and amplitude of oscillations of intracellular free calcium levels.
Figure 4. Glucose-induced [Ca+2]i changes in pancreatic islets after 72 hours of dexamethasone and Dexa plus NAC treatment. Bars represent mean values of at least five independent experiments.
![Figure 4. Glucose-induced [Ca+2]i changes in pancreatic islets after 72 hours of dexamethasone and Dexa plus NAC treatment. Bars represent mean values of at least five independent experiments.](/cms/asset/f64129f9-0e56-406b-92bd-1c5f9bed27ed/yrer_a_11679302_f0004_b.jpg)
Effect of dexamethasone and NAC on the expressions of genes involved in oxidative stress and exocytosis
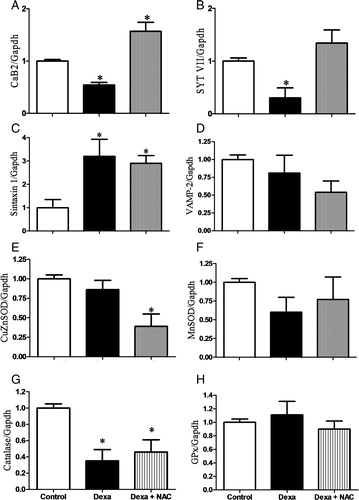
mRNA levels of genes coding for components of both the exocytotic and the calcium sensing machinery were analysed by real-time PCR. Dexamethasone significantly decreased the expression of the calcium channel beta 2 subunit (CaB2) and synaptotagmin VII (SYT VII) genes, compared to controls (A and 5B, respectively). Concomitant incubation of pancreatic islets with dexamethasone plus NAC strongly increased the expression of both genes. Syntaxin 1 expression was increased in Dexa and Dexa plus NAC groups when compared with controls, whereas vesicle-associated membrane protein 2 (VAMP-2) gene expression was not modulated. In addition to the genes that might directly act in the exocytotic pathway, we analysed genes of selected antioxidant enzymes involved in ROS generation/detoxification. Dexamethasone slightly decreased gene expression of catalase without modifying the expression of Cu/ZnSOD (copper zinc superoxide dismutase soluble), MnSOD (manganese superoxide dismutase mitochondrial), and GPx. Similarly, the Dexa plus NAC group had lower catalase expression and no changes in GPx and MnSOD enzymes. However, the Dexa plus NAC group showed lower Cu/ZnSOD expression.
Figure 5. Effects of Dexa and NAC treatment on the gene expressions of (A) calcium channel, voltage-dependent, CaB2, (B) SYT VII, (C) syntaxin 1, (D) VAMP-2, (E) Cu/ZnSOD, (F) MnSOD, (G) catalase, and (H) GPx. After reverse transcription, real-time analyses were performed using Gapdh as an internal control. The experiments were performed after 72 hours of culture. N= 6–8 independent experiments. *P < 0.05, ANOVA followed by Bonferroni.
Discussion
GC mediate toxicity on targeting cells through complex signal cascades, which affect the gene expression profile and are critical for cell death and survival.Citation11 Dexamethasone can be toxic to insulin-producing cells in vitro, through induction of oxidative stress.Citation7 This may explain why GC are neither a preventive immunosuppressant strategy to treat autoimmune diabetes nor an adjuvant for pancreatic islets transplantation. Based on our previous results,Citation7 we hypothesize that excessive H2O2 generated after GC exposure in vitro might be deleterious to beta cells and could thus impair the insulin secretion process. It is thus conceivable that antioxidants might be beneficial in the diabetes context and in the treatment of GC toxicity. To address this question, we analysed whether NAC could counteract the deleterious effects of dexamethasone on pancreatic islets. Our results provide evidence that dexamethasone signals in pancreatic islets through increases in ROS, modulating the expression of genes linked to the insulin exocytosis and to the antioxidant system. The use of NAC improves pancreatic islet function and viability after dexamethasone exposure.
Dexamethasone inhibits insulin release from pancreatic islets in vitroCitation12 (and also this study, A). The impairment in NAD(P)H generation in dexamethasone-treated islets, as shown in this study, might contribute to this damage, since it may indicate both an imbalance in the redox state within the cells and an impaired mitochondrial function. It has been recently proposed that NADPH may couple the Ca2+ levels inside the cells to insulin secretion, an effect that involves the NADPH-dependent protein glutaredoxin-1.Citation13 Our results also show that NAC counteract dexamethasone-dependent decrease in NAD(P)H and restores insulin secretion capacity. Therefore, disturbance in the NAD(P)H levels within cells is one line of interpretation that can partly explain the decrease in insulin secretion in the context of GC toxicity on pancreatic islets, and the recovery of this capacity after NAC treatment is possible because NAC restores the NAD(P)H levels. Since NAC is a precursor of glutathioneCitation14 the enzymatic capacity of enzymes that use glutathione as a cofactor, like GPx and peroxiredoxins (PRDX), are improved. In the diabetes context, it is extremely important, because GPx and PRDX are mainly responsible for H2O2 metabolization as beta cells do not possess significant amounts of catalase.Citation1 Hence, NAC allows cells to cope with higher amounts of H2O2 without losing insulin secretion capacity.
Our results show that while dexamethasone decreases insulin secretion at stimulatory conditions (high-glucose concentration), no changes in basal insulin secretion, intracellular Ca2+ levels and total insulin content are observed after dexamethasone treatment. These results are in line with previous observations showing that the rise in cytoplasmic Ca2+, glucose oxidation and insulin content are not lower in dexamethasone-treated pancreatic islets after a load of glucose.Citation12 Therefore, the deleterious effects of GCs on insulin release by pancreatic islets might reside in distal steps of the secretion process and the decrease in SYT VII, an important calcium sensor involved in stimulated insulin secretion, might be one of the factors responsible for this impairment of insulin release after dexamethasone treatment. Previous studies have already shown that SYT VII-null mutant mice are glucose intolerant and have impaired insulin secretion in vivo, but at the same time islet architecture, insulin content, and the rise in the intracellular calcium in response to glucose are preserved in these animals,Citation15,Citation16 which reinforces the dexamethasone-mediated decrease of SYT VII as a potential target responsible for impaired insulin secretion. Additionally, our results show that NAC treatment does not switch back the increase in Syntaxin 1 gene expression induced by dexamethasone. Syntaxin 1 is involved in the fusion of exocytotic granules to the plasma membraneCitation17 and therefore this increase might be an attempt of the cells to improve pancreatic islet secretion capacity, which might not be enough in cells exposed to dexamethasone alone. As a final outcome, the concomitant NAC treatment is able to recover the insulin secretion capacity of pancreatic islets exposed to dexamethasone.
It is interesting to note that VAMP-2 expression is not modulated either by Dexa or by Dexa plus NAC, indicating that docking granules are probably not affected. Since NAC improves insulin secretion in these islets and, at the same time, increases the expression of genes related to calcium signalling, which are impaired by dexamethasone, it is tempting to speculate that the expression of genes involved in exocytosis might be modulated by the redox status in pancreatic islet.
The antioxidant enzymes neutralize the free radicals formed during cell metabolism,Citation18 and a decrease or increase in antioxidant enzymes activity could be responsible for the excessive ROS generation and, therefore, cell damage.Citation19 The Cu/ZnSOD is a cytoplasmatic/peroxisomal enzyme implicated in the detoxification of superoxide radicals, leading to H2O2 formation.Citation19–Citation21 Therefore, high SOD activity could lead to an excess of intracellular H2O2 when cells are exposed to superoxide generators and at the same time have small capacity for H2O2 metabolization, as is the case of beta cells exposed to dexamethasone,Citation7 since they express low levels of GPx and particularly catalase, two main enzymes in charge of neutralizing peroxides.Citation1 Hence, the lower expression of Cu/ZnSOD in the Dexa plus NAC-treated group could result in lower intracellular H2O2 formation and, therefore, lower toxicity to pancreatic islets.Citation7 It is important to note that we cannot make a final statement on the source of superoxide in this context, although peroxisomal xanthine oxidase might be a plausible candidate.Citation7,Citation20,Citation21
NAC improves insulin signalling in muscle and liver of animal models of insulin resistanceCitation22 and improves glucose uptake in dexamethasone-treated insulin-resistant adipocytes.Citation23 Here, we show that NAC ameliorates dysfunction of islets caused by the diabetogenic GC dexamethasone. All these noxious effects related to diabetes are, in principle, somewhat dependent on excessive ROS generation.Citation7,Citation23 Hence, antioxidant therapy using NAC may be an efficient strategy to block the deleterious effects of GCs. It is of note that other compounds also protect beta cells against GC toxicity, like exendin-4,Citation24 which raises the possibility that they also act through decreasing ROS generation. The very low toxicity of NAC when administered orally and the fact that glutathione-dependent enzymes (GPx and PRDX) are present in several subcellular compartments of pancreatic beta cells are additional advantages of the drug.Citation14 Finally, NAC alone seems not to alter basal or glucose-stimulated insulin secretion in a normal context, what is further evidence of the tolerability of the drug, although we must be cautious with the limitations of in vitro experiments.
In conclusion, in our experimental conditions dexamethasone increases the ROS and decreases viability and glucose-stimulated insulin secretion in cultured pancreatic rat islets. These effects can be efficiently counteracted after NAC treatment. The NAC action is mediated in part through improvement of NAD(P)H production and through modulation of genes involved in insulin secretion and antioxidants enzymes, but not through changes in intracellular calcium handling. These results may contribute to the better understanding of the role of ROS in the mechanisms of the deleterious effects of GC on beta cells, and might provide the basis to delay the onset of autoimmune diabetes by concomitant administration of immunosuppressant doses of dexamethasone alongside NAC.
Conflict of interest statement: The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
Acknowledgments
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Brazil), Fundação Carlos Chagas de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, Brazil) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil). L.P. Roma was the recipient of a CAPES post-graduated fellowship (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil).
References
- Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans 2008;36:343–7.
- Souza KL, Elsner M, Mathias PC, Lenzen S, Tiedge M. Cytokines activate genes of the endocytotic pathway in insulin-producing RINm5F cells. Diabetologia 2004;47:1292–302.
- Souza KL, Gurgul-Convey E, Elsner M, Lenzen S. Interaction between pro-inflammatory and anti-inflammatory cytokines in insulin-producing cells. J Endocrinol 2008;197:139–50.
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 1996;20:463–6.
- Li N, Frigerio F, Maechler P. The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem Soc Trans 2008;36:930–4.
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes 2007;56:1783–91.
- Roma LP, Bosqueiro JR, Cunha DA, Carneiro EM, Gurgul-Convey E, Lenzen S, et al. Protection of insulin-producing cells against toxicity of dexamethasone by catalase overexpression. Free Radic Biol Med 2009;47:1386–93.
- Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 2002;96:23–43.
- Delaunay F, Khan A, Cintra A, Davani B, Ling ZC, Andersson A, et al. Pancreatic beta cells are important targets for the diabetogenic effects of glucocorticoids. J Clin Invest 1997;100:2094–8.
- McMahon M, Gerich J, Rizza R. Effects of glucocorticoids on carbohydrate metabolism. Diabetes Metab Rev 1988;4:17–30.
- Sun Y, Tao YG, Kagan BL, He Y. Modulation of transcription parameters in glucocorticoid receptor-mediated repression. Mol Cell Endocrinol 2008;295:59–69.
- Lambillotte C, Gilon P, Henquin JC. Direct glucocorticoid inhibition of insulin secretion. An in vitro study of dexamethasone effects in mouse islets. J Clin Invest 1997;99:414–23.
- Reinbothe TM, Ivarsson R, Li DQ, Niazi Q, Jing X, Zhang E, et al. Glutaredoxin-1 mediates NADPH-dependent stimulation of calcium-dependent insulin secretion. Mol Endocrinol 2009;23:893–900.
- Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther 2008;8:1955–62.
- Li Y, Wang P, Xu J, Gorelick F, Yamazaki H, Andrews N, et al. Regulation of insulin secretion and GLUT4 trafficking by the calcium sensor synaptotagmin VII. Biochem Biophys Res Commun 2007;362:658–64.
- Gauthier BR, Wollheim CB. Synaptotagmins bind calcium to release insulin. Am J Physiol Endocrinol Metab 2008;295:E1279–86.
- Cunha DA, Roma LP, Boschero AC. Prolactin modulates the association and phosphorylation of SNARE and kinesin/MAP-2 proteins in neonatal pancreatic rat islets. Mol Cell Endocrinol 2007;273:32–41.
- Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J 2007;401:1–11.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007;39:44–84.
- Bonekamp NA, Volkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors 2009;35:346–55.
- Angermuller S, Islinger M, Volkl A. Peroxisomes and reactive oxygen species, a lasting challenge. Histochem Cell Biol 2009;131:459–63.
- Blouet C, Mariotti F, Azzout-Marniche D, Mathe V, Mikogami T, Tome D, et al. Dietary cysteine alleviates sucrose-induced oxidative stress and insulin resistance. Free Radic Biol Med 2007;42:1089–97.
- Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006;440:944–8.
- Ranta F, Avram D, Berchtold S, Dufer M, Drews G, Lang F, et al. Dexamethasone induces cell death in insulin-secreting cells, an effect reversed by exendin-4. Diabetes 2006;55:1380–90.