
Abstract
Objective:
To broaden the ethnic groups in which tapentadol IR is evaluated for treating acute postoperative pain to include Asians.
Methods:
In this phase 3, multicenter, double-blind, randomized study, 352 Korean adults with moderate-to-severe pain following hallux valgus surgery received tapentadol IR 50 or 75 mg or placebo orally every 4–6 hours for 72 hours. Patients requesting other (rescue) analgesics during this period were discontinued for lack of efficacy. The primary endpoint, sum of pain intensity difference (SPID) over 48 hours, was evaluated based on the difference between tapentadol IR and placebo in least squares (LS) mean change from baseline using analysis of covariance (ANCOVA). Secondary endpoints included the time to first rescue medication use and the distribution of responder rates.
Results:
A treatment effect, favoring tapentadol IR, was observed for SPID48 (p < 0.001 for both doses vs. placebo, ANCOVA). The between-group difference (vs. placebo) in LS means of SPID48 was 76.4 (95% CI: 51.0, 101.7) for tapentadol IR 50 mg and 90.6 (95% CI: 65.1, 116.1) for tapentadol IR 75 mg. Time to first rescue medication use was delayed for tapentadol IR (p < 0.001 for both doses vs. placebo; log-rank test). The distribution of responders at 12, 24, 48, and 72 hours favored tapentadol IR (p ≤ 0.001 for both doses vs. placebo; Cochran–Mantel–Haenszel test). Dizziness, nausea, and vomiting were each reported in ≥10% tapentadol-treated patients and at an incidence ≥2-fold higher vs. placebo. The study findings may be limited by study drug dosing every 4 to 6 hours and frequent monitoring during treatment, neither of which mimic pain treatment in clinical practice. However, any potential bias based on this systematic monitoring of patients would be mitigated by the randomized, double-blind nature of the study, with all treatment groups similarly affected by such biases, if any.
Conclusions:
Tapentadol IR reduced acute pain intensity, significantly more than placebo, after orthopedic surgery in Korean patients.
Clinical trial registration:
NCT01516008.
Introduction
Acute pain following surgery is common and associated with important clinical sequelae. Between one and three of every four patients report poorly controlled, moderate to severe pain following surgeryCitation1–6. Ineffective postoperative pain management can increase the risk of a diverse array of complications (e.g., deep vein thrombosis, pulmonary embolism, pneumonia, coronary ischemia, ileus, poor wound healing, insomnia, chronic pain and associated disabilities)Citation7–9, delay discharge from hospital or ambulatory surgery center, result in unscheduled hospital admission/readmission, and prolong postoperative recoveryCitation10–13.
Opiates are the standard of care for moderate to severe postoperative painCitation14. Most opioids used in clinical practice produce analgesia by activating µ-opioid receptors on neurons within the pain transmission pathwayCitation15. However, these treatments are commonly associated with adverse effects, including nausea, vomiting, constipation, dizziness, and somnolenceCitation16, which can lead to undertreatment of the pain, in an attempt to minimize these events.
Tapentadol is a centrally active analgesic with a dual mode of action (i.e., µ-opioid receptor agonism and norepinephrine uptake inhibition)Citation17, distinguishing it from other commercially available opioids. Tapentadol is approved in various regions of the world, such as the US, the EU, Australia, and Canada, as an immediate-release (IR) formulation for the relief of acute pain in adults, as an extended-release (ER) formulation for the management of chronic pain in adults, and, in the US, for neuropathic pain associated with diabetic peripheral neuropathy. Tapentadol is also approved as an oral solution (OS) for the relief of acute pain in adults (US and EU). Clinical trials of patients with various types of moderate to severe acute pain have shown that tapentadol IR provides analgesia comparable to that of the pure μ-opioid agonist, oxycodone IR, with improved gastrointestinal tolerability (lower incidence of nausea, vomiting, constipation)Citation18–22.
Inter-ethnic differences in the pain experience and its treatment have been reported. In some studies, patients of Asian ethnicity described greater postoperative pain intensity, made fewer demands for analgesia, and experienced different rates of opioid-related adverse events (e.g., less respiratory depression, more pruritus), compared to white patientsCitation23–26. Since published studies of tapentadol IR have included primarily white, black, and Hispanic patients, we investigated tapentadol IR’s efficacy and tolerability in Asian patients undergoing orthopedic (hallux valgus) surgery. The results indicate that tapentadol IR is efficacious for reduction of acute postoperative pain intensity in a broader range of ethnic groups than heretofore reported.
Patients and methods
The study design and methods of this phase 3 study conducted in South Korea were similar to those of previously conducted studiesCitation18,Citation19, the key features of which are summarized below.
Ethical practices
An independent ethics committee at each participating hospital reviewed and approved the study protocol. The study was conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, consistent with Good Clinical Practices and applicable regulatory requirements. All patients provided written informed consent before their study participation commenced. The study is registered at clinicaltrials.gov, NCT 01516008.
Patients
Study participants were South Korean men and women aged 20 to 80 years old, inclusive, who were undergoing primary unilateral first metatarsal hallux valgus surgery, which included a distal Chevron osteotomy. Eligibility criteria required that patients were healthy or medically stable, having no history of seizure disorder, active infection, severe renal insufficiency or moderately/severely impaired liver function based on laboratory tests, or acute gout/pseudogout during the 6 months before screening, among others. If female, eligible patients were postmenopausal, surgically sterile, or practicing an effective method of birth control and were not pregnant or lactating; if male and sexually active, eligible patients agreed to use an approved method of birth control (to prevent pregnancy in their female partner). In addition, they had not been treated with systemic corticosteroids within the 4 weeks before screening or anticonvulsants, monoamine oxidase inhibitors (MAOIs), tricyclic antidepressants (TCAs), or neuroleptics within 2 weeks before randomization. Patients were eligible if they had received selective serotonin reuptake inhibitor (SSRI) treatment at a stable dose for at least 30 days before screening.
Study design
This phase 3, randomized, double-blind, placebo-controlled, parallel-group, multicenter study was conducted at 17 sites in South Korea between 11 January 2012 and 2 February 2013. The study consisted of four periods: (1) a screening period during which patients were initially screened for study eligibility (using the criteria described above), (2) a surgical period, (3) a qualification period during which patients’ eligibility for randomization into the double-blind treatment period was assessed, and (4) a 72 hour, double-blind, inpatient treatment period.
In the immediate preoperative period (approximately 1 hour prior to surgery), patients who met study selection criteria received surgical anesthesia utilizing a popliteal sciatic block (PSB). After localization of the sciatic nerve was confirmed, 0.5% ropivacaine was injected to achieve surgical anesthesia and a catheter was placed in the popliteal space for delivery of postoperative anesthesia utilizing a continuous PSB (CPSB) infusion. Hallux valgus surgery was then performed.
Upon completion of surgery and arrival to the postanesthesia care unit (PACU), patients received CPSB infusion of 0.5% mepivacaine (rate maximum of 14 ml/hour), and boluses (maximum of three 10 ml boluses/hour) as needed, through the indwelling catheter into the popliteal space to control postoperative pain and limit the need for supplemental analgesia. If needed, supplemental analgesia (i.e., paracetamol 650 mg orally [PO] and/or ketorolac 30 mg IV every 4–6 hours, or alternatively, fentanyl 100 µg IV) was allowed until the beginning of the qualification period. The continuous infusion of 0.5% mepivacaine was terminated and the catheter was removed at 3 am on the first postoperative day, which marked the beginning of the qualification period. During this period the research staff regularly assessed patients for an increase in pain intensity sufficient to qualify the patient for entry into the double-blind treatment period. Patients were qualified to enter the double-blind treatment period when they rated their pain intensity as ≥4 on an 11 point (0 to 10) numerical rating scale (NRS) after CPSB termination. However, patients could enter the double-blind treatment period no earlier than 10 hours after the first surgical incision yet still be within 9 hours of termination of the CPSB at 3 am on postoperative day 1.
Qualified patients were then randomized to one of three treatment groups – tapentadol IR 50 mg, tapentadol IR 75 mg, or placebo – in a 1:1:1 ratio based on a computer-generated randomization schedule. Randomization was balanced using randomly permuted blocks and was stratified by study site. The total double-blind treatment period was 72 hours in duration. Study drug was administered PO every 4 to 6 hours, with or without food. To enhance retention in the study, patients were allowed to take their second dose of study drug at any time between 1 and 6 hours after the first dose of study drug, only on day 1, in the event of a pain flare. All subsequent doses of study drug were taken every 4 to 6 hours from the time of the previous dose. Thus, each patient could receive up to 19 total doses of study drug during the 72 hour double-blind treatment, including up to seven doses on the first day, and six doses each on days 2 and 3.
The use of analgesic medication other than the assigned study drug was prohibited during the double-blind treatment period, with the exception of low-dose aspirin (≤325 mg/day) taken for cardiac or vascular event prophylaxis. Patients who requested additional analgesic medication (i.e., rescue medication) in addition to study drug for pain relief during the double-blind treatment period were discontinued from the study for lack of efficacy. Metoclopramide 5 to 10 mg IV or intramuscularly (IM), as needed, was allowed for treatment (not prophylaxis) of nausea and vomiting.
Pain assessments during the double-blind treatment period
Using an 11 point NRS (i.e., 0 = no pain to 10 = pain as bad as you can imagine), patients rated their current pain intensity by answering the question, ‘What is your pain level at this time?’, at the following time points: predose, 0.5, 1, 2, 4, 12, 24, 48, and 72 hours after the first dose of study drug and at predose, 0.5, 1, 2, and 4 hours after the second and all subsequent doses of study drug. Patients also rated their level of pain relief at these same time points by responding to the question, ‘How much relief have you had from your starting pain?’, using a 5 point NRS (0 = none to 4 = complete). Pain assessments were avoided between the hours of 11 pm and 5 am to allow patients to sleep, with the exception of the 12, 24, 48, and 72 hour assessments relative to initial study drug administration, which were made regardless of the time of day.
At the conclusion of the double-blind treatment period (or at premature discontinuation), patients rated their overall status since beginning study medication (i.e., global impression of change [PGIC]) using a 7 point NRS (1 = very much improved to 7 = very much worse).
Safety assessments
Adverse events were monitored throughout the study, beginning at screening through the end of the double-blind treatment period (or for 48 hours after the last dose of study drug for patients who discontinued early). Other standard safety assessments (i.e., clinical laboratory tests [hematology, chemistry, urinalysis], 12-lead electrocardiogram (ECG), vital signs, pulse oximetry, physical examination) were also conducted at prespecified time points during the study.
Data analysis
Sample size determination
Based on previously conducted hallux valgus studiesCitation18,Citation19, a between-group difference of 62.4 (1.3 on a 0–10 scale) and a standard deviation (SD) of 134.4 (2.8 on a 0–10 scale) for the sum of pain intensity difference (SPID) over the first 48 hours of treatment with study drug (SPID48) were assumed for sample size calculations. A more conservative SD was assumed compared to the previous studies given the increased number and diversity of study sites. Given these assumptions, a sample size of 116 subjects per group (348 total) would provide 90% power to show at least one tapentadol IR group was statistically different from the placebo group at an overall significance level of 0.05 (0.025 for each comparison).
Efficacy endpoints and analyses
The primary efficacy endpoint was SPID48. Secondary efficacy endpoints included: time to first use of rescue medication; distribution of responder rates based on percentage change in pain intensity from baseline at 12, 24, 48, and 72 hours; PGIC at 72 hours; SPID at 12, 24, and 72 hours; total pain relief (TOTPAR) based on the 5 point NRS pain relief assessment; and the sum of TOTPAR and SPID (SPRID) at 12, 24, 48, and 72 hours.
SPID was calculated as the weighted sum of the pain intensity difference (i.e., difference between baseline pain intensity at the qualifying period and current pain intensity) collected up to the specified time point. The weights were taken as the number of hours elapsed since the previous pain measurement. TOTPAR was calculated in the same manner as SPID using pain relief assessments instead of pain intensity assessment. The last observation carried forward (LOCF) imputation rule was applied for any patient who prematurely discontinued from the double-blind treatment period. Intermittent missing data were imputed using linear interpolation.
Statistical analyses of SPID, TOTPAR, and SPRID were performed using an analysis of covariance (ANCOVA) model with treatment and study site as factors and baseline pain intensity as a covariate. Pairwise comparisons of each tapentadol IR dose versus placebo were estimated based on between-group differences of least squares means and 95% confidence intervals (CI) around the differences. The p-values of the pairwise comparisons for the primary efficacy parameter, SPID48, were adjusted for multiplicity based on the Hochberg procedureCitation27. The p-values for the other SPID parameters and for the TOTPAR and SPRID parameters were not adjusted for multiplicity.
A Kaplan–Meier plot of time to first rescue medication for each treatment group is presented. Each tapentadol IR group was compared to the placebo group using a log-rank test stratified by site. The Hochberg procedureCitation27 was applied to the log-rank test p-values to adjust for multiplicity.
Cumulative distribution curves (‘responder curves’; i.e., the percentage of patients with different degrees of reduction in pain intensity) for each tapentadol IR group were compared to the curve for the placebo group using the log-rank test stratified by site. For the predefined response thresholds of at least 30% and 50% reductions in pain intensity, the tapentadol IR groups were compared to the placebo group using a Cochran–Mantel–Haenszel test stratified by site. In these responder analyses, 0% reduction in pain intensity was assumed for patients who discontinued or used rescue medication prior to the 48 hour time point, whose pain intensity worsened compared to baseline, or whose pain intensity rating was missing at that time.
The distribution of PGIC ratings for each tapentadol IR group was compared to that for the placebo group using the Cochran–Mantel–Haenszel test controlling for site.
Results
Patients
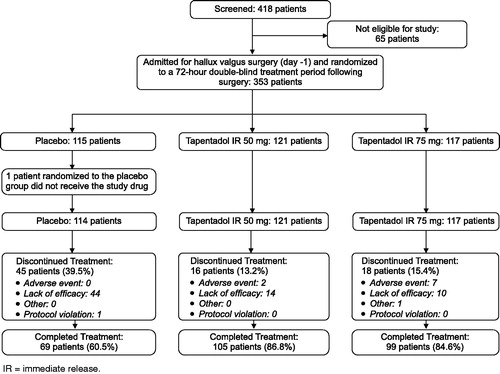
A total of 418 Korean patients were screened for the study, of whom 353 qualified patients were randomized into the double-blind treatment period and 352 received at least one dose of study drug: 114 in the placebo control group, 121 patients in the tapentadol IR 50 mg group, and 117 patients in the tapentadol IR 75 mg group (). All 352 randomized and treated patients were included in the intent-to-treat (ITT) analysis set for efficacy and safety.
Figure 1. Subject disposition.
The three treatment groups were similar based on demographic and other baseline characteristics (). The study population was composed primarily of women (94.0%). The mean (SD) age was 51.3 (12.33) years. The mean (SD) length of surgery was 1.0 (0.39) hour, time from end of surgery to end of CPSB was 13.0 (2.12) hours, and time from end of CPSB to first dose of study drug was 3.1 (1.89) hours. The majority of patients (68.8%) reported severe pain (i.e., pain intensity ≥6 on 11 point NRS) at the baseline pain assessment (taken within 30 minutes prior to randomization).
Table 1. Demographic and baseline characteristics (ITT data set).
Efficacy results
The majority of tapentadol IR treated patients (86.8% for 50 mg; 84.6% for 75 mg) completed the 72 hour double-blind treatment, compared to 60.5% of patients in the placebo group. The most common reason for premature discontinuation of study drug was lack of efficacy (38.6% of patients in the placebo group and 11.6% and 8.5% of patients in the tapentadol IR 50 mg and 75 mg groups, respectively).
A significant improvement in pain intensity was observed in both tapentadol IR dose groups based on the primary efficacy variable, SPID48 (p < 0.001 vs. placebo for both comparisons; ). The between-group differences (vs. placebo) in least squares mean of SPID48 were 76.4 (95% CI: 51.0, 101.7) and 90.6 (95% CI: 65.1, 116.1) for tapentadol IR 50 mg and 75 mg, respectively. (Notably, the larger the value of SPID48, the greater the reduction in pain.) Similar to the SPID48 results, the secondary endpoints of SPID at 12, 24, and 72 hours also showed significant improvement in pain intensity in the tapentadol IR groups versus placebo (p < 0.001 at each time point for both tapentadol IR dose groups; ).
Table 2. Summary of pain intensity and pain relief variables.
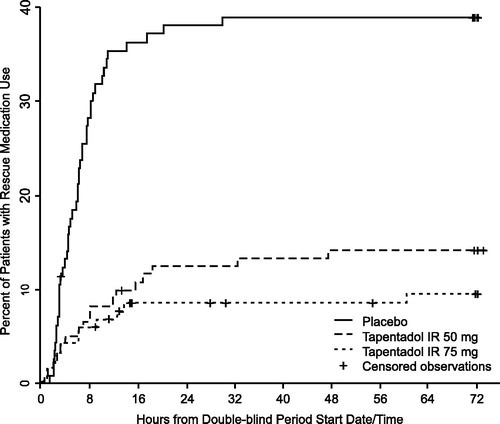
Time to first use of rescue medication was significantly delayed in the tapentadol IR 50 and 75 mg groups compared to the placebo group (p < 0.001; ). More placebo patients (38.6%) required rescue medication for pain relief during the double-blind treatment period, leading to mandatory discontinuation of study drug, than did patients treated with tapentadol IR (14.0% 50 mg; 9.4% 75 mg).
Figure 2. Time to first use of rescue medication.
Across all levels of percentage improvement in pain intensity at 48 hours, a numerically higher percentage of patients in both tapentadol IR dose groups met specific levels of improvement compared to the placebo group (). A significant between-group difference (p < 0.001), favoring tapentadol IR over placebo, was observed for the overall distribution of responder rates at 48 hours. Likewise, the distribution of responders at 12, 24, and 72 hours favored tapentadol IR (p ≤ 0.001 for both dose groups versus placebo).
Figure 3. Distribution of responder rates based on pain intensity at 48 hours.
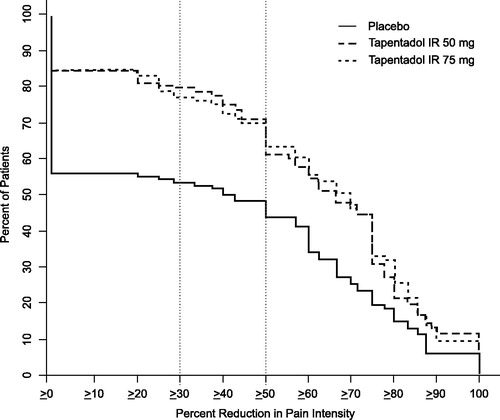
The proportions of patients with ≥30% and ≥50% improvement in pain intensity were significantly higher in the tapentadol IR groups compared with the placebo group at 12, 24, 48, and 72 hours (p < 0.05, ≥50% improvement at 24 hours for 50 mg group; p ≤ 0.001 for all other comparisons and time intervals). In the tapentadol IR groups, 79.3% (50 mg) and 76.9% (75 mg) of patients reported at least a 30% reduction in pain intensity at 48 hours, versus 53.5% for placebo. At least 50% reduction in pain intensity at 48 hours was noted for 71.5%, 70.1%, and 48.2% of patients in the tapentadol IR 50 mg, tapentadol IR 75 mg, and placebo groups, respectively.
The results from the SPID analyses are supported by the results of TOTPAR analyses and SPRID analyses over 12, 24, 48, and 72 hours (). Pain relief was significantly greater in the tapentadol IR treatment groups compared with placebo based on TOTPAR scores and SPRID scores over all these time intervals (p < 0.001 for both tapentadol dose groups and all time intervals).
A significant between-group difference, favoring tapentadol IR (p < 0.001 for both the 50 mg and for 75 mg groups versus placebo), was observed for the distribution in PGIC. The percentage of patients who rated their status as ‘much improved’ or ‘very much improved’ on the PGIC at the end of double-blind treatment was 81.0% for tapentadol IR 50 mg, 83.8% for tapentadol IR 75 mg, and 52.6% for placebo.
Safety results
The proportion of patients with at least one treatment emergent adverse event was numerically higher in the tapentadol IR 50 mg group (56.2%, 68/121) and tapentadol IR 75 mg group (72.6%, 85/117) than in the placebo group (35.1%, 40/114). The most common treatment-emergent adverse events (i.e., reported in ≥5% of patients in any treatment group) were (in decreasing order of incidence for the tapentadol groups combined) nausea, dizziness, vomiting, constipation, and pyrexia (). In all treatment groups, the majority of treatment-emergent adverse events were rated as mild to moderate in intensity. Adverse events led to the discontinuation of 2 (1.7%) patients in the tapentadol IR 50 mg group (due to nausea), 7 (6.0%) patients in the tapentadol IR 75 mg group (due to vomiting [n = 6], nausea [n = 5], dizziness [n = 3], cold sweat [n = 1], and/or hypotension [n = 1]), and no patients in the placebo group. No patient in the placebo and tapentadol 75 mg groups experienced a treatment-emergent serious adverse event. Two patients in the tapentadol 50 mg group each experienced one serious adverse event. The serious events of essential tremor and procedural site reaction occurred 18 and 13 days, respectively, after completion of the study treatment; both of these events resolved and were considered not related to the study drug by the investigators.
Table 3. Treatment-emergent adverse events reported in ≥5% of patients in any treatment group.
No unexpected findings were reported during assessments of clinical laboratory tests, 12-lead ECG, vital signs, pulse oximetry, and physical examination.
Discussion
Adequate postsurgical pain relief is essential, as uncontrolled postoperative pain can increase the risk of complicationsCitation7–9 and delay recovery from surgeryCitation10,Citation11.
We evaluated tapentadol IR for effects on moderate to severe pain after hallux valgus surgery, which is an established pain model for assessments of analgesia, the results of which can be generalized to other surgical proceduresCitation28–31. The model is ideal for evaluating analgesia for a number of reasons. The pain model employs standardized surgical and anesthetic procedures, with less interpatient variability in painCitation28,Citation29,Citation31. Furthermore, patients undergoing hallux valgus surgery experience a predictable and sustained level of moderate to severe pain following the procedure that requires analgesic treatment for several daysCitation28,Citation29,Citation31. The procedure does not involve the viscera, reducing the likelihood of gastrointestinal and respiratory dysfunction due to surgery. Also, the cause of pain during postoperative ambulation can be easily determined.
In our study of Asian patients, significant between-group differences, favoring tapentadol IR versus placebo, were observed for the primary and secondary pain intensity/relief endpoints (all p < 0.001 vs. placebo). The results of pain intensity and pain relief assessments were supported by the study’s other measurements of analgesia efficacy, including time to first use of rescue medication and PGIC, underscoring the robust nature of the study findings. Our results are in line with those of studies of tapentadol IR for treating acute pain in primarily non-Asian patients, showing statistically significant separation from placeboCitation18,Citation19,Citation22. Using similar study methods, Daniels et al. reported a treatment effect with tapentadol IR, based on mean SPID48 values (primary endpoint) of 119.1 for 50 mg and 139.1 for 75 mg (versus 24.5 for placebo; p ≤ 0.001 for each tapentadol dose group vs. placebo) in a study population of primarily white, black, and Hispanic patientsCitation19. The investigators also reported statistically significant between-group differences for pain relief, as assessed by TOTPAR score at time points between 12 and 72 hoursCitation19, that were comparable to those observed in our Asian patients.
High placebo effect is a common observation in pain studies. The placebo response rate in Asian patients treated in this study was 60.5% (i.e., 69 of 114 patients in the placebo group completed the entire 72 hour study), similar to what has been observed by others studying pain following hallux valgus surgery. Daniels et al. reported a 50% placebo response in a study population of primarily white, black, and Hispanic patientsCitation19. In another study conducted by the same research group, in which (non-Asian) patients were allowed to take two doses of acetaminophen in the first 12 hours of the double-blind treatment period, the investigators reported a 71% placebo responseCitation18. Completion rates of up to 97% have been reported for placebo-treated patients who participated in studies utilizing the same postoperative acute pain modelCitation32,Citation33.
The most common treatment-emergent adverse events observed in this study (i.e., nausea, dizziness, vomiting, constipation) are consistent with the tolerability profile of a central-acting analgesic. The rates of gastrointestinal adverse events reported by Asian patients were generally of the same magnitude as has been reported for 50 and 75 mg dosing of tapentadol IR in patients of non-Asian ethnicity. In two such studies cited aboveCitation18,Citation19, the rates were 34–35% and 36–38% for nausea in the 50 mg and 75 mg dose groups, 12–18% and 21–28% for vomiting, and 7–8% and 1–5% for constipation in the respective dose groups. The incidence of the central nervous system event of dizziness was also reported at similar rates by non-Asian patients (15–16% for 50 mg and 22–25% for 75 mg)Citation18,Citation19 to those in our study by Asian patients.
Our study findings may be limited by dosing of study drug every 4 to 6 hours and frequent monitoring during treatment, neither of which mimic pain treatment in clinical practice. However, any potential bias based on this systematic monitoring of patients would be mitigated by the randomized, double-blind nature of the study, with all treatment groups similarly affected by such biases, if any. The study population was composed primarily of women, which reflects the fact that hallux valgus deformity is more problematic for womenCitation34.
In summary, tapentadol IR (50 and 75 mg) reduced acute pain intensity and relieved acute pain, significantly more so than placebo, after orthopedic surgery in Asian patients. The magnitude of improvement in postoperative pain intensity and pain relief achieved with tapentadol IR in patients of Asian ethnicity is comparable to that observed in similarly designed studies that enrolled primarily white, black, and Hispanic patients. Tapentadol IR was well tolerated in the Asian population; the overall safety profile was consistent with that in previous studies of non-Asian patients.
Transparency
Declaration of funding
This study was funded by Janssen Research & Development LLC. The funder was involved in the design, data analysis, and reporting of this study.
Author contributions: H.Y.R., E.J.L., H.L., K.K., and D.S. were involved in study design, medical monitoring, and data review and analysis for the research study. H.S.L., J.S.K., and Y.K.L. were principal investigators of the clinical study. K.K. performed the statistical analysis. All authors contributed to writing drafts of the article. All authors approved the final version of the article, including the authorship list.
Declaration of financial/other relationships
Y.K.L., J.S.K., and H.S.L. have disclosed that they were investigators for this study but have no other significant relationships with or financial interests in any commercial companies related to this study or article. H.Y.R. and E.J.L. have disclosed that they are employees of Jan-Cil Korea, Seoul, South Korea. K.K. and H.L. have disclosed that they are employees of Janssen Research & Development LLC, Titusville, NJ, USA. D.S. has disclosed that he was an employee of Janssen Research & Development LLC, Titusville, NJ, USA at the time this study was conducted and manuscript was written; he is now retired.
CMRO peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Acknowledgments
Writing support was provided by Sandra Norris PharmD of the Norris Communications Group LLC, and funded by Janssen Research & Development. Wendy P. Battisti PhD (of Janssen Research & Development LLC) provided additional editorial assistance.
The authors thank the study participants of this study, without whom the study would never have been accomplished, and the investigators, all from South Korea, for their participation in the study: Byungki Cho MD PhD; Woo Jin Choi MD PhD; Hyung Jin Chung MD PhD; Heui Chul Gwak MD; Hyuk-Soo Han MD PhD; Bio Jeong MD PhD; Honggeun Jung MD PhD; Jin Su Kim MD PhD; Sang Woo Kim MD; Dong Yeon Lee MD PhD; Keun Bae Lee MD PhD; Woochun Lee MD PhD; Hyung Taek Park MD; Yong Wook Park MD; and Ki-Sun Sung MD PhD.
References
- Apfelbaum JL, Chen C, Mehta SS, Gan TJ. Postoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanaged. Anesth Analg 2003;97:534-40
- Beauregard L, Pomp A, Choinière M. Severity and impact of pain after day-surgery. Can J Anaesth 1998;45:304-11
- Dolin SJ, Cashman JN, Bland JM. Effectiveness of acute postoperative pain management: I. Evidence from published data. Br J Anaesth 2002;89:409-23
- McGrath B, Elgendy H, Chung F, et al. Thirty percent of patients have moderate to severe pain 24 hr after ambulatory surgery: a survey of 5,703 patients. Can J Anaesth 2004;51:886-91
- McHugh GA, Thoms GM. The management of pain following day-case surgery. Anaesthesia 2002;57:270-5
- Rawal N, Hylander J, Nydahl PA, et al. Survey of postoperative analgesia following ambulatory surgery. Acta Anaesthesiol Scand 1997;41:1017-22
- Carr DB, Goudas LC. Acute pain. Lancet 1999;353:2051-8
- MacIntyre PE, Schug SA, Scott DA, et al.; on behalf of the Working Party of the Australian and New Zealand College of Anaesthetists. Acute Pain Management: Scientific Evidence, 3rd edn. Melbourne, Australia: Australian and New Zealand College of Anaesthetists and Faculty of Pain Medicine, 2010. Available at: http://www.nhmrc.gov.au/guidelines/publications/cp104 [Last accessed 29 April 2014]
- Perkins FM, Kehlet H. Chronic pain as an outcome of surgery. A review of predictive factors. Anesthesiology 2000;93:1123-33
- Awad IT, Chung F. Factors affecting recovery and discharge following ambulatory surgery. Can J Anaesth 2006;53:858-72
- Chung F, Ritchie E, Su J. Postoperative pain in ambulatory surgery. Anesth Analg 1997;85:808-16
- Coley KC, Williams BA, DaPos SV, et al. Retrospective evaluation of unanticipated admissions and readmissions after same day surgery and associated costs. J Clin Anesth 2002;14:349-53
- Gold BS, Kitz DS, Lecky JH, Neuhaus JM. Unanticipated admission to the hospital following ambulatory surgery. JAMA 1989;262:3008-10
- American Society of Anesthesiologists Task Force on Acute Pain Management. Practice guidelines for acute pain management in the perioperative setting: an updated report by the American Society of Anesthesiologists Task Force on Acute Pain Management. Anesthesiology 2012;116:248-73
- Chevlen E. Opioids: a review. Curr Pain Headache Rep 2003;7:15-23
- Wheeler M, Oderda GM, Ashburn MA, Lipman AG. Adverse events associated with postoperative opioid analgesia: a systematic review. J Pain 2002;3:159-80
- Tzschentke TM, Christoph T, Kögel B, et al. (−)-(1R,2R)-3-(3-Dimethylamino-1-ethyl-2-methyl-propyl)-phenol hydrochloride (tapentadol HCl): a novel μ opioid receptor agonist/norepinephrine reuptake inhibitor with broad-spectrum analgesic properties. J Pharmacol Exp Ther 2007;323:265-76
- Daniels S, Casson E, Stegmann JU, et al. A randomized, double-blind, placebo-controlled phase 3 study of the relative efficacy and tolerability of tapentadol IR and oxycodone IR for acute pain. Curr Med Res Opin 2009;25:1551-61
- Daniels SE, Upmalis D, Okamoto A, et al. A randomized, double-blind, phase III study comparing multiple doses of tapentadol IR, oxycodone IR, and placebo for postoperative (bunionectomy) pain. Curr Med Res Opin 2009;25:765-76
- Hale M, Upmalis D, Okamoto A, et al. Tolerability of tapentadol immediate release in patients with lower back pain or osteoarthritis of the hip or knee over 90 days: a randomized, double-blind study. Curr Med Res Opin 2009;25:1095-104
- Hartrick C, Van Hove I, Stegmann JU, et al. Efficacy and tolerability of tapentadol immediate release and oxycodone HCl immediate release in patients awaiting primary joint replacement surgery for end-stage joint disease: a 10-day, phase III, randomized, double-blind, active- and placebo-controlled study. Clin Ther 2009;31:260-71
- Stegmann JU, Weber H, Steup A, et al. The efficacy and tolerability of multiple-dose tapentadol immediate release for the relief of acute pain following orthopedic (bunionectomy) surgery. Curr Med Res Opin 2008;24:3185-96
- Faucett J, Gordon N, Levine J. Differences in postoperative pain severity among four ethnic groups. J Pain Symptom Manage 1994;9:383-9
- Houghton IT, Aun CS, Gin T, Lau JT. Inter-ethnic differences in postoperative pethidine requirements. Anaesth Intensive Care 1992;20:52-5
- Konstantatos AH, Imberger G, Angliss M, et al. A prospective cohort study comparing early opioid requirement between Chinese from Hong Kong and Caucasian Australians after major abdominal surgery. Br J Anaesth 2012;109:797-803
- Zhou HH, Sheller JR, Nu H, et al. Ethnic differences in response to morphine. Clin Pharmacol Ther 1993;54:507-13
- Hochberg Y. A sharper Bonferroni procedure for multiple significance testing. Biometrika 1988;75:800-3
- Desjardins PJ, Shu VS, Recker DP, et al. A single preoperative oral dose of valdecoxib, a new cyclooxygenase-2 specific inhibitor, relieves post-oral surgery or bunionectomy pain. Anesthesiology 2002;97:565-73
- Thipphawong JB, Babul N, Morishige RJ, et al. Analgesic efficacy of inhaled morphine in patients after bunionectomy surgery. Anesthesiology 2003;99:693-700, discussion 6A
- Desjardins PJ, Black PM, Daniels S, et al. A randomized controlled study comparing rofecoxib, diclofenac sodium, and placebo in post-bunionectomy pain. Curr Med Res Opin 2004;20:1523-37
- Desjardins PJ, Traylor L, Hubbard RC. Analgesic efficacy of preoperative parecoxib sodium in an orthopedic pain model. J Am Podiatr Med Assoc 2004;94:305-14
- Apfelbaum JL, Desjardins PJ, Brown MT, Verburg KM. Multiple-day efficacy of parecoxib sodium treatment in postoperative bunionectomy pain. Clin J Pain 2008;24:784-92
- Pollak R, Raymond GA, Jay RM, et al. Analgesic efficacy of valdecoxib for acute postoperative pain after bunionectomy. J Am Podiatr Med Assoc 2006;96:393-407
- Coughlin MJ, Jones CP. Hallux valgus: demographics, etiology, and radiographic assessment. Foot Ankle Int 2007;28:759-77