Abstract
Recent research has identified an important role for a cystine–glutamate antiporter (system Xc) in the biology of malignant brain tumors. This transporter is effectively inhibited by sulfasalazine, a drug widely used to treat a number of chronic inflammatory conditions such as Crohn’s disease. Preclinical data show that sulfasalazine is an effective inhibitor of tumor growth and tumor-associated seizures. These studies suggest that the cystine–glutamate antiporter is a valuable drug target for which tumor-specific drugs can be generated. In the meantime, sulfasalazine may be considered as adjuvant treatment for malignant gliomas.
In spite of decades of research, primary brain tumors, that is, gliomas, carry a dismal prognosis and have few treatment options. Surgery, radiation and temozolomide chemotherapy are the standard of care, yet for high-grade gliomas these therapies only provide an average patient survival of 12 – 14 months following diagnosis. In many glioma patients, unexplained seizures are an early symptom, with 80% of patients suffering epileptic seizures over the course of their illness. While tumor-associated seizures present a serious co-morbidity, the underlying cause is not well understood. Recent research suggests that the seizures may be caused by the neurotransmitter l-glutamate, which is released from the tumor and overactivates excitatory amino acid receptors present on peritumoral neurons Citation[1].
This conclusion is supported by several independent studies. Glioma cells in culture assiduously release glutamate Citation[2] and only those cells that secrete glutamate produce tumors when implanted into host animals Citation[3]. These gliomas also induce excitotoxic pathology and edema Citation[4]. In glioma patients, glutamate sampled by microdialysis reached 100 – 400 μM, approximately 100-fold above normal cerebrospinal fluid (CSF) levels Citation[5] and sufficient to cause widespread glutamate-mediated excitotoxicity Citation[2]. It has been hypothesized that glutamate release by gliomas serves to vacate space for the growing tumor by destroying tumor-associated neurons and possibly glial cells as well. However, glutamate has also been shown to act as a pleiotropic growth factor that supports tumor growth and invasiveness through activation of Ca2+ permeable AMPA (2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid) receptors, activation of AKT, MAPK and focal adhesion kinases Citation[6].
The search for the mechanism(s) through which glutamate is released from gliomas has converged on system Xc (SXC), a heterodimeric antiporter in which the subunit responsible for transport activity (xCT) is an SCL7A11 family member. SXC is highly expressed in gliomas and its expression is enhanced under hypoxic conditions typical of tumor tissue Citation[7]. The primary role for SXC in normal and cancerous tissue has been linked to the import of cystine (the oxidized form of cysteine) as a precursor for glutathione (g-glutamyl-cysteinyl-glycine; GSH) synthesis as well as for balancing the levels of cystine and cysteine Citation[8]. GSH is the major free thiol in mammalian cells and serves as an important intracellular reducing agent. Intracellular GSH protects tumor cells not only from endogenously generated free radicals but also from radicals generated in conjunction with radiotherapy. Furthermore, GSH is often required for the successful removal of xenobiotics, including chemotherapeutics, via the efflux of drug conjugates through the multi-drug resistance transporters. Therefore, enhanced concentrations of GSH, typical of many cancers, contribute to both radio- and chemo-resistance. Since the supply of cystine via SXC can be rate limiting for GSH production, SXC becomes an ‘Achilles heel’ for cellular protection in these tumors and hence a potentially valuable antitumor drug target.
Studies of native and recombinantly expressed SXC transporters have identified a number of glutamate and cystine analogs that block SXC uptake assays, acting as either alternative substrates or non-transportable inhibitors. These include ibotenate, quisqualate Citation[9] and a number of phenylglycine Citation[2] derivatives of which S-4-carboxyphenylglycine (S-4CPG) is the most potent. Computational modeling has been used to generate ligand-based pharmacophore models that provide insight into the requisite positioning of the backbone and functional groups (e.g., carboxylate, amino and aryl moieties) needed to effectively bind to SXC Citation[10]. When extrapolated from the ligand overlays to the protein transporter itself, the modeling suggests that variations in the exact positioning of the carboxylate and amino groups of the ligands are tolerated within the binding site, which is surprising considering the site equally accommodates glutamate and cystine as substrates. From a functional perspective, it appears that while the site consists of a larger number of contact points, ranging from those that interact with the charged carboxylate and amino groups to those that bind lipophilic aryl groups on novel inhibitors, the individual ligands need only to contact a subset of the domains in order to effectively bind and block SXC activity. For example, the increased potency observed when aryl groups are appropriately positioned on an isoxazole ring, demonstrates that interacting with the one or both of the lipophilic domains can compensate for the less than optimal binding of the carboxylate and amino group mimics Citation[11]. Developing ligands that interact with these lipophilic domains provides a strategy not only to increase potency, but to also insure that the compounds can bind to the transporter but be inactive as substrates (i.e., be translocated).
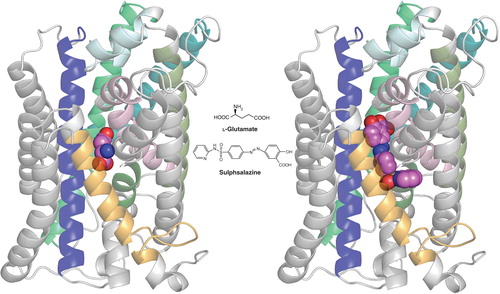
More recently, sulfasalazine (SAS, tradename: Azulfidine) has emerged as an effective clinically approved SXC inhibitor Citation[12]. This drug has been a mainstay in the treatment of Crohn’s disease and ulcerative colitis and is now more broadly used to treat chronic inflammation in the gut, joints and retina. SAS was designed to be cleaved in the intestine by azoreductases into the antibiotic 5-aminopyridine and the anti-inflammatory aspirin-derivative 5-aminosalicylic acid (5-ASA) to target the chronic bacterial infection and inhibition of cyclooxygenases, respectively. Studies have since shown that SAS is also effective in inhibiting the nuclear transcription factor NF-kB and as such, has been used for a number of disease conditions where NF-kB involvement was suspected. In vitro SXC competition assays confirm SAS to be among the most potent inhibitors identified, exhibiting an IC50 in the low µM range that is at par with S-4-CPG. When assessed within ligand pharmacophore models, SAS was predicted to interact with a number of defined domains, even though it lacks an obvious biostere mimicking the prototypical -amino acid scaffold. Its postulated contact with the previously discussed lipophilic domains would be consistent with its action as a non-transportable inhibitor. More recently, a protein homology model of SXC has been generated by threading the xCT sequence over the crystal structure of the related ApcT transporter from Methanococcus jannaschi Citation[13]. While still very preliminary, this homology model has provided a template for initial docking studies. As shown in , the postulated binding site for SAS extends beyond that of glutamate, with the distal pyridine group helping to define the boundaries of one of two proposed lipophilic domains adjacent to the substrate site.
Figure 1. Homology model of xCT with docked ligands. The model was established using the crystal structure coordinates of the amino acid, polyamine and organocation transporter (ApcT) from the RCSB protein databank (3GIA). Protein sequences of human xCT and ApcT were aligned using ClustalW after which the sequences were threaded and the pictured homology model of xCT generated using the default automodel process of MODELLER v9.9. Left shows the xCT model with glutamate docked, right shows it with sulfasalazine docked. The ligand (hot pink) structures were energy minimized using Tripos Sybyl (MMFF forcefield) and docked into the xCT homology model using CCDC GOLD and default settings (GoldScore) system. Modeling was carried out in the University of Montana Molecular Computational Core Facility, in collaboration with S. Patel, N. Natale, M. Braden and J. Gerdes. TMDs 2, 4, 6, 9, 11 and 12 of xCT have either been removed or colored light grey to permit a better view of ligand docking. TMDs shown in color include TMI (light pink), TM3 (dark blue), TM5 (teal), TM7 (olive), TM8 (bright green) and TM10 (gold). Note that only xCT is shown, as it is the subunit responsible for substrate transport. CD98, the regulatory subunit is required for membrane association and is not illustrated.
Importantly, for SAS to inhibit SXC the molecule must be intact, as neither of its cleavage products shows inhibitory activity at SXC Citation[14]. Note however that 5-ASA, which is also used clinically under the name Mesalamine inhibits the activity of NF-kB. This of course poses a challenge for the use of SAS as inhibitor of SXC when systemic or even central nervous system (CNS) administration is required. Bioavailability studies in humans suggest that only approximately 12% of the drug escapes colonic cleavage after oral administration and enters the systemic circulation. How much of the drug enters the brain is unknown, although in the context of gliomas SAS must cross the blood–brain barrier (BBB), or enter the brain through a compromised BBB, since the tumor responds to SAS treatment. Tumor growth is reduced by 80% in vivo following i.p. administration of SAS in tumor-bearing mice Citation[15]. The drug also appears to act rapidly as acute drug administration inhibits peritumoral seizures within minutes. A second consideration pertains to the half-life of the SAS once in the system. Reported data from rodents suggest a half-life between 80 and 180 min Citation[16] albeit in humans half-life in serum was reported to be 10.2 h Citation[17]. Consistent with the former, the antiseizure effects in mice lasted only between 2 and 3 h Citation[1].
Given the important role of SXC in glioma progression and the absence of any effective drugs, the exploratory use of SAS is warranted. Studies to date have shown promise in animal models of glioma to both inhibit tumor growth Citation[15] and epileptic events attributable to glutamate release Citation[1]. These epileptic events present a common and early symptom in patients with glioma and in many instances seizures continue to occur spontaneously, a condition referred to as tumor-associated epilepsy. Until recently, it had been suspected that compression of brain tissue by an expanding tumor mass initiates these seizures. Now it appears more likely that glutamate release via SXC plays at least a contributory role. The observed inhibition of epileptic events by SAS is exciting, as the drug can be readily used in clinical studies given it is FDA approved. Indeed, SAS has already been used in a small Phase I study that was terminated because the drug showed no apparent benefit Citation[18]. Patients enrolled in this study were, however, in very poor health (average Karnofsky score = 50) and had only weeks to live, making these results difficult to interpret. Thus, for a limited Phase I safety study the conclusion of a lack of efficacy is misleading. We believe that SAS should be re-examined in adjuvant use, possibly with radiation and/or temozolomide in glioma patients who are in better health (Karnofsky > 70), through a well-powered study to achieve unequivocal data on its therapeutic efficacy. In light of its short half-life, it behooves us to consider SAS a lead compound to develop more specific and potent inhibitors with increased bioavailability. Several groups have embarked on generating such compounds and early indications suggest that SXC-specific high-affinity drugs that are not subject to bacterial degradation can be generated Citation[11,19]. Until such novel compounds gain clinical approval, however, we would suggest it appropriate to pursue SAS as adjuvant chemotherapeutic reagent in glioma patients.
Expert opinion
Seizures are a common co-morbidity in brain tumor patients. Previously these had been attributed to irritation or compression of brain tissue by the growing tumor. Instead, considerable evidence now supports the notion that epilepsy in patients with primary brain tumors is not caused exclusively by tumor growth, but also by the release of glutamate through SXC that subsequently activate peritumoral neuronal glutamate receptors. SXC can be effectively inhibited in vivo using SAS, one of the more potent non-transportable substrates yet identified. Although SAS has other targets, including NF-kB it appears to preferentially affect glutamate release from tumors through an action on SXC. Peritumoral seizures also indicate active tumor expansion in which the glutamate is likely creating additional room for the tumor expansion by triggering excitotoxic neuronal necrosis. This may be important to the biology of these cancers, as their growth is otherwise constrained by the skull. Hence, the opportunity exists to slow tumor growth and reduce tumor-associated epilepsy using SAS, which is already a clinically approved drug to treat Crohn’s disease. Previously, a limited Phase I study focused on reducing NF-kB activity was carried out with SAS in a small number of glioma patients. This study was halted owing to a lack of efficacy for risk reduction with no potential for benefit. Unfortunately, the poor neurological status of these study subjects likely prevented the potential beneficial effects of SAS as a SXC blocker to be observed and highlights the need for such studies in less compromised patients. Recent human microdialysis data showing high peritumoral glutamate burden in patients with gliomas, together with convergent studies in mouse models in vivo suggesting a reduction of excitotoxicity, edema and seizure burden suggest the use of SAS as an early intervention strategy in patients. Ideally, SAS should be considered in conjunction with known disease-altering strategies, most notably radiation and/or temozolomide. Radiation effects in particular may be further enhanced because the inhibition of cystine uptake should deplete cellular GSH level and enhance glioma sensitivity to ionizing radiation.
Declaration of interest
The authors state no conflict of interest. This work was supported by the NIH (RO1 NS052634).
Bibliography
- Buckingham SC, Campbell SL, Haas BR, Glutamate release by primary brain tumors induces epileptic activity. Nat Med 2011;17(10):1269-74
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res 1999;59:4383-91
- Takano T, Lin JH, Arcuino G, Glutamate release promotes growth of malignant gliomas. Nat Med 2001;7:1010-15
- Savaskan NE, Heckel A, Hahnen E, Small interfering RNA-mediated xCT silencing in gliomas inhibits neurodegeneration and alleviates brain edema. Nat Med 2008;14:629-32
- Marcus HJ, Carpenter KL, Price SJ, Hutchinson PJ. In vivo assessment of high-grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol 2010;97:11-23
- de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia 2011;59(8):1181-9
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 1999;274:11455-8
- Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol 2008;215:593-602
- Powis G, Briehl M, Oblong J. Redox signalling and the control of cell growth and death. Pharmacol Ther 1995;68:149-73
- Bridges RJ, Natale NR, Patel SA. System x(c) (-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol 2012;165:20-34
- Patel SA, Rajale T, O'Brien E, Isoxazole analogues bind the system xc- transporter: structure-activity relationship and pharmacophore model. Bioorg Med Chem 2010;18:202-13
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 2001;15:1633-40
- Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a Na+-independent amino acid transporter. Science 2009;325:1010-14
- Chung WJ, Sontheimer H. Sulfasalazine inhibits the growth of primary brain tumors independent of nuclear factor-kappaB. J Neurochem 2009;110:182-93
- Chung WJ, Lyons SA, Nelson GM, Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci 2005;25:7101-10
- Zheng W, Winter SM, Mayersohn M, Toxicokinetics of sulfasalazine (salicylazosulfapyridine) and its metabolites in B6C3F1 mice. Drug Metab Dispos 1993;21:1091-7
- Azadkhan AK, Truelove SC, Aronson JK. The disposition and metabolism of sulphasalazine (salicylazosulphapyridine) in man. Br J Clin Pharmacol 1982;13:523-8
- Robe PA, Martin DH, Nguyen-Khac MT, Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer 2009;9:372
- Shukla K, Thomas AG, Ferraris DV, Inhibition of xc transporter-mediated cystine uptake by sulfasalazine analogs. Bioorg Med Chem Lett 2011;21:6184-7